")
Back to Journals » Open Access Journal of Clinical Trials » Volume 9
Efficacy and safety of two fast-absorbing formulations of paracetamol in combination with caffeine for episodic tension-type headache: results from two randomized placebo- and active-controlled trials
Authors Yue Y, Reed KD, Shneyer L, Liu DJ
Received 2 February 2017
Accepted for publication 11 April 2017
Published 26 June 2017 Volume 2017:9 Pages 41—57
DOI https://doi.org/10.2147/OAJCT.S133629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Greg Martin
Yong Yue,1 Kenneth D Reed,1 Lucy Shneyer,2 Dongzhou J Liu3
1GlaxoSmithKline Consumer Healthcare, Parsippany, NJ, 2Shneyer Statistics LLC, Denville, NJ, 3GlaxoSmithKline, Collegeville, PA, USA
Objectives: Two randomized placebo-controlled trials evaluated whether combining rapid-acting paracetamol formulations with caffeine resulted in faster/greater relief of episodic tension-type headache (ETTH) compared with placebo and over-the-counter (OTC) analgesics. Both studies were prematurely terminated.
Materials and methods: In the single-blind crossover study 1, adults with ETTH (n=66) received three of the following in random sequence: paracetamol 1,000 mg with sodium bicarbonate 650 mg and caffeine 130 mg; paracetamol 1,000 mg; ibuprofen 400 mg; and placebo. In the double-blind parallel-group study 2, adults with ETTH (n=157) were randomly assigned 2:2:1 to treat up to three headaches with paracetamol with Optizorb technology 1,000 mg plus caffeine 130 mg; ibuprofen 400 mg; and placebo.
Results: In study 1, the primary outcome – mean time to perceptible pain relief – was 36.7, 38, 48.9, and 42.7 minutes in the paracetamol–sodium bicarbonate–caffeine, ibuprofen, paracetamol, and placebo groups, respectively; differences were not statistically significant. In study 2, the weighted sum of pain intensity (scale of 0 [no pain] to 4 [severe pain]) difference from the time of treatment to hour 4, the primary outcome, showed numerically favorable differences for paracetamol with Optizorb–caffeine compared with ibuprofen (difference in least square means −0.3, 95% confidence interval −1.05 to 0.45) and placebo (−0.47, 95% confidence interval −1.36 to 0.42). In both studies, secondary outcomes suggested faster and greater relief with rapid paracetamol–caffeine compared with placebo and paracetamol; a few of these outcomes achieved statistical significance at ~45–90 minutes in study 1. Adverse events were mostly mild and consistent with known safety profiles of OTC analgesics and caffeine.
Conclusion: Firm conclusions regarding the speed and efficacy of rapid-acting paracetamol formulations plus caffeine compared with placebo and traditional OTC analgesics for ETTH cannot be drawn, because the studies were terminated early. Some encouraging trends seen suggest this combination deserves further investigation.
Keywords: paracetamol, caffeine, ibuprofen, tension-type headache, pain
Introduction
Tension-type headache (TTH) is the most common headache disorder worldwide.1 Up to 38% of adults are affected by episodic TTH (ETTH) annually, with peak prevalence occurring in people between 30 and 39 years of age and those with higher levels of education.2 International Classification of Headache Disorders diagnostic criteria for ETTH include bilateral pressing or tightening nonpulsating pain of mild–moderate intensity that lasts 30 minutes to 7 days.3 ETTH may be accompanied by either photophobia or phonophobia (but not nausea), and is not aggravated by physical activity.3 ETTH is further subdivided into infrequent ETTH (ten or more episodes occurring <1 day/month average) and frequent ETTH (ten or more episodes occurring 1–14 days per month on average for at least 3 months).3 Activation of myofascial nociceptors may contribute to muscle pain and acute TTH, and after repeated episodes the central nervous system may become sensitized, resulting in chronic headaches.4
ETTH is usually self-treated with over-the-counter (OTC) analgesics, such as paracetamol (acetaminophen) or nonsteroidal anti-inflammatory drugs (NSAIDs).5,6 Paracetamol is one of the most commonly used analgesic medications worldwide.7 A Cochrane review of 23 studies of paracetamol for treatment of frequent ETTH showed that a single dose of paracetamol 1,000 mg provides modest pain relief compared with placebo (relative risk of being pain-free at 2 hours 1.3, 95% confidence interval [CI], 1.1–1.4), with a safety profile similar to that of placebo.7 Findings from a meta-analysis of six studies of paracetamol compared with NSAIDs in patients with ETTH suggest that the efficacy of paracetamol is equivalent to NSAIDs, with a better tolerability profile than high-dose NSAIDs.8
Efficacy and time to onset of action are important considerations when choosing a medication for relief of ETTH.6 Efforts have been made to further enhance paracetamol’s efficacy and time to onset in treating ETTH. For example, when paracetamol is formulated with sodium bicarbonate, the rate of absorption is considerably increased,9 and onset of action begins within 15 minutes of dosing.10 In addition, GlaxoSmithKline Consumer Healthcare (Dungarvan, Ireland) has developed a formulation of paracetamol using proprietary Optizorb technology, which has a faster rate of absorption and less interpatient variability compared with standard paracetamol (data on file). Adding caffeine as an adjuvant to common analgesics, including paracetamol, has been shown to provide a small but significant increase in relief from various types of acute pain.11 More specifically, randomized double-blind studies have found that caffeine enhances the analgesic efficacy of paracetamol in patients with ETTH.12
Here, we report results from two multicenter, randomized, placebo-controlled studies of patients with ETTH to evaluate the efficacy of a combination of paracetamol and caffeine using two different formulations designed to enable faster absorption of paracetamol in combination with caffeine: one that included sodium bicarbonate (study 1) and one that used paracetamol with Optizorb (study 2). Key objectives of these studies were to compare the efficacy and speed of pain relief with these two new rapid-acting paracetamol-plus-caffeine formulations versus placebo and ibuprofen in ETTH.
Materials and methods
Study design and procedures
Study 1 (ClinicalTrials.gov identifier: NCT01755702) was a Phase IIB, multicenter, randomized, single-blind, partial-dummy, four-way crossover study in participants with ETTH conducted from July 2009 to March 2010 at seven clinic sites in the USA. Study 2 (ClinicalTrials.gov identifier: NCT01842633) was a Phase III, multicenter, randomized, three-arm, parallel-group, double-blind, double-dummy, placebo-controlled study in participants with ETTH conducted from April 2013 to March 2015 at 20 study sites in the US.
In both studies, participants who met initial eligibility criteria at screening entered a run-in phase lasting 2–4 weeks in study 1 and 4 weeks in study 2, during which they characterized their headache-pain intensity and self-treated with their usual OTC analgesics, recording scores in an “eDiary”. To characterize pain intensity, study 1 used a 5-point categorical scale (0 no headache, 1 mild, 2 moderate, 3 moderately severe, 4 severe), and study 2 used a 4-point categorical scale (0 no headache, 1 mild, 2 moderate, 3 severe). After the run-in phase, those who met additional eligibility criteria were entered into the treatment phase, which lasted up to 8 weeks in study 1 and up to 6 weeks in study 2. All eligibility criteria are described in the “Study populations” section.
Subjects entering the treatment phase were sequentially assigned a randomization number; the randomization schedules were computer-generated by the Biostatistics and Data Management Department of GlaxoSmithKline Consumer Healthcare (Parsippany, NJ, US). During the treatment phase, subjects characterized the pain intensity of all headache episodes using the same rating scale.
In study 1, subjects sequentially treated each of the first three headache episodes that met qualifying criteria programmed into the eDiary (defined below) with a single dose of an assigned treatment. Each patient’s treatment assignment consisted of three of the following four regimens in random sequence: two paracetamol 500 mg–sodium bicarbonate 325 mg–caffeine 65 mg caplets plus two placebo ibuprofen caplets; two paracetamol 500 mg caplets plus 2 placebo paracetamol–sodium bicarbonate–caffeine caplets; two ibuprofen 200 mg caplets plus 2 placebo paracetamol–sodium bicarbonate–caffeine caplets; and two placebo paracetamol–sodium bicarbonate–caffeine caplets plus two placebo ibuprofen caplets. The study design was partial dummy, because it used placebos matched to paracetamol–sodium bicarbonate–caffeine and ibuprofen but not to standard paracetamol. Investigators were fully blinded, but participants were partially blinded, because each received one instead of two different placebos per treatment. Subjects were given a treatment kit containing their assigned three treatments in three separate envelopes.
In study 2, subjects treated the first three headache episodes that met the qualifying criteria (defined below) each with a single dose of their one assigned study medication. Therefore, in contrast to study 1, where subjects used three different treatments once each to treat a single headache, participants in study 2 used the same treatment regimen to treat up to three headaches. Study 2 subjects were randomly assigned at a ratio of 2:2:1 to two rapid-acting paracetamol (Panadol with Optizorb technology; GlaxoSmithKline Consumer Healthcare, Dungarvan, Ireland) 500 mg plus caffeine 65 mg caplets plus two placebo ibuprofen caplets; two ibuprofen 200 mg caplets plus two placebo paracetamol with Optizorb–caffeine caplets; or two placebo paracetamol with Optizorb–caffeine caplets plus two placebo ibuprofen caplets.
In both studies, the eDiaries contained a series of questions aimed at identifying headaches that qualified for study treatment. Qualifying headaches were required to be of at least moderate intensity (ie, pain intensity ≥2). Participants were instructed not to treat headaches with study treatment if they had signs/symptoms consistent with migraine or had consumed food or caffeine within 2 hours, rescue medication within 18 hours, or unapproved OTC or prescription medication in the past 48 hours. Headaches that occurred during menstruation were also excluded from treatment. Participants in both studies also had to agree that they would be able to adhere to study restrictions and complete headache assessments over the next 4 hours before being prompted to administer study treatment.
Following each administration of study treatment, participants assessed headache-pain intensity and pain relief (assessment times, study 1: 15, 30, 45, 60, 90, 120, and 240 minutes posttreatment; study 2: 10, 15, 20, 25, 30, 40, 50, 60, 90, 120, 180, and 240 minutes posttreatment). Pain intensity was assessed using the same 5-point (study 1) and 4-point (study 2) categorical rating scales described for the run-in period. The pain-relief scale (PRS) in both studies consisted in 0 representing no relief, 1 a little relief, 2 some relief, 3 a lot of relief, and 4 complete relief. Such rating scales are well accepted and widely used in the pain literature.
In study 1, time to first perceptible pain relief was assessed using a stopwatch included in the eDiary. In study 2, time to first perceptible pain relief was determined by asking participants at the first assessment time when PRS score was ≥1 (at least a little relief) to estimate the time to the nearest minute since the previous assessment when at least a little headache relief was first achieved. In study 2, at 120 minutes posttreatment, participants assessed whether or not they had achieved complete headache relief (yes/no). In both studies, at approximately 4 hours posttreatment of each qualifying headache (or at the time of initial rescue-medication use), participants evaluated their global impression of treatment in the eDiary using a 5-point scale where 0 represented very poor, 1 poor, 2 neutral, 3 good, and 4 very good.
Use of rescue and other medications
During the run-in and treatment phases of both studies, subjects were permitted to use aspirin 1,000 mg as needed as rescue medication for headaches that did not respond adequately to study medication within 2 hours and to treat any headaches that did not qualify for use of the blinded study medication. Participants were instructed not to treat a qualifying headache with study medication within 48 hours of treating a previous qualifying headache; in such cases, they could use rescue medication.
Participants were to refrain from taking OTC or prescription medications, except for oral contraceptives, hormone-replacement therapy, and medications deemed appropriate by the study investigator, unless necessary to treat a new medical condition. Participants were required to maintain their normal routines regarding tobacco use and caffeine intake to avoid withdrawal headaches, and to abstain from consuming alcohol during the first 4 hours of treating each qualifying headache with study medication. Study 1 also required participants to avoid eating for 2 hours after treating a qualifying headache.
Ethical considerations
Both studies were approved by an institutional review board or independent ethics committee at each study site (Thomas Jefferson University Institutional Review Board, Philadelphia, PA; Western Institutional Review Board, Olympia, WA; Biomedical Research Alliance of New York, Lake Success, NY) and were conducted in accordance with requirements specified in the Declaration of Helsinki. All participants provided written informed consent.
Study populations
The patient populations in both studies were similar and largely recruited via local advertising. Both studies enrolled men and women aged 18–65 years with ETTH who were in good general health and able/willing to comply with study procedures and restrictions. Enrollment was limited to those with body mass index 18–35 kg/m2 in study 1 and 18–33 kg/m2 in study 2. In addition, ETTH had to occur on average ≥2 days per month in study 1 and 1–14 days per month in study 2, had to have an onset at age <50 years and ≥12 months prior to study initiation, and had to occur frequently during the 3 months prior to the study (defined as four or more and ten or fewer episodes per month in study 1 and two or more and 14 or fewer episodes per month in study 2). Study 1 required that headaches be bilateral, nonpulsatile, tight band, pressing, or tightening, with no exacerbation by exercise, in accordance with the International Headache Society (IHS) classification of ETTH.3,13 In both studies, the subject’s typical ETTH had to be at least moderate in severity, last ≥4 hours if untreated, and respond to OTC analgesics in ≤2 hours. Females of childbearing potential had to be using a reliable method of contraception.
Both studies excluded women who were pregnant or breast-feeding and persons with chronic TTH (ie, >15 headaches per month for 3 months) or hypersensitivity, allergy, intolerance, or contraindications to any study-medication ingredient. Additional exclusion criteria included history of migraine (more than two episodes per month in study 1, more than one episode per month in study 2); inability to differentiate between migraine and TTH; need for migraine prophylaxis; current psychiatric disease requiring treatment, cognitive disorder, or chronic pain disorder; current or recent (≤3 months) use of medications or herbal supplements that could interfere with study assessments; and alcohol or substance abuse within the last 2 years. Study 1 also excluded persons with a history of severe-headache episodes with neurological disability, and study 2 excluded people with a history of alcohol use that exacerbated headaches.
Subjects in both studies still had to meet these criteria and also meet additional criteria after the run-in phase, in order to undergo randomization and enter the treatment phase. In study 1, during the run-in period, they had to have had at least two headache episodes, at least one of which met the qualifying criteria. The qualifying headache also had to be at least somewhat relieved by the subject’s usual OTC analgesic within 2 hours of administration. In addition, subjects had to have ≥60% compliance with the eDiary and had to have adhered to the restrictions outlined in the “Use of rescue and other medications” section. A protocol amendment to study 1 allowed for exceptions to the 60%-compliance requirement at the investigator’s discretion if the subject showed improvement in compliance, underwent supplemental eDiary training, and provided reassurance that he/she would fulfill the expectations.
In study 2, participants who had had one or more qualifying headaches, were not suspected of having chronic TTH, demonstrated proficiency using the eDiary, and confirmed receiving at least “some” headache pain relief from their usual OTC analgesics within 2 hours of treatment for at least 50% of their qualifying headaches during the run-in phase were eligible for randomization. In addition, they had to exhibit at least 70% compliance with the eDiary during the run-in; subjects with <80% compliance during the run-in and treatment phases received supplemental training.
Study outcomes
In study 1, the primary end point was time (in minutes) to first perceptible pain relief (score of 1 on the 5-point PRS) with paracetamol–sodium bicarbonate–caffeine versus placebo. In study 2, the primary end point was the weighted sum of pain-intensity difference (SPID) from the time of treatment to hour 4 (SPID0–4) for paracetamol with Optizorb–caffeine versus placebo. In study 2, pain-intensity difference (PID) was calculated by subtracting the baseline pain-intensity value from the postbaseline values. SPID was calculated as the products of PID and the amount of time between the current and previous time points (ie, ΣPID × [timet − timet–1]).
Secondary end points in study 1 included PID from baseline to each assessment time (calculated by subtracting the postbaseline pain-intensity value from the baseline value); PRS at each assessment time; SPID, total pain relief (TOTPAR), and area under the time–response curve for change in headache intensity and headache relief (SPRID) at 60, 90, 120, and 240 minutes; number of headaches resolved at 1 and 2 hours before any rescue medication, time to rescue medication, and global impression of treatment. SPID was calculated as the sum of the products of PID with the amount of time between the current and previous time points (ie, ΣPID × [timet – timet–1]). TOTPAR was the sum of the products of PRS with the amount of time between the current and previous time points (ie, ΣPRSt × [timet – timet–1]). SPRID was calculated as the sum of TOTPAR and SPID. The number of resolved headaches was calculated as a proportion of subjects with complete relief (PRS 4) and a rating of “no headache” (pain-intensity rating 0) divided by the total number of subjects.
Secondary end points in study 2 included SPID from time of treatment to hours 1, 2, and 3, time to perceptible pain relief (PRS ≥1); time to meaningful pain relief (PRS ≥2); PID and PRS at each assessment time; TOTPAR and SPRID from time of treatment to hours 1, 2, 3, and 4; the proportion of participants with complete relief at 1 hour (based on PRS 4) and 2 hours (based on “yes” answer to “Do you have complete relief?”) posttreatment; time to and rate of rescue-medication use; and global impression of treatment. TOTPAR was the sum of the products of PRS with the amount of time between the current and previous time points in fractions of an hour (ie, ΣPRSt × [timet – timet–1]/60). As in study 1, SPRID was sum of TOTPAR and SPID.
Because of the difference in how PID was calculated between the 2 studies, it should be noted that positive PID and SPID values in study 1 and negative PID and SPID values in study 2 are indicative of favorable reductions in pain.
In both studies, safety was assessed based on the frequency and severity of treatment-emergent adverse events (TEAEs), serious TEAEs, and their relationship to study medication.
Statistical analyses
The safety population in both studies consisted of all participants who were randomized and received any study medication. The intent-to-treat (ITT) population was also defined in the same way in both studies, and consisted of participants who received at least one dose of study treatment for a qualifying headache and had at least one postbaseline efficacy assessment.
Sample-size calculations were performed in both studies to determine the number of evaluable participants needed to provide 80% power to show significant differences between treatment groups for the primary end points. For study 1, it was estimated that 450 subjects would need to be screened to enroll and randomize 300 and have at least 240 be evaluable. For study 2, it was estimated that 550 participants would need to enter the run-in phase to have 290–300 qualified to be randomized to treatment and 265 evaluable study completers. However, enrollment in both studies was terminated before the planned numbers of participants were randomized. Study 1 was stopped due to business reasons; study 2 was stopped based primarily on enrollment difficulties and impending expiration of drug supplies.
In study 1, Kaplan–Meier curves were generated for time to first perceptible headache-pain relief (PRS ≥1). Differences in time to first perceptible headache-pain relief between treatment groups were compared using a Cox proportional-hazard model with treatment as a factor and baseline pain intensity as a covariate. An analysis of covariance (ANCOVA) model was used for analysis of PRS, headache-pain intensity, SPID, TOTPAR, and area under the time–response curve. The model included factors for participants and period as random effects and for treatment formulations as a fixed effect; baseline pain intensity and site were included as covariates. All analyses were carried out using SAS version 8.2.
In study 2, results for each relevant outcome were averaged over all qualifying headaches for each patient. ANCOVA was used to obtain least squares (LS) means for all SPID, PID, PRS, TOTPAR, and SPRID outcomes; treatment and pooled study sites were fixed effects, and baseline pain intensity was a covariate. An ANCOVA model with treatment and pooled site as fixed factors was used for analysis of the global impression of treatment. Based on an amendment to the statistical analysis plan resulting from the study’s early termination, efficacy was determined based on the 95% CIs for the differences in LS means from the ANCOVA analyses, rather than the originally planned inferential statistics (P-values). Time to first perceptible relief (PRS ≥1) and time to meaningful relief (PRS ≥2) were analyzed using a Cox proportional-hazard model with treatment group, site, and baseline pain intensity as covariates; hazard ratios and 95% CIs were used for treatment comparisons. Kaplan–Meier curves were also generated for these outcomes. The proportion of participants with complete headache relief at 1 and 2 hours posttreatment was analyzed using a 2×2 χ2 test. All analyses were conducted using SAS version 9.2. In both studies, all available data were included for subjects who withdrew prematurely; after rescue medication use, no subsequent PRS was included in the analyses.
Results
Subject flow and baseline characteristics
Study 1 was terminated after 228 people were screened and 66 randomly assigned to treatment (Figure 1A). All 66 treated at least one headache and were included in the safety and ITT populations (28% of planned evaluable participants). In the second study, 365 people were screened and 165 randomly assigned to treatment (Figure 1B). Of those, 157 treated at least one qualifying headache and were included in the safety and ITT populations (59% of planned evaluable participants).
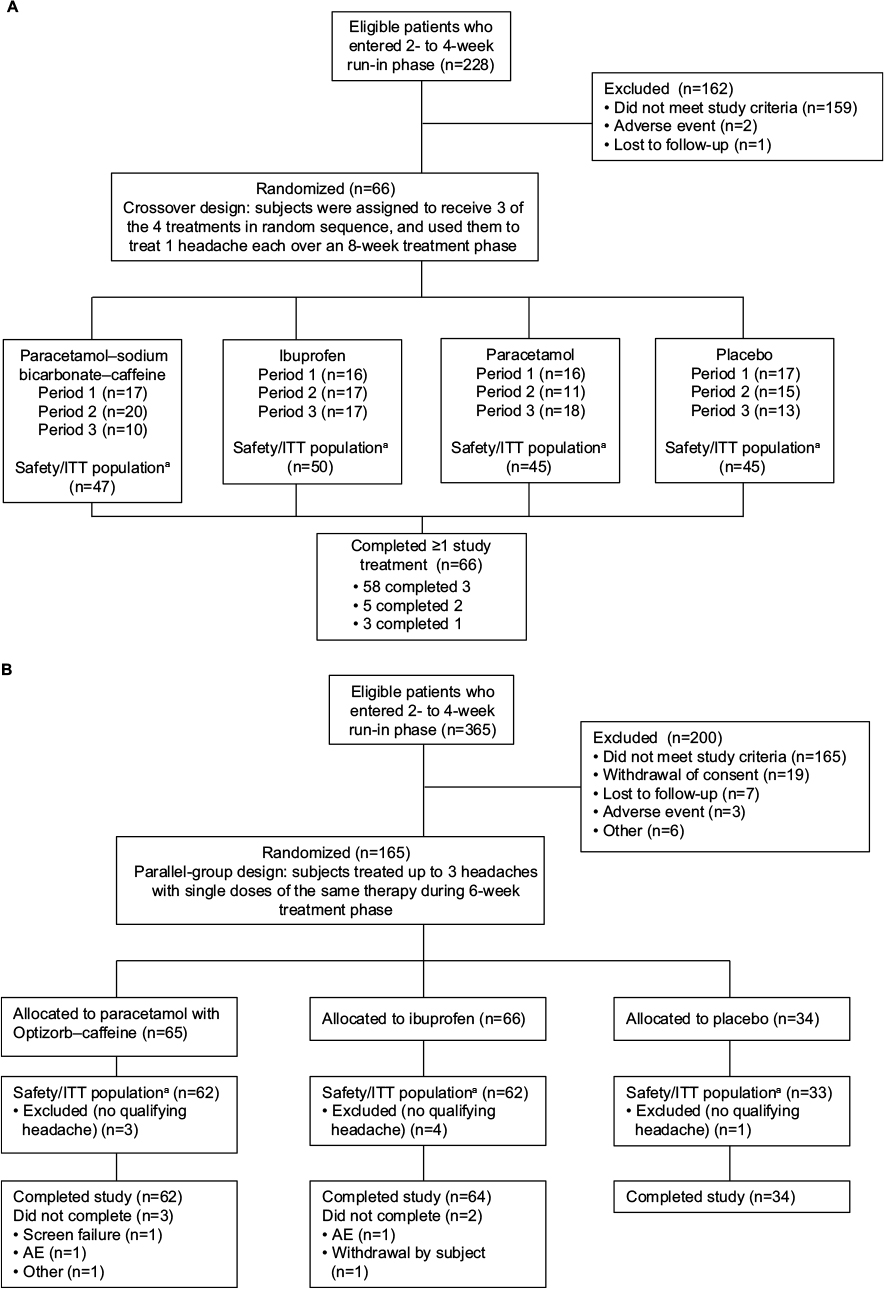 | Figure 1 (A) Subject disposition, study 1; (B) subject disposition, study 2. Notes: aIn both studies, the safety population consisted of all participants who were randomized and received any study medication. In study 1, the intent-to-treat (ITT) population consisted of participants who received at least one study treatment and had at least one postbaseline efficacy assessment. In study 2, the ITT population consisted of those who had at least one evaluable headache during the treatment phase, received at least one dose of study medication, and had at least one postbaseline pain-intensity assessment. Abbreviations: ITT, intent to treat; AE, adverse event. |
In study 1, the mean age of participants was 42 years, 67% were female, 56% were white, and 44% were African-American (Table 1). Mean pain intensity and the proportion with moderate versus severe pain severity at baseline were similar across the four treatment arms. In study 2, mean age was 39 years, 72% were female, 89% were white, 9% were African-American, and 1% each were Asian or of multiple races (Table 2). Demographic and baseline clinical characteristics were similar across treatment groups.
 | Table 1 Demographics and baseline characteristics, study 1, safety/ITT population Notes: aParticipants rated their baseline headache severity using the eDiary on a 5-point categorical scale (0, no headache; 1, mild headache; 2, moderate headache; 3, moderately severe headache; 4, severe headache); bmoderate group included those with pain rating of 2, severe group those with pain rating of 3 or 4; headaches with ratings of 0 or 1 were not considered qualifying headaches for treatment. Abbreviations: ITT, intent to treat; SD, standard deviation. |
 | Table 2 Demographics and baseline characteristics for study 2, safety/ITT population Notes: aParticipants rated their baseline headache severity using the eDiary on a 4-point categorical scale (0, no headache; 1, mild headache; 2, moderate headache; 3, severe headache). Headaches with ratings of 0 or 1 were not considered qualifying headaches for treatment. Abbreviations: ITT, intent to treat; SD, standard deviation. |
Study 1 efficacy
For the primary outcome (Figure 2), mean (median) time to perceptible pain relief (PRS 1) was 36.7 (30), 38 (30), 48.9 (30), and 42.7 (30) minutes in the paracetamol–sodium bicarbonate–caffeine, ibuprofen, paracetamol, and placebo groups, respectively. There were no clinically relevant differences between groups (hazard ratio for paracetamol–sodium bicarbonate–caffeine versus paracetamol 1.32, 95% CI 0.86–2.01; hazard ratio for paracetamol–sodium bicarbonate–caffeine versus placebo 1.25, 95% CI 0.83–1.9).
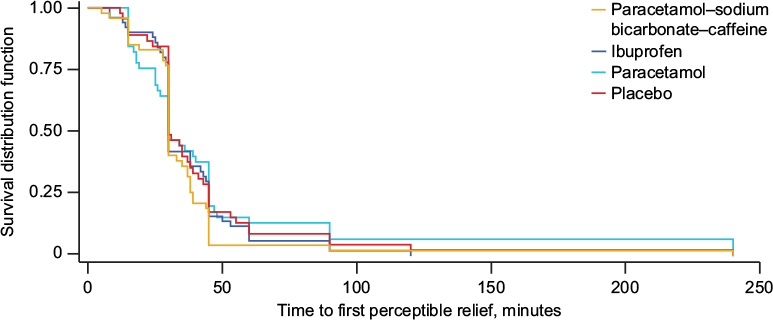 | Figure 2 Time to perceptible pain relief (primary outcome), Kaplan–Meier curve, study 1, intent-to-treat population. Notes: Hazard ratios (95% confidence interval) for comparison with placebo were 1.25 (0.83–1.9) for paracetamol–sodium bicarbonate–caffeine, 1.08 (0.72–1.61) for ibuprofen, and 0.95 (0.62–1.45) for paracetamol. |
There were some indications of greater headache relief and reduced headache severity with paracetamol–sodium bicarbonate–caffeine compared with placebo and standard paracetamol in the early phase of headache, demonstrated by multiple measurements. PID scores (Table S1) were greater (indicating greater reductions from baseline in pain severity) with paracetamol–sodium bicarbonate–caffeine versus placebo at 45 minutes (LS mean difference between treatments 0.45, 95% CI 0.11–0.8; P=0.0103) and 60 minutes (0.4, 95% CI 0.07–0.72; P=0.0176) and with paracetamol–sodium bicarbonate–caffeine versus paracetamol at 60 minutes (0.41, 95% CI 0.08–0.73; P=0.0153). A greater difference in PRS (Table S1) between paracetamol–sodium bicarbonate–caffeine and placebo was observed at 60 minutes (LS mean difference 0.59, 95% CI 0.14–1.03; P=0.0109). PRS was also greater with paracetamol–sodium bicarbonate–caffeine compared with paracetamol at 45 minutes (LS mean difference 0.42, 95% CI 0.02–0.82; P=0.0394) and at 60 minutes (0.78, 95% CI 0.33–1.23; P=0.0009). Trends in headache relief were supported by results for SPID (Figure 3) and TOTPAR (Figure 4); results for SPRID showed a pattern consistent with results for SPID and TOTPAR. Paracetamol–sodium bicarbonate–caffeine showed greater differences compared with both placebo and paracetamol on these three outcomes at 0–60 and 0–90 minutes.
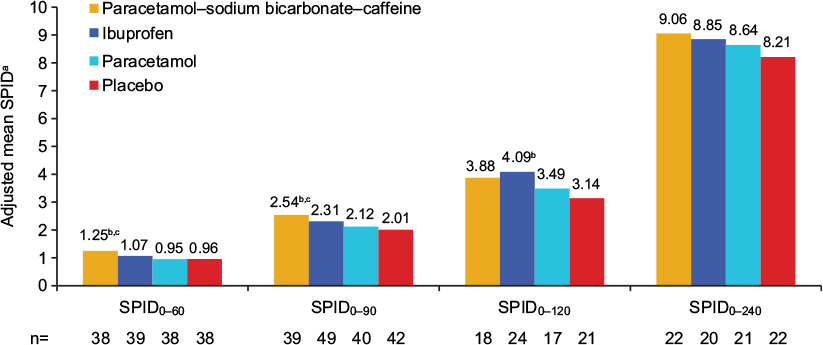 | Figure 3 Adjusted mean SPID0–60, SPID0–90, SPID0–120, and SPID0–240 (secondary outcomes), study 1, ITT population. Notes: aAdjusted means are least squares means from ANCOVA adjusted for baseline pain intensity. Positive values indicate reduction in pain. bP<0.05 vs placebo; cP<0.05 vs paracetamol. Abbreviations: SPID, sum of pain-intensity difference (numeric subscript ranges indicate minutes); ITT, intent to treat; ANCOVA, analysis of covariance. |
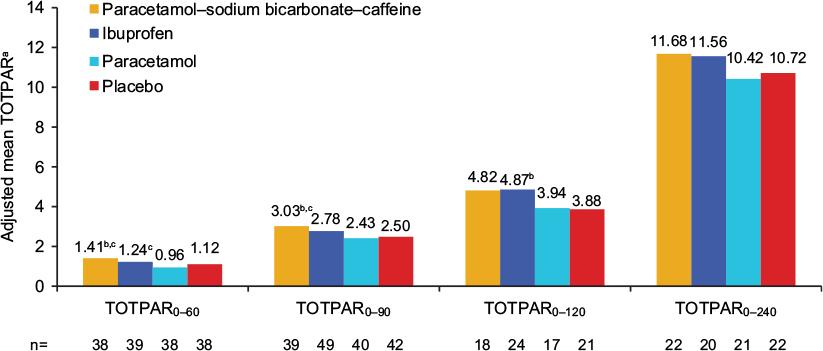 | Figure 4 Adjusted mean TOTPAR (secondary outcome), study 1, ITT population. Notes: aAdjusted means are least squares means from ANCOVA adjusted for baseline pain intensity. bP<0.05 vs placebo; cP<0.05 vs paracetamol. Abbreviations: TOTPAR, total pain relief (numeric subscript ranges indicate minutes); ITT, intent to treat; ANCOVA, analysis of covariance. |
At 1 hour after treatment administration, the proportion of participants who reported their headaches were completely resolved (PRS 4 + pain-intensity rating 0) with paracetamol–sodium bicarbonate–caffeine was more than double that with other treatments (Table 3). Only a small number of participants (three or fewer in each treatment group) required rescue medication during the study. Global impression of treatment-response results were similar across treatment groups, with the majority (≥70%) of participants in each group rating their response as “good” or “very good” (Table 3).
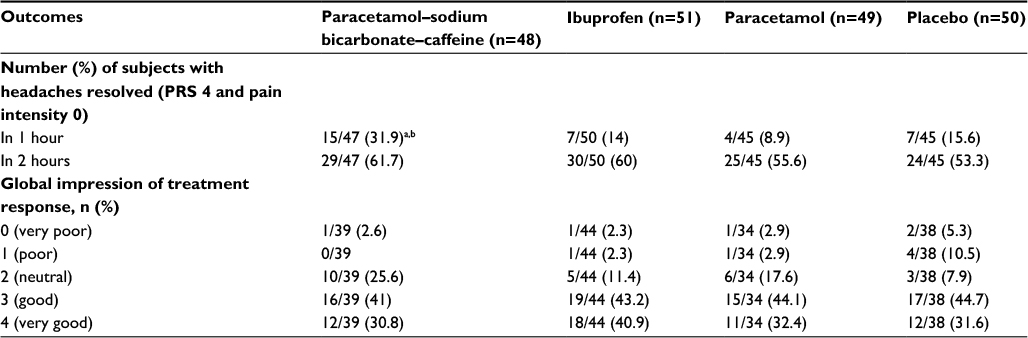 | Table 3 Number of subjects with headache resolution and global impression of treatment (secondary efficacy outcomes), study 1, ITT population Notes: aP<0.05 vs paracetamol; bP<0.05 vs ibuprofen. Numbers of patients shown in the column headings represent the full ITT population; sample sizes for the individual end points varied, due to missing data at some time points for some outcomes. Abbreviations: ITT, intent to treat; PRS, pain-relief score. |
Study 2 efficacy
Results for SPID0–4, the primary efficacy outcome, are summarized for paracetamol with Optizorb–caffeine compared with ibuprofen and placebo (Figure 5). Similar results were observed for SPID0–1, SPID0–2, and SPID0–3 (Figure 5). PID results at each individual time point (data not shown) showed a pattern consistent with those summarized for SPID.
 | Figure 5 Adjusted mean SPID0–1, SPID0–2, SPID0–3 (secondary outcomes), and SPID0–4 (primary outcome), study 2, ITT population. Notes: aAdjusted means and confidence limits for treatment differences from ANCOVA model with treatment and pooled site as fixed factors and baseline intensity as covariate. Negative values indicate reduction in pain. Treatment differences shown are for paracetamol with Optizorb–caffeine versus either ibuprofen or placebo such that a negative result favors paracetamol with Optizorb–caffeine. Abbreviations: SPID, sum of pain-intensity difference (numeric subscript ranges indicate hours); ITT, intent to treat; ANCOVA, analysis of covariance; CI, confidence interval. |
TOTPAR scores (Figure 6) are summarized for paracetamol with Optizorb–caffeine, ibuprofen, and placebo. PRS scores at each individual time point (data not shown) showed a pattern generally consistent with those summarized for TOTPAR, and SPRID scores showed a pattern consistent with SPID and TOTPAR.
 | Figure 6 Adjusted mean TOTPAR0–1, TOTPAR0–2, TOTPAR0–3, and TOTPAR0–4 (secondary outcomes), study 2, ITT population. Notes: aAdjusted means and confidence limits for treatment differences from ANCOVA model with treatment and pooled site as fixed factors and baseline intensity as covariate. Treatment differences shown are for paracetamol with Optizorb–caffeine versus either ibuprofen or placebo such that a positive result favors paracetamol with Optizorb–caffeine. Abbreviations: TOTPAR, total pain relief (numeric subscript ranges indicate hours); ITT, intent to treat; ANCOVA, analysis of covariance; CI, confidence interval. |
Survival curves were generated for time to first perceptible relief (Figure 7A) and time to meaningful pain relief (Figure 7B) for paracetamol with Optizorb–caffeine (mean 37.3 and 65.1 minutes, respectively), ibuprofen (mean 43.3 and 68.8 minutes, respectively), and placebo (mean 40.6 and 74.4 minutes, respectively).
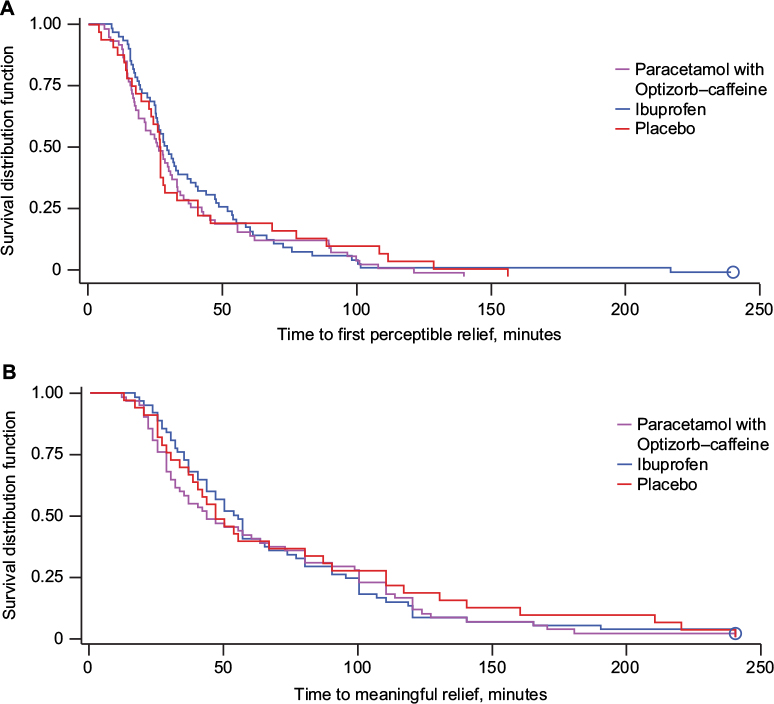 | Figure 7 (A) Time to perceptible pain relief and (B) time to meaningful pain relief (both secondary outcomes), Kaplan–Meier curves, study 2, intent-to-treat population. Notes: Time to perceptible relief: hazard ratio (95% confidence interval [CI]) for comparison with placebo was 1.12 (0.72–1.73) for paracetamol with Optizorb–caffeine and 0.97 (0.62–1.52) for ibuprofen; hazard ratio (95% CI) for paracetamol with Optizorb–caffeine compared with ibuprofen was 1.17 (0.81–1.67). Time to meaningful relief: hazard ratio (95% CI) for comparison with placebo was 1.11 (0.72–1.73) for paracetamol with Optizorb–caffeine and 1.09 (0.69–1.73) for ibuprofen; hazard ratio (95% CI) for paracetamol with Optizorb–caffeine compared with ibuprofen was 1.03 (0.71–1.48). Circles represent censored data. |
In the paracetamol with Optizorb–caffeine, ibuprofen, and placebo groups, respectively, 29%, 27.4%, and 18.2% had complete headache relief (PRS 4) at 1 hour after dosing (Table 4). Fewer participants in the paracetamol with Optizorb–caffeine group required rescue medication than in the ibuprofen and placebo groups (Table 4). Time to rescue-medication use was similar in all groups (Table 4). Global impression of treatment-response scores were numerically higher with paracetamol with Optizorb–caffeine than with ibuprofen and placebo (Table 4).
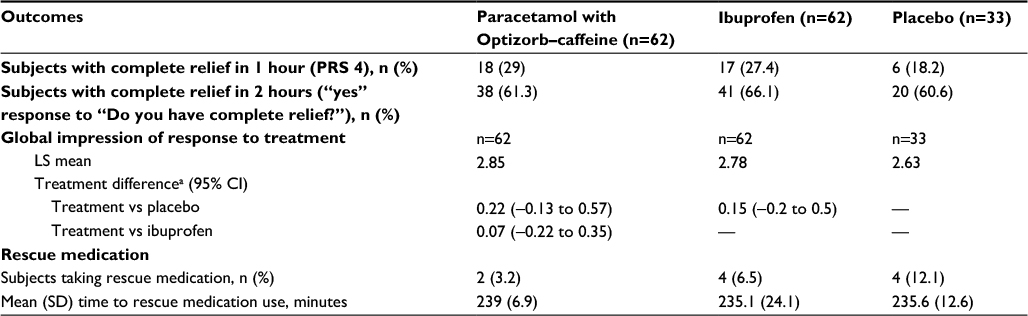 | Table 4 Number of subjects with complete relief, global impression of treatment, and use of rescue medication (secondary efficacy outcomes), study 2, ITT population Note: aDifference between the first-named treatment group and the second-named treatment group such that a positive result favors the first treatment. Abbreviations: ITT, intent to treat; PRS, pain-relief score; LS, least squares; CI, confidence interval; SD, standard deviation. |
Safety
There were nine TEAEs reported in study 1: two in the paracetamol–sodium bicarbonate–caffeine group, four in the ibuprofen group, none in the paracetamol group, and three in the placebo group (Table 5). Seven were mild, and two (oral pain during ibuprofen and plantar fasciitis during placebo) were moderate in intensity. Three mild gastrointestinal TEAEs were the only TEAEs considered to be related to treatment, including one case each of upper-abdominal pain and dyspepsia with ibuprofen and one case of dry mouth with placebo. The only serious AE, which consisted of viral hyperhidrosis associated with heart-rate increase and nausea, was observed before the start of study treatment; the participant who experienced this event was not randomized to treatment.
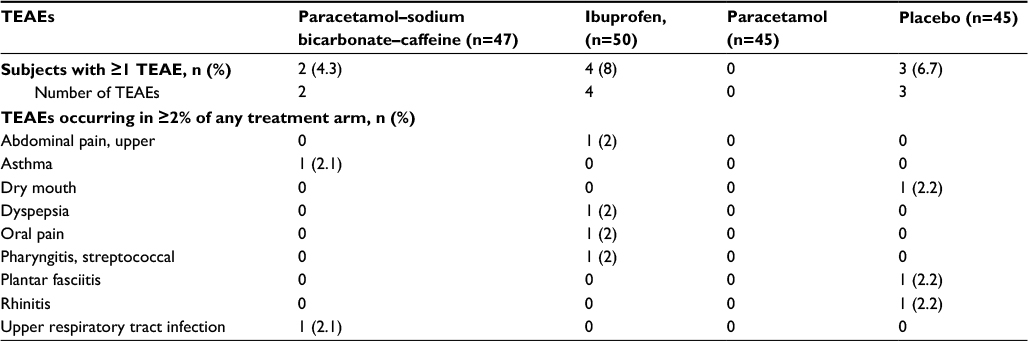 | Table 5 TEAEs in study 1, safety population Abbreviation: TEAEs, treatment-emergent adverse events. |
In study 2, eight subjects experienced a total of 12 TEAEs, including four in the paracetamol with Optizorb–caffeine group, three in the ibuprofen group, and one in the placebo group (Table 6). All were mild, except one moderate case of paranasal sinus discomfort in the ibuprofen group. Two subjects had TEAEs that were considered treatment-related: one participant in the paracetamol with Optizorb–caffeine group had mild jitteriness and one in the ibuprofen group experienced mild insomnia. No severe or serious TEAEs were reported in study 2.
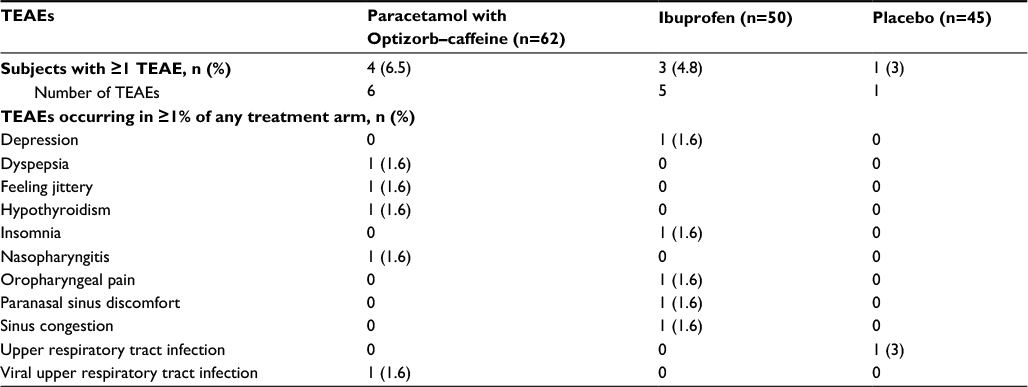 | Table 6 TEAEs in study 2, safety population Abbreviation: TEAEs, treatment-emergent adverse events. |
Discussion
Due to the underenrollment and early termination of these two studies, findings can only be considered exploratory. Trends observed in both studies suggest that adding caffeine to rapid-acting formulations of paracetamol might decrease the time to first perceptible pain relief compared with paracetamol and ibuprofen. In study 1, many of the secondary efficacy outcomes, including PID, PRS, TOTPAR, SPID, and SPRID, showed potentially clinically relevant differences in favor of paracetamol–sodium bicarbonate–caffeine compared with placebo and standard paracetamol at approximately 45–90 minutes, and there were two to three times as many patients achieving complete relief of headache at 1 hour with the combination treatment compared with either paracetamol or ibuprofen. In study 2, paracetamol with Optizorb–caffeine was consistently associated with numerical improvements in time to onset of relief and the majority of efficacy assessments compared with ibuprofen and placebo.
Recent guidance from the IHS recommends using pain-free rate at 2 hours as the primary efficacy measure in ETTH studies, and reporting number needed to treat (NNT) for that outcome as well.14 A recent meta-analysis of randomized controlled trials of ETTH found that few trials adhered to this recommendation (although many were conducted before it was issued), such that only ten of 41 trials reported rates of patients who were pain-free at 2 hours.15 The authors concluded that sufficient evidence demonstrating efficacy on this outcome was available only for paracetamol 1,000 mg, ibuprofen 400 mg, and ketoprofen 25 mg.
In both of our studies, the proportion of patients with complete headache relief after 2 hours was reported as a secondary outcome. In study 1, approximately 61.7% of subjects were headache-free at 2 hours after paracetamol–sodium bicarbonate–caffeine compared with 60% with ibuprofen, 55.6% with paracetamol, and 53.3% with placebo. Although the rate of headache relief at 2 hours was not significantly different between paracetamol–sodium bicarbonate–caffeine and placebo, there was a trend suggesting potentially better relief at 2 hours after taking the active treatment than placebo in study 1. In study 2, complete relief after 2 hours was achieved by 61.3% with paracetamol with Optizorb–caffeine, 66.1% with ibuprofen, and 60.6% with placebo; differences were not statistically significant. The percentage of participants with complete relief at 2 hours was higher for all treatments in our studies than reported in several recent Cochrane meta-analyses, in which 23.6% of patients treated with paracetamol 1,000 mg, 22.7% treated with ibuprofen 400 mg, 27.5% treated with ketoprofen 25 mg, and 16%–18.9% treated with placebo were pain-free at 2 hours.7,16,17 The differences between our studies and the ones in these analyses may be attributable to differences in study design, patient population, eDiary use, or other factors.
The NNT for the pain-free at 2 hours outcome with paracetamol–sodium bicarbonate–caffeine in study 1 was 12, which is similar to NNTs reported in meta-analyses for paracetamol (NNT 22),7 ibuprofen (NNT 14),11 and ketoprofen (NNT 9).17 The NNT for paracetamol with Optizorb–caffeine in study 2 could not be calculated, because the absolute risk difference from placebo was <1%. In comparing the NNTs from our studies with those in the meta-analyses, it is important to take into account that the placebo response rates in our studies were substantially higher than those previously reported. The individual studies included in the previous meta-analyses had placebo response rates of 1.3%–28.7% for the percentage who were pain-free at 2 hours7,11,17 compared with 53.3% and 60.6% in the current studies.
Importantly, because our studies used a rapid-dissolving analgesic, pain-free at 2 hours may not be an optimal assessment, because it fails to capture more rapid pain relief. As noted in both the IHS guidance and a recent meta-analysis, short measurement intervals and early assessments (eg, 1-hour outcomes) may be necessary when evaluating drugs in which speed of onset is of interest.14,15 In study 1, 31.9% of subjects treated with paracetamol–sodium bicarbonate–caffeine (NNT 6), 14% of those treated with ibuprofen, 8.9% of those treated with paracetamol, and 15.6% of those treated with placebo had complete headache resolution at 1 hour. In study 2, complete headache relief was achieved at 1 hour in 29% of those treated with paracetamol with Optizorb–caffeine (NNT 9), 27.4% of those treated with ibuprofen, and 18.2% of those in the placebo group. In the recent Cochrane meta-analysis of paracetamol in ETTH, the proportion pain-free or with only mild pain at 1 hour was 6% for paracetamol and 5.1% with placebo.7 In a meta-analysis of ibuprofen studies, 6.3% of ibuprofen-treated patients and 5.8% of placebo-treated patients were pain-free at 1 hour.16 It is difficult to compare NNTs from our results with the existing literature, because the Cochrane meta-analyses for ibuprofen and paracetamol did not calculate NNTs for pain-free at 1 hour (the risk difference from placebo was <1%), and no data on this outcome were available for ketoprofen.7,11,17
Ours are the first published studies to combine rapid-acting paracetamol formulations with caffeine in the treatment of ETTH. Previous studies have reported enhanced efficacy when caffeine was added to standard paracetamol or ibuprofen. For example, in a pooled analysis of two randomized, double-blind, double-dummy, two-period crossover trials in adults with ETTH, 1,000 mg paracetamol plus 130 mg caffeine showed significantly better efficacy on SPID, maximum PID, TOTPAR, maximum pain relief, and duration of time with pain at least half gone compared with both placebo and 1,000 mg paracetamol alone (all P<0.001).12 An Italian randomized, double-blind, double-dummy, crossover trial in patients with TTH found that 1,000 mg paracetamol plus 130 mg caffeine was as effective as 550 mg naproxen sodium and more effective than placebo with regard to PID, TOTPAR, and proportion of subjects requiring rescue medication.18 Another randomized, double-blind, crossover trial in adults with nonmigraine headaches found that 65 or 130 mg caffeine alone, 648 mg paracetamol alone, or 648 mg paracetamol plus either 65 or 130 mg caffeine all significantly reduced pain (as reported on 100 mm visual analogue scale) compared with placebo at 120 minutes, whereas 130 mg caffeine with or without paracetamol also produced significant pain reduction versus placebo at 30 and 60 minutes postdose (all P<0.05).19 Finally, a randomized, double-blind, parallel-group trial in adults with TTH found that 400 mg ibuprofen plus 200 mg caffeine was significantly more effective with regard to peak PID, peak pain relief, PRS scores at most time points, SPID0–4, SPID0–6, TOTPAR0–6, and overall global rating than 400 mg ibuprofen alone, 200 mg caffeine alone, and placebo (all P<0.05).20,21
In addition, efficacy has been shown for the triple combination of caffeine, aspirin, and paracetamol. In a pooled analysis of four randomized, double-blind, double-dummy, two-period crossover trials in adults with ETTH, 500 mg paracetamol–500 mg aspirin–130 mg caffeine also showed significantly better efficacy on SPID, maximum PID, TOTPAR, maximum pain relief, and duration of time with pain at least half gone compared with both placebo and 1,000 mg paracetamol alone (all P<0.001).12 A second analysis of the same four studies reported that the triple combination was also associated with a significantly (P<0.0001) greater percentage of pain-free subjects at 2 hours (28.5%) compared with acetaminophen alone (21%) or placebo (18%); this was also true in the subset of subjects with severe pain at baseline (percentage pain-free at 2 hours 20.2% vs 12.1% [P<0.0001] and 10.8% [P=0.0003], respectively).22 In addition, a randomized, double-blind, placebo-controlled trial also supported a more rapid onset and greater pain relief with paracetamol–aspirin–caffeine compared with aspirin, paracetamol, caffeine, and placebo alone in the treatment of a variety of headache types.23 Therefore, the patterns of response seen in our two studies are consistent with the overall positive effects of caffeine when added to other analgesics for treatment of ETTH, based on the current literature in this area.
It is important to recognize that both studies were stopped before the number of planned subjects was enrolled; therefore, definitive conclusions about the efficacy of rapid-acting formulations of paracetamol and caffeine compared with placebo, ibuprofen, and paracetamol without caffeine cannot be drawn from these data. Study 1 was stopped due to business reasons, with only 28% of the planned number of evaluable participants. Study 2, which was stopped based primarily on enrollment difficulties and impending expiration of drug supplies, terminated with only 57% of the planned number of evaluable participants. People with ETTH often self-treat with OTC products that provide effective relief of headache pain;5,6 therefore, many do not seek medical attention, which makes enrollment in ETTH trials challenging. Furthermore, because subjects with chronic tension headaches are not as responsive to OTC headache medicines, a run-in period is essential for future studies to ensure that only those with ETTH are included. This can prove to be an impediment to enrollment, due to the increased study duration. However, it should be noted that subjects who met the criteria after the run-in period were highly likely to complete the study. The high rate of retention was in part due to the use of eDiaries that subjects had to complete on a daily basis, which kept them actively engaged. Another reason for incomplete enrollment in these studies is that the planned duration of the enrollment period proved insufficient. ETTH efficacy studies are uncommon; therefore, there was little information to guide the study team in determining what a realistic rate of enrollment was. A range of site and physician types participated in the conduct of these studies, because it was unclear to the study developers which types of physicians and sites were likely to be high enrollers. Future studies of ETTH will need to overcome these enrollment issues with better screening procedures and site identification, and longer enrollment periods.
In both studies, the reference arm containing standard ibuprofen showed limited effect compared with placebo (Tables 4 and 5). Reasons for this finding are unclear, and the fact that even the active control failed to show a response suggests possible flaws in the trial design that may have contributed to the limited response seen with the study treatments. Ibuprofen has been shown to be effective compared with placebo in some24 but not all20 studies of ETTH. High failure rates, even for analgesic products generally accepted as efficacious, are not uncommon in pain trials, and according to some experts the fault lies with the trials rather than with the treatment.25,26 Reasons may include larger-than-expected placebo response and poor subjective patient reporting.25 The self-limiting nature of TTH often results in a high placebo response rate, adding to the difficulties of detecting a treatment effect.20 In the case of ETTH, the headaches typically resolve within 30 minutes to a week, even in the absence of treatment.3
All study treatments in both studies were generally well tolerated, with largely mild TEAEs and no unexpected safety signals. The formulation used in study 1 contained sodium bicarbonate, whereas the formulation in study 2 did not. Oral sodium bicarbonate is sometimes used as an antacid, and is also used in the treatment of peptic ulcers, metabolic acidosis, acidosis in renal tubular disorders, and uric acid crystallization in gout.27 Oral administration of sodium bicarbonate may cause gastric distension, flatulence, belching, retention of sodium and water, and edema.27 It was well tolerated in the small number of subjects treated in study 1, but a longer-term assessment with a larger population would be necessary to characterize the safety and tolerability profile of this formulation fully.
Neither study included rapid-acting paracetamol without caffeine as a comparator, so it is not possible to determine whether any added benefits stemmed from the more rapidly absorbed paracetamol, the addition of caffeine, or both. Different formulations of fast-absorbing paracetamol were used in the two trials; positive trends were seen with both, and in the absence of comparative data, conclusions cannot be drawn regarding the benefits of one such formulation over another. An additional limitation of study 1 was that no placebo corresponding to paracetamol was included, so subjects were not fully blinded.
In conclusion, in these 2 studies that were terminated early, trends suggest the possibility that rapid-acting formulations of paracetamol plus caffeine might shorten time to onset and improve relief of ETTH compared with traditional OTC analgesics. Therefore, these formulations warrant further investigation in larger, fully powered studies if enrollment challenges can be overcome.
Acknowledgments
This study was sponsored by GlaxoSmithKline Consumer Healthcare, Parsippany, NJ, USA. Medical writing and editorial assistance were provided by Peloton Advantage, Parsippany, NJ and were funded by GlaxoSmithKline Consumer Healthcare.
Disclosure
YY was an employee of GlaxoSmithKline Consumer Healthcare at the time of this study. KDR is an employee of and LS a contractor for GlaxoSmithKline Consumer Healthcare. DJL is an employee of GlaxoSmithKline.
References
Crystal SC, Robbins MS. Epidemiology of tension-type headache. Curr Pain Headache Rep. 2010;14(6):449–454. | ||
Schwartz BS, Stewart WF, Simon D, Lipton RB. Epidemiology of tension-type headache. JAMA. 1998;279(5):381–383. | ||
International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808. | ||
Bendtsen L, Ashina S, Moore A, Steiner TJ. Muscles and their role in episodic tension-type headache: implications for treatment. Eur J Pain. 2016;20(2):166–175. | ||
Bendtsen L, Jensen R. Treating tension-type headache: an expert opinion. Expert Opin Pharmacother. 2011;12(7):1099–1109. | ||
Bendtsen L. Treatment guidelines: implications for community-based headache treatment. Int J Clin Pract Suppl. 2015;69(182):13–16. | ||
Stephens G, Derry S, Moore RA. Paracetamol (acetaminophen) for acute treatment of episodic tension-type headache in adults. Cochrane Database Syst Rev. 2016;(6):CD011889. | ||
Yoon YJ, Kim JH, Kim SY, Hwang IH, Kim MR. A comparison of efficacy and safety of non-steroidal anti-inflammatory drugs versus acetaminophen in the treatment of episodic tension-type headache: a meta-analysis of randomized placebo-controlled trial studies. Korean J Fam Med. 2012;33(5):262–271. | ||
Rostami-Hodjegan A, Shiran MR, Ayesh R, et al. A new rapidly absorbed paracetamol tablet containing sodium bicarbonate – I: a four-way crossover study to compare the concentration-time profile of paracetamol from the new paracetamol/sodium bicarbonate tablet and a conventional paracetamol tablet in fed and fasted volunteers. Drug Dev Ind Pharm. 2002;28(5):523–531. | ||
Burnett I, Schachtel B, Sanner K, Bey M, Grattan T, Littlejohn S. Onset of analgesia of a paracetamol tablet containing sodium bicarbonate: a double-blind, placebo-controlled study in adult patients with acute sore throat. Clin Ther. 2006;28(9):1273–1278. | ||
Derry CJ, Derry S, Moore RA. Caffeine as an analgesic adjuvant for acute pain in adults. Cochrane Database Syst Rev. 2014;(12):CD009281. | ||
Migliardi JR, Armellino JJ, Friedman M, Gillings DB, Beaver WT. Caffeine as an analgesic adjuvant in tension headache. Clin Pharmacol Ther. 1994;56(5):576–586. | ||
Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders, 2nd ed. Cephalalgia. 2004;24 (Suppl 1):9–160. | ||
Bendtsen L, Bigal ME, Cerbo R, et al. Guidelines for controlled trials of drugs in tension-type headache: second edition. Cephalalgia. 2010;30(1):1–16. | ||
Moore RA, Derry S, Wiffen PJ, Straube S, Bendtsen L. Evidence for efficacy of acute treatment of episodic tension-type headache: methodological critique of randomised trials for oral treatments. Pain. 2014;155(11):2220–2228. | ||
Derry S, Wiffen P, Moore RA, Bendtsen L. Ibuprofen for acute treatment of episodic tension-type headache in adults. Cochrane Database Syst Rev. 2015;(7):CD011474. | ||
Veys L, Derry S, Moore RA. Ketoprofen for episodic tension-type headache in adults. Cochrane Database Syst Rev. 2016;9:CD012190. | ||
Pini LA, Del Bene E, Zanchin G, et al. Tolerability and efficacy of a combination of paracetamol and caffeine in the treatment of tension-type headache: a randomised, double-blind, double-dummy, cross-over study versus placebo and naproxen sodium. J Headache Pain. 2008;9(6):367–373. | ||
Ward N, Whitney C, Avery D, Dunner D. The analgesic effects of caffeine in headache. Pain. 1991;44(2):151–155. | ||
Diamond S, Balm TK, Freitag FG. Ibuprofen plus caffeine in the treatment of tension-type headache. Clin Pharmacol Ther. 2000;68(3):312–319. | ||
Diamond S, Freitag FG. The use of ibuprofen plus caffeine to treat tension-type headache. Curr Pain Headache Rep. 2001;5(5):472–478. | ||
Diener HC, Gold M, Hagen M. Use of a fixed combination of acetylsalicylic acid, acetaminophen and caffeine compared with acetaminophen alone in episodic tension-type headache: meta-analysis of four randomized, double-blind, placebo-controlled, crossover studies. J Headache Pain. 2014;15:76. | ||
Diener HC, Pfaffenrath V, Pageler L, Peil H, Aicher B. The fixed combination of acetylsalicylic acid, paracetamol and caffeine is more effective than single substances and dual combination for the treatment of headache: a multicentre, randomized, double-blind, single-dose, placebo-controlled parallel group study. Cephalalgia. 2005;25(10):776–787. | ||
Nebe J, Heier M, Diener HC. Low-dose ibuprofen in self-medication of mild to moderate headache: a comparison with acetylsalicylic acid and placebo. Cephalalgia. 1995;15(6):531–535. | ||
Usdin S. Shaking the cup for pain. BioCentury. 2011;May 30:13–14. | ||
Dworkin RH, Turk DC, Peirce-Sandner S, et al. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. Pain. 2012;153(6):1148–1158. | ||
National Center for Biotechnology Information. Sodium bicarbonate. 2016. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/516892. Accessed March 15, 2017. |
Supplementary material
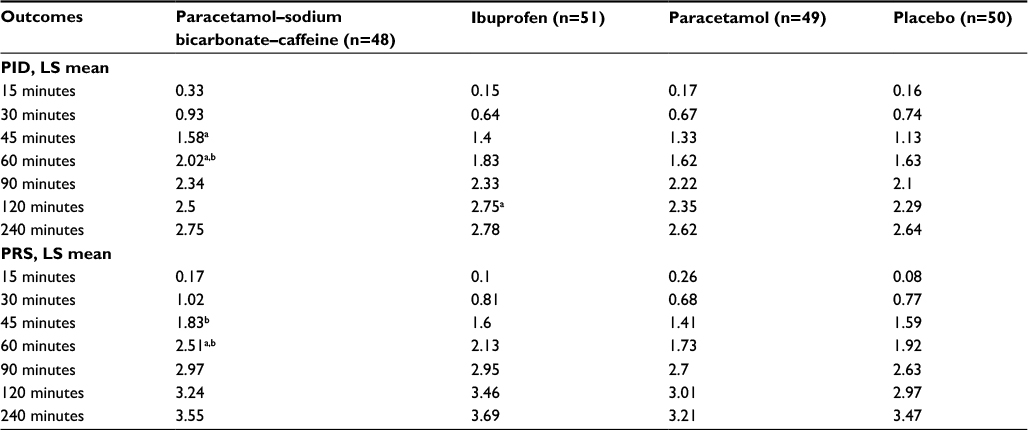 | Table S1 Additional secondary efficacy outcomes from study 1, ITT population Notes: LS means are from the analysis-of-covariance model adjusted for baseline pain intensity. Patient numbers shown in the column headings represent the full ITT population; sample sizes for the individual end points varied, due to missing data at some time points for some outcomes. aP<0.05 vs placebo; bP<0.05 vs paracetamol. Abbreviations: ITT, intent to treat; PID, pain-intensity difference; LS, least squares; PRS, pain-relief score. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.