")
Back to Journals » Patient Preference and Adherence » Volume 8
Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis
Authors Takeda Y, Tsujino K, Kijima T, Kumanogoh A
Received 5 November 2013
Accepted for publication 23 January 2014
Published 21 March 2014 Volume 2014:8 Pages 361—370
DOI https://doi.org/10.2147/PPA.S37233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Yoshito Takeda,1 Kazuyuki Tsujino,2 Takashi Kijima,1 Atsushi Kumanogoh1
1Department of Respiratory Medicine, Allergy and Rheumatic Diseases, Osaka University Graduate School of Medicine, Suita, Osaka, Japan; 2Department of Respiratory Medicine, Kinki Central Hospital of the Mutual Aid Association of Public School Teachers, Itami, Hyogo, Japan
Abstract: Idiopathic pulmonary fibrosis (IPF) is a devastating chronic fibrotic lung disease. Although the precise cause of the disease is still unknown, recent studies have shown that the pathogenesis of pulmonary fibrosis involves multiple mechanisms, with abnormal behavior of alveolar epithelial cells considered a primary event. Pirfenidone is a multifunctional, orally available small molecule with anti-fibrotic, anti-inflammatory, and antioxidative activities, and has been shown to be a modulator of cytokines and growth factors, including TGF-ß1, TNF-α, bFGF, IFN-γ, IL-1ß, and IL-18 in animal models. Although its precise mechanism of action is not currently clear, pirfenidone is considered to exert inhibitory effects on multiple pathways involved in the pathogenesis of IPF. Two randomized placebo-controlled clinical trials in Japan demonstrated that pirfenidone significantly reduced the rate of decline of vital capacity in IPF patients. A Phase III study showed a significant increase in progression-free survival of patients in pirfenidone-treated groups compared to the placebo group. These results paved the way for the approval of pirfenidone for the treatment of IPF patients in Japan in 2008. The promising results of the Phase II study in Japan led to a larger international Phase III trial (CAPACITY). Subsequently, pirfenidone has also been approved in the European Union, South Korea, and Canada to date. Pirfenidone treatment is generally tolerated. Major adverse events are gastrointestinal symptoms, including decreased appetite, abdominal discomfort and nausea, photosensitivity, and fatigue, but many of these are mild and manageable. Clinical experience has shown that reduction in pirfenidone dose and the supportive use of gastrointestinal drugs are effective ways to manage these symptoms. Thus, pirfenidone treatment provides a means of intervention in the clinical course of IPF, and is a promising candidate for improving patient prognosis. For future development, it is important to establish the appropriate modality of treatment with pirfenidone and/or novel potential drugs.
Keywords: pirfenidone, safety, efficacy, anti-fibrotic drugs
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating chronic fibrotic lung disease of unknown etiology. IPF is characterized by progressive deposition of collagen and other extracellular matrix (ECM) molecules.1 Previously, IPF was viewed as a “smoldering” inflammatory response that ultimately led to chronic lung injury with subsequent fibrosis. However, inflammation is no longer regarded as crucial to the etiology of IPF, largely because current anti-inflammatory therapies for IPF have provided little benefit for patients.2 Instead, it has become clear that abnormal behavior of alveolar epithelial cells (AECs) is the primary event in the development of pulmonary fibrosis.3,4 The disease process is initiated through repetitive injury of AECs, causing AEC activation, which in turn leads to the recruitment of immune cells and fibroblasts within the lung microenvironment. Aberrantly activated AECs, in cooperation with migrated immune cells and fibroblasts, secrete and activate latent TGF-β1, as well as other pro-fibrotic factors, which promote the differentiation of fibroblasts and AECs to myofibroblasts, resulting in overproduction of ECM in the lung. Epithelial–mesenchymal transition (EMT) may also be a source of ECM-producing (myo)fibroblasts in IPF.
Accumulating evidence suggests that both intrinsic factors in host lungs (ie, genetic predisposition), and extrinsic factors that accelerate injury of AECs play an etiologic role in IPF. Genetic predisposition is evidenced by the association of genes with disease, including telomerase reverse transcriptase (TERT) and MUC5B.5–9 Close attention has also been paid to β1 integrins in AECs and fibroblasts, focusing on their potential roles in pulmonary fibrosis. Integrin-related tetraspanin CD151 is essential for AECs to maintain epithelial integrity via firm adhesion to the basement membrane. The deletion of CD151 promotes mesenchymal-like changes and activation of TGF-β signaling in AECs in mice, and downregulation of CD151 in AECs of IPF patients is considered to be the result of an extrinsic factor, rather than a genetic factor.10
Until pirfenidone was approved in 2008, no drug was proven to be effective to treat IPF, although several clinical trials of potential agents have been conducted.11 Novel treatments for IPF, including bosentan (dual endothelin receptor antagonist), imatinib (tyrosine kinase inhibitor), sildenafil (phosphodiesterase type-5 inhibitor), etanercept (antibody against tumor necrosis factor (TNF)-α), and interferon (IFN)-γ-1b have been evaluated in randomized clinical trials, but none of these have demonstrated a statistically significant treatment effect on the primary endpoint. Furthermore, a recent randomized trial (PANTHER-IPF [Prednisone, Azathioprine, and N-Acetylcysteine: A Study That Evaluates Response in Idiopathic Pulmonary Fibrosis]) demonstrated that triple therapy with prednisone, azathioprine, and N-acetylcysteine (NAC) was not beneficial for IPF patients, and instead, increased the risk of death and hospitalization as compared with placebo.12
Pirfenidone is the first anti-fibrotic drug approved for the treatment of IPF. The approval of pirfenidone in 2008 brought hope for both IPF patients and physicians, with the anticipation that the drug could intervene in the disease course of IPF, and thus improve prognosis for this devastating disease in the future. Here, we review the efficacy and safety of pirfenidone for the treatment of IPF. In addition, the current understanding of the mechanisms of action of pirfenidone is described.
Mechanism of action
Pirfenidone is an orally available pyridone analog (5-methyl-1-phenyl-1H-pyridin-2-one) (Figure 1). It is multifunctional, having anti-fibrotic, anti-inflammatory, and antioxidant activities. Pirfenidone was discovered by an American pharmacologist, Solomon Margolin (1920–2008) in the course of a synthetic study.13 It had initially been developed as an anti-inflammatory agent, but the focus was then shifted to developing pirfenidone as an anti-fibrotic agent, since it was found to have anti-fibrotic activity in a canine lung infection model,14,15 as well as reducing fibrosis and improving lung function in a hamster model of bleomycin-induced lung injury.16,17 The anti-fibrotic action of pirfenidone has also been reported in several animal models of progressive fibrotic disorders of other organs, including the kidney, liver, and heart.18,19 In addition to its clinical efficacy for IPF, as described in detail below, a randomized and placebo-controlled study showed that pirfenidone also has potential benefit in diabetic nephropathy.20
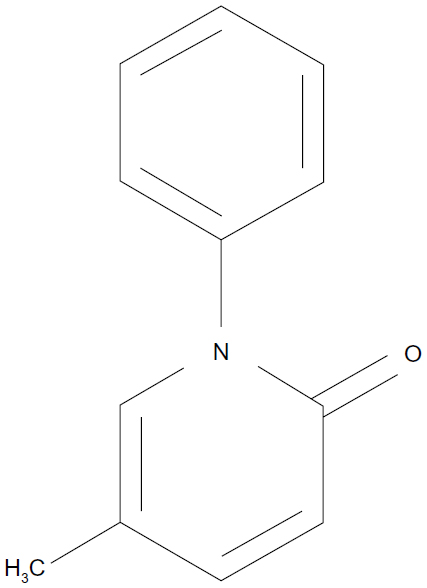 | Figure 1 Pirfenidone (5-methyl-1-phenyl-1H-pyridin-2-one). |
Pirfenidone can modulate the expression of pro-fibrotic factors and proinflammatory cytokines, and potentially suppresses the production of reactive oxygen species (ROS) (Figure 2). The anti-inflammatory activity of pirfenidone has been demonstrated in a septic shock model in mice.21,22 Pirfenidone was found to protect mice from endotoxin-induced lethal shock through the inhibition of TNF-α production. Pirfenidone inhibited TNF-α production by post-transcriptional regulation of the TNF-α gene.23 Pirfenidone also enhanced the production of IL-10 in this model. These effects on TNF-α and IL-10 are also considered to be involved in the anti-fibrotic activities of pirfenidone.
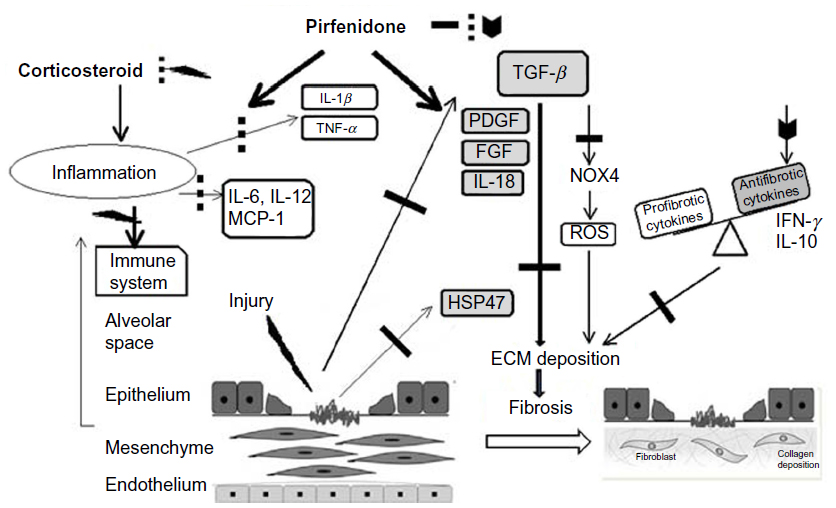 | Figure 2 Potential mechanisms for the suppression of fibrogenesis by pirfenidone. |
Oku et al investigated the anti-fibrotic effect of pirfenidone in bleomycin-induced lung fibrosis in mice using prednisolone as a reference agent.24 Mice were injected intravenously (iv) with bleomycin (10 mg/kg) for 5 consecutive days. In this model, lung inflammation occurred and peaked on day 10 after the first injection of bleomycin. Following inflammation, lung fibrosis was initiated and progressed gradually. Both pirfenidone and prednisolone suppressed lung inflammation in this model, but only pirfenidone significantly suppressed the subsequent lung fibrosis as evaluated by hydroxyproline content and histological fibrotic score. Pirfenidone suppressed the elevation of fibrogenic factors such as transforming growth factor (TGF)-β1 and basic fibroblast growth factor (bFGF). Furthermore, pirfenidone prevented the lowering of IFN-γ, which is known to be an anti-fibrotic cytokine through inhibition of TGF-β1 production and is also involved in type 1 helper T cell/type 2 helper T cell (Th1/Th2) balance. Prednisolone, however, did not suppress the changes in these factors. On the other hand, proinflammatory cytokines in the lungs, such as IL-1β, IL-6, and monocyte chemoattractant protein (MCP)-1 were suppressed by both agents. In addition, it was reported that pirfenidone inhibited platelet-derived growth factor (PDGF) production in bleomycin-induced lung fibrosis in hamsters.25
The anti-fibrotic activity of pirfenidone is mainly considered to be due to the suppression of TGF-β. TGF-β is one of the most studied pro-fibrotic cytokines.26 In the lung, TGF-β is produced by a wide variety of cell types, including alveolar macrophages, neutrophils, activated alveolar epithelial cells, endothelial cells, fibroblasts, and myofibroblasts. TGF-β induces the proliferation of macrophages and fibroblasts via PDGF expression. In these cells, TGF-β also stimulates the expression of a number of other proinflammatory and fibrogenic cytokines, such as TNF-α, IL-1β, and IL-13, thereby further enhancing and perpetuating the fibrotic response.
A recent report implied that pirfenidone suppressed the formation of a multi-molecular complex known as the NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome.27 This led to attenuation of the expression of IL-1β, which induces inflammatory and pro-fibrotic responses.28 That study also reported that pirfenidone reduced left ventricular remodeling and subsequent fibrosis in a mouse model of cardiac fibrosis induced by thoracic aortic constriction (TAC). TAC induced an elevation in inflammatory cell infiltration, and the production of ROS and inflammatory mediators including c-Jun, NLRP3, and IL-1β, but pirfenidone attenuated these effects. The upregulation of NLRP3 following TAC suggests that inflammasome formation may play a role in post-TAC remodeling and fibrosis. Mitochondrial production of ROS activates the NLRP3 inflammasome, and induces the synthesis and secretion of cytokines IL-1β and IL-18, which in turn leads to increased expression of pro-fibrotic factors such as TGF-β1 and PDGF. Inhibition of inflammasome formation might play a role in the anti-fibrotic and anti-inflammatory activities of pirfenidone in pulmonary fibrosis, although it remains to be seen whether this is a primary or secondary effect of pirfenidone.29
The anti-fibrotic effect of pirfenidone is in part mediated by a reduction in oxidative stress. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals.30 Although pirfenidone is ineffective as a scavenger of superoxide radicals, it reduced the oxidative stress induced by toxic hydroxyl radicals produced during differentiation of human lung fibroblasts as well as in bleomycin-induced pulmonary fibrosis in mice, potentially through inhibition of Nox4 (NADPH oxidase isoform 4) and Nox1.31
Pirfenidone was also found to inhibit the responder frequency of T cell receptor (TCR)-stimulated CD4+ cells in vitro and in vivo and to significantly diminish Th2 cytokines in the bronchoalveolar lavage fluid from animals given chronic allergen challenge to the airway using ovalbumin.32–34 Okazaki et al reported that pirfenidone ameliorates isolated cough, based on increased cough reflex sensitivity in guinea pigs, and suggested that it potentially relieves chronic cough in IPF patients.34 Pirfenidone also inhibits the expression of heat shock protein (HSP)-47 which is a collagen-specific molecular chaperone involved in the intracellular processing of procollagen.35,36 Furthermore, pirfenidone might suppress epithelial–mesenchymal transition (EMT).36,37
Although the key target molecule for pirfenidone is not yet clear and further research is needed to elucidate its precise mechanism of action, through its multifunctional activity, pirfenidone appears to exert inhibitory effects on multiple pathways that lead to the development of IPF.
Efficacy
Clinical trials of pirfenidone for IPF
Two open-label trials and four double-blind trials have been carried out with pirfenidone for IPF.38–42 The first double-blind trial was a Phase II study in Japan.40 This study was stopped early (at 9 months) due to an increased incidence of acute exacerbations in patients treated with placebo, while none occurred in the pirfenidone group. The primary endpoint (lowest saturation of peripheral oxygen [SpO2] under exertion) was not significantly different between patients treated with pirfenidone (1,800 mg/day) and patients treated with placebo at 9 months, but a significant reduction in the decline in vital capacity (VC) was shown in the pirfenidone group, suggesting potential efficacy for IPF patients.
A Phase III clinical trial of pirfenidone has also been conducted in Japan.41 The trial included 275 patients with IPF (20–75 years of age). Inclusion criteria were based on: 1) oxygen desaturation of ≥5% difference between resting SpO2 and the lowest SpO2 during a 6-minute steady-state exercise test (6MET), and 2) a lowest SpO2 during the 6MET of ≥85% while breathing air. Patients were randomized to three groups; high dose (1,800 mg/day), low dose (1,200 mg/day), and placebo, at a ratio of 2:1:2 for a period of 52 weeks. The primary endpoint was the change in VC from baseline at 52 weeks. This study demonstrated that the decline in VC was significantly smaller (P=0.0416) in the high-dose pirfenidone group (−0.09 L) than in the placebo group (−0.16 L). The absolute difference in VC was 70 mL and the relative difference was 44%. The decline in VC in the low-dose pirfenidone group (−0.08 L) was also significantly reduced (P=0.0394) compared to that in the placebo group (Figure 3). This study also showed that pirfenidone significantly (P=0.0280) increased progression-free survival (PFS) (Figure 4). PFS was defined as the time until death and/or ≥10% decline in VC from baseline. Other secondary endpoints (SpO2, serum markers), however, did not reach statistical significance among the groups.
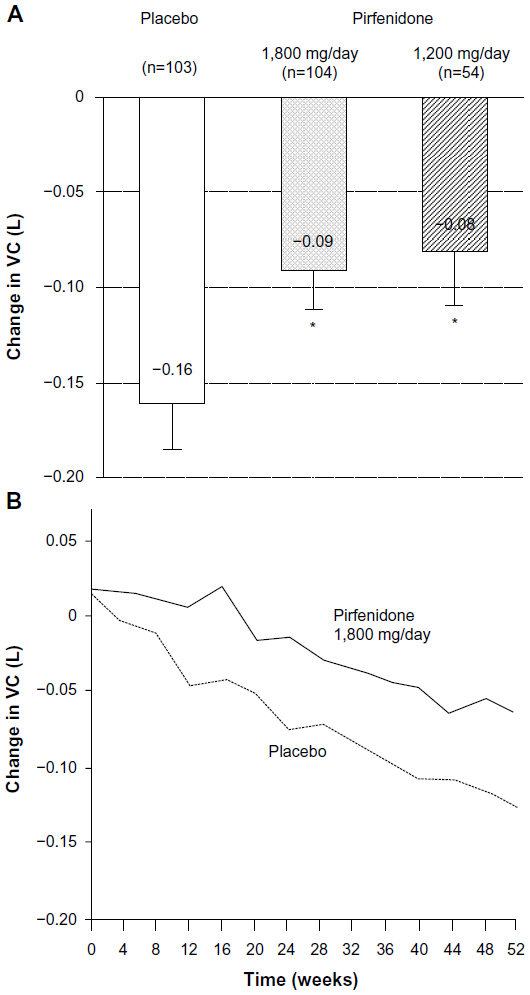 | Figure 3 Change in vital capacity at 52 weeks (a primary endpoint) (A) and serial changes in VC in (1,800 mg/day pirfenidone (-) and placebo group (---) over a 52-week period) (B), in a Phase III study of IPF patients in Japan *P<0.05 compared with placebo group. |
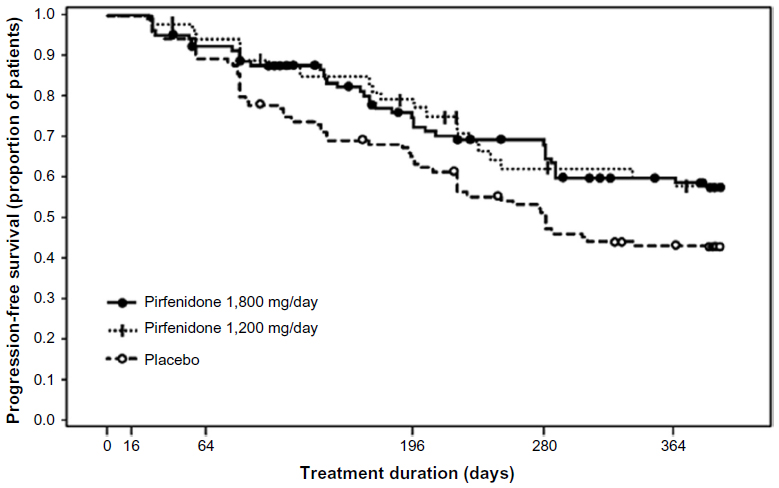 | Figure 4 Pirfenidone increased PFS in 1,800 mg/day and 1,200 mg/day pirfenidone groups as compared to the placebo group in a Phase III study of IPF patients in Japan. |
Two international clinical trials (CAPACITY [Clinical Studies Assessing Pirfenidone in idiopathic pulmonary fibrosis: Research of Efficacy and Safety Outcomes]-1, -2) were conducted in North America, Australia, and Europe.42 The decline in percentage predicted forced vital capacity (FVC) at 72 weeks was significantly reduced in pirfenidone-treated (2,400 mg/day) patients compared to those treated with placebo in CAPACITY-2, while the difference in FVC change at 72 weeks was not significant in CAPACITY-1. Pooled analysis of the two trials showed that pirfenidone significantly improved 6-minute walking distance.43 Applications for the approval of pirfenidone were submitted with the results of the CAPACITY trials to the European and US authorities. The data from the CAPACITY trials were assessed together with the data from the Japanese Phase III study by the European Medicines Agency (EMA), and pirfenidone was approved in the EU in March 2011. However, the US Food and Drug Administration (FDA) requested an additional trial, and that trial, the ASCEND (Assessment of Pirfenidone to Confirm Efficacy and Safety in IPF) study, is currently in progress. The enrollment of IPF patients (555 patients) was completed in January 2013, and top-line results from the study are expected to be available in the second quarter of 2014.
Meta-analysis (Cochrane review)
A recent Cochrane review summarized the studies of ten non-steroid agents for the treatment of IPF and identified IFN-γ-1b and pirfenidone as suitable for analysis.44 The meta-analysis combining the data from two IFN-γ-1b randomized controlled trials showed no significant difference between IFN-γ-1b and placebo in the clinical endpoint of overall survival (Figure 5A). Since meta-analysis of three Phase III studies of pirfenidone showed an increase in PFS (Figure 5B) and a suppression of decline in VC, pirfenidone is the only drug to show a significant effect on IPF to date.
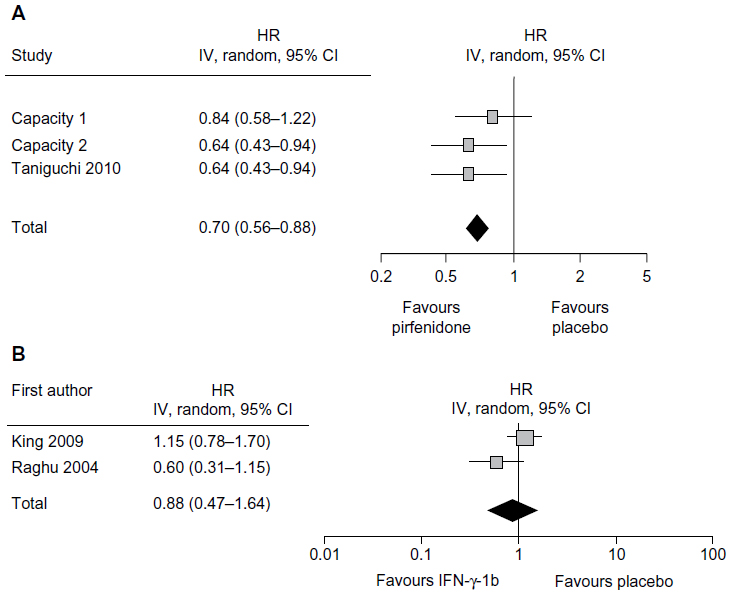 | Figure 5 Forest plot of pirfenidone (A) or interferon-γ-1b (B) versus placebo in improving PFS in IPF. |
Among prognostic factors for IPF, a decline in VC of 10% or more within 12 months is considered to be of most prognostic value.45 Marginal decline (≥5%) in VC was also reported to be associated with poor prognosis in IPF.46 Recently, there have been active debates on the most clinically meaningful endpoint for clinical trials of therapeutic agents for IPF.47–49 Although it has been proposed that all-cause mortality and all-cause hospitalization are the most appropriate endpoints,47 there are some limitations that should be considered.48 For clinical trials to be adequate for evaluation of mortality as an endpoint, in terms of statistical power, they need to include a high number of patients (>2,500) and require a much longer follow-up time (>3 years).50 In addition, patients will not agree to stay in a trial for a long period of time, in either the placebo or the treatment arm, if their disease progresses. For these reasons, a surrogate marker for mortality is needed and serial changes in VC or FVC are currently considered to be the preferred option. It has also been proposed that PFS could be a clinically meaningful endpoint.51
Sub-analysis of the Phase III clinical trial in Japan
Sub-analyses of the Japanese Phase III trial of pirfenidone have been carried out. Azuma et al reported that patients with a percentage predicted VC ≥70%, and a SpO2 at exertion <90% at baseline most likely benefited from pirfenidone.52 In such patients, pirfenidone significantly suppressed the worsening of subjective symptoms of cough and dyspnea as compared to placebo. Furthermore, Taniguchi et al showed that 70% of patients who had not worsened by 3 months, using a threshold of 5% change in VC, continued not to worsen by 12 months after beginning pirfenidone treatment.53 This finding suggests that VC changes (≥5%) at 3 months may be indicative for making clinical decisions on the continuation of pirfenidone treatment.
According to an exploratory analysis of the Japanese Phase III trial, pirfenidone appears to be more efficacious in the early stage than in the advanced stage of IPF.52 Therefore, an early and accurate diagnosis of IPF is critical for ensuring early intervention with pirfenidone, in order to suppress IPF progression at the early stage. With early intervention, improvements in the long-term clinical outcome of this devastating disease might be anticipated. Recently, Cottin and Cordier pointed out the value of “Velcro crackles” for early diagnosis,54 although the assessment of Velcro-like crackling sounds upon lung auscultation is already routinely used for the diagnosis in Japan. There are several parameters which are informative in determining when or whether to initiate treatment, including subjective symptoms, desaturation of oxygen, and decline in VC (>5%) within 3–6 months. However, with respect to early treatment, it must be taken into account that IPF is a heterogeneous disease, and that while many patients progress, some do not. Heterogeneous components of the disease may affect the response to treatment with pirfenidone, although factors which predict the response to pirfenidone have not yet been elucidated.
Safety
Phase II and Phase III clinical trials in Japan showed that pirfenidone was relatively well tolerated in patients with IPF.40,41 Major adverse events in the pooled pirfenidone groups from these two trials were photosensitivity reaction (51.7%), decreased appetite (23.0%), abdominal discomfort (14.0%), and nausea (12.1%). In the laboratory tests, elevation of γ-GTP was reported in 20.0% of patients receiving pirfenidone. Adverse events with an incidence ≥5% are listed in Table 1. International multi-center Phase III trials (CAPACITY)42 also showed that pirfenidone was well tolerated for IPF patients. The type and frequency of adverse events were generally consistent with those in Japanese clinical trials. There have been no reports on any ethnic differences in the pharmacology of pirfenidone.
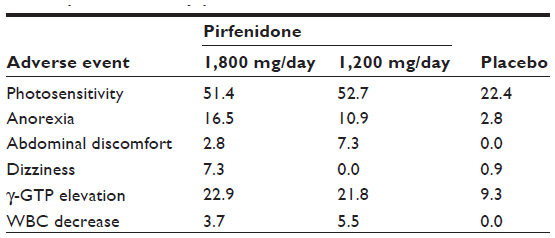 | Table 1 Incidence of major adverse events in Phase III clinical trial of pirfenidone in Japan |
The photosensitivity reaction was generally mild-to-moderate in severity, and only in 3% (n=5) of patients in pirfenidone groups (n=164) did these result in treatment discontinuation in the Phase III trial in Japan.41 Photosensitivity reactions are minimized by adequate UV protection, including avoiding exposure to direct sunlight and use of protective sunscreens. Recently, Okuda et al reported the safety profile of pirfenidone in 76 patients with IPF in clinical practice.55 The most frequent adverse event was anorexia (decreased appetite) with an incidence of 42.1%, although the grade of anorexia in most patients was mild (two patients with grade 3 and others with grade 1 or 2) (Table 2). They reported that anorexia improved in 84% of affected patients following reduction of the pirfenidone dose. On the other hand, the incidence of photosensitivity reaction in clinical practice was 18.4%, which was lower than that reported in Phase II and III clinical trials in Japan.40,41 The incidences of other adverse events in clinical practice were similar to those in clinical trials in Japan.
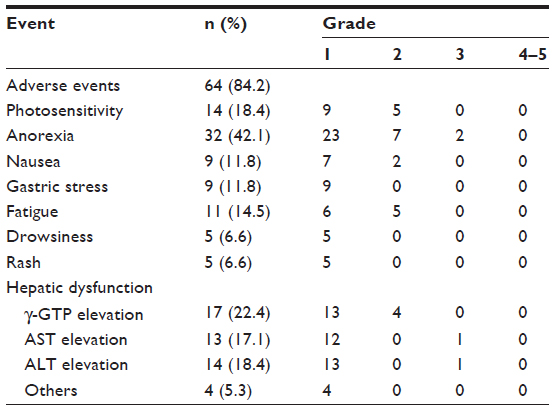 | Table 2 Adverse events of pirfenidone in 76 IPF patients in clinical practice |
Findings from clinical trials and clinical practice indicate that one of the most important issues for the continuation of pirfenidone therapy is the management of gastrointestinal symptoms, including decreased appetite, abdominal discomfort, and nausea, in order to gain maximal benefit from pirfenidone.
It might be an important factor for safety that pirfenidone has little suppressive effect on humoral and cellular immunity.56 Increased susceptibility to infection is anticipated to be less of a concern with clinical use of pirfenidone than with corticosteroids, and indeed this is considered to be one of the benefits of pirfenidone.
Routine toxicity screening performed during preclinical development showed that pirfenidone induced photogenotoxicity in an in vitro assay using Chinese hamster ovary (CHO) cells. Because the concentration of pirfenidone required for photogenotoxicity was near the upper limit estimated from pharmacokinetics in healthy humans, a warning for an increased risk of skin cancer is described in the package insert supplied with pirfenidone in Japan. There have been no reports of skin cancer determined to be caused by pirfenidone since its launch in Japan, nor in the RECAP trial (CAPACITY extension study) or during subsequent clinical use of pirfenidone in the EU. Increased risk of skin cancer is not described in the package insert for pirfenidone in the EU. Therefore, although the observation period and the number of cases treated are still limited, the increase in risk of the development of skin cancer when using pirfenidone is considered to be very low.
Current clinical status of pirfenidone for IPF
As of September 2013, in addition to Japan, pirfenidone has been approved for the treatment of IPF in the European Union, South Korea, and Canada. It is estimated that more than 5,000 patients with IPF are currently prescribed pirfenidone in Japan. Actual clinical experience of pirfenidone use is being reported, and new knowledge is being obtained regarding its effectiveness and safety. Iwasawa et al retrospectively examined the changes in the extent of fibrosis in IPF patients using high resolution computed tomography (HRCT), with both visual scoring by a radiologist and computer-aided analysis, over 1 year in clinical practice.57 The study included 38 cases of patients taking pirfenidone and 40 age-matched control patients with IPF but not taking pirfenidone. They reported that pirfenidone significantly reduced the mean increase in fibrosis scores compared with control patients (P<0.001). Okuda et al reported clinical experiences of pirfenidone use in 76 Japanese patients with mild-to-severe IPF, and suggested that pirfenidone also had beneficial effects in some patients with severe and/or progressive disease.55
In Japan, post-marketing surveillance (PMS) was conducted for all patients who initiated pirfenidone therapy within approximately 1 year after its launch (December 2008–October 2009). More than 1,400 patients with IPF were registered and were followed for a maximum of 2 years. The results from an interim analysis of 973 patients were presented at the European Respiratory Society (ERS) congress in 2012.58 While patients enrolled in the clinical trials in Japan and in the CAPACITY trials had mainly mild-to-moderate disease severity, the PMS showed that pirfenidone was also being prescribed to a substantial number of patients with severe IPF. The most frequent reason for the discontinuation of pirfenidone was gastrointestinal symptoms, the incidence of which was similar to that in the clinical trials in Japan. In addition to the use of gastrointestinal drugs, reduction of pirfenidone dose improved gastrointestinal symptoms as described above. The incidence of photosensitivity reaction in the PMS was 15%, significantly lower than in the clinical trials in Japan (51.7%), but consistent with the incidence in clinical practice as reported by Okuda et al.55 This discrepancy in frequency is considered to be mainly the result of clinicians alerting patients to the need for UV protection. In terms of pirfenidone efficacy, analysis of VC changes suggested that the decline in VC might be suppressed in the majority of patients who were treated with pirfenidone for more than 6 months. This is the first official PMS for pirfenidone. The findings of the PMS suggest that controlling gastrointestinal symptoms is important in ensuring that therapy can be continued and maximal benefit from pirfenidone can be gained.
Summary and future directions
Pirfenidone has been used for the treatment of IPF in Japan for more than 5 years, and it is now also available for IPF patients in several countries in the EU, in South Korea, and in Canada. A PMS in Japan and clinical experiences in “real world” settings have demonstrated the benefit/risk profile of pirfenidone for the treatment of IPF. The profiles of adverse events in clinical practice were generally consistent with those in clinical trials, and serious adverse events were rare. In a 2011 statement from ATS/ERS/JRS/ALAT, pirfenidone received a “weak no” (ie, against the use of these treatments for most patients) recommendation.59 However, randomized controlled trial data published since that time indicate that reassessment of the treatment guidelines for IPF may be warranted. Combination treatment with prednisone, azathioprine, and NAC, as well as treatment with warfarin, was given a “weak no” recommendation; however, this should be reassessed given the safety concerns brought to light by the PANTHER-IPF trial.12 The “weak no” recommendation for the use of pirfenidone should also be reevaluated given the promising findings from the three Phase III trials mentioned earlier. Recently, recommendation for the use of pirfenidone was reassessed in several European countries. In German guidelines published early in 2013, pirfenidone received a “weak yes” for the treatment of IPF patients with mild-to-moderate severity.60 Spanish guidelines were also published in August 2013 and pirfenidone is recommended as a first-line treatment for mild-to-moderate IPF patients.61
More effective therapy would ensure that the benefits for patients with IPF are maximized. Approaches to increasing therapeutic efficacy might include the selection of patients positive for markers which are predictive of drug response, or combination drug treatment with pirfenidone and NAC or other promising new drugs,62 or even with corticosteroids and/or immunosuppressants for patients with severe disease or with inflammatory components. BIBF 1120 (nintedanib) is a kinase inhibitor originally developed as an agent for cancer treatment, which simultaneously inhibits three receptor families implicated in angiogenesis: PDGF, vascular endothelial growth factor (VEGF), and FGF.63 A Phase II 12-month, randomized and placebo-controlled study of BIBF 1120 (TOMORROW [To Improve Pulmonary Fibrosis With BIBF1120] study) was conducted to evaluate its safety and efficacy in IPF.64 Although this study did not reach significance for its primary endpoint (decline of VC over a 12-month period, tested by closed testing procedure), the annual rate of decline in FVC in the group receiving 300 mg/day of BIBF 1120 was 0.06 L, as compared with 0.19 L in the placebo group. Moderate gastrointestinal symptoms and liver toxicity were reported in the high-dose (300 mg/day BIBF 1120) arm compared with placebo. Currently, two multinational Phase III studies of BIBF 1120 are being carried out to determine its efficacy and safety for the treatment of IPF.
In terms of the future development of IPF treatment, establishing the appropriate modality of treatment with pirfenidone and/or new potential novel drugs will contribute to the effective treatment of IPF patients.
Acknowledgments
We thank Dr Hisashi Oku and Dr Shuichiro Inagaki (Shionogi & Co, Ltd) for helpful discussions and critical reading of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. | |
Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. | |
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. | |
Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–731. | |
Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. | |
Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–7557. | |
Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45(10):654–656. | |
Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. | |
Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–2339. | |
Tsujino K, Takeda Y, Arai T, et al. Tetraspanin CD151 protects against pulmonary fibrosis by maintaining epithelial integrity. Am J Respir Crit Care Med. 2012;186(2):170–180. | |
Lota HK, Wells AU. The evolving pharmacotherapy of pulmonary fibrosis. Expert Opin Pharmacother. 2013;14(1):79–89. | |
Raghu G, Anstrom KJ, King TE Jr, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. | |
Azuma A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2012;6(2):107–114. | |
Margolin SB, inventor; John H, assignee. Composition for reparation and prevention of fibrotic lesions. United States patent US 5716632. February 10, 1998. | |
Margolin SB, Lefkowitz S. Pirfenidone: a novel pharmacologic agent for prevention and resolution of lung fibrosis. FASEB J. 1994;8:A382. | |
Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125(6):779–785. | |
Schelegle ES, Mansoor JK, Giri S. Pirfenidone attenuates bleomycin-induced changes in pulmonary function in hamsters. Proc Soc Exp Biol Med. 1997;216(3):392–397. | |
Macías-Barragán J, Sandoval-Rodríguez A, Navarro-Partida J, Armendáriz-Borunda J. The multifaceted role of pirfenidone and its novel targets. Fibrogenesis Tissue Repair. 2010;3:16. | |
Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20(120):85–97. | |
Sharma K, Ix JH, Mathew AV, et al. Pirfenidone for diabetic nephropathy. J Am Soc Nephrol. 2011;22(6):1144–1151. | |
Cain WC, Stuart RW, Lefkowitz DL, Starnes JD, Margolin S, Lefkowitz SS. Inhibition of tumor necrosis factor and subsequent endotoxin shock by pirfenidone. Int J Immunopharmacol. 1998;20(12):685–695. | |
Oku H, Nakazato H, Horikawa T, Tsuruta Y, Suzuki, R. Pirfenidone suppresses tumor necrosis factor-α, enhances interleukin-10 and protects mice from endotoxic shock. Eur J Pharmacol. 2002;446(1–3):167–176. | |
Nakazato H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur J Pharmacol. 2002;446(1–3):177–185. | |
Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–408. | |
Gurujeyalakshmi G, Hollinger MA, Giri SN. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am J Physiol. 1999;276:L311–318. | |
Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. 2012;9(3):111–116. | |
Artlett CM. Inflammasomes in wound healing and fibrosis. J Pathol. 2013;229(2):157–167. | |
Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. 2013;126(1):1–11. | |
Roche P, Czubryt MP. Pirfenidone and the inflammasome: Getting to the heart of cardiac remodeling. Cardiology. 2013;126(1):59–61. | |
Misra HP, Rabideau C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem. 2000;204:119–126. | |
Gaggini F, Laleu B, Orchard M, et al. Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg Med Chem. 2011;19(23):6989–6999. | |
Hirano A, Kanehiro A, Ono K, et al. Pirfenidone modulates airway responsiveness, inflammation, and remodeling after repeated challenge. Am J Respir Cell Mol Biol. 2006;35:366–377. | |
Visner GA, Liu F, Bizargity P, et al. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation. 2009;88(3):330–338. | |
Okazaki A, Ohkura N, Fujimura M, Katayama N, Kasahara K. Effects of pirfenidone on increased cough reflex sensitivity in guinea pigs. Pulm Pharmacol Ther. 2013;26(5):603–608. | |
Kakugawa T, Mukae H, Hayashi T, et al. Pirfenidone attenuates expression of HSP47 in murine bleomycin-induced pulmonary fibrosis. Eur Respir J. 2004;24(1):57–65. | |
Hisatomi K, Mukae H, Sakamoto N, et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med. 2012;12:24. | |
Yang Y, Ye Y, Lin X, Wu K, Yu M. Inhibition of pirfenidone on TGF-beta2 induced proliferation, migration and epithlial-mesenchymal transition of human lens epithelial cells line SRA01/04. PLoS One. 2013;8(2):e56837. | |
Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label Phase II study. Am J Respir Crit Care Med. 1999;159:1061–1069. | |
Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T. Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med. 2002;41(12):1118–1123. | |
Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(9):1040–1047. | |
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis: a Phase III clinical trial in Japan. Eur Respir J. 2010;35(4):821–829. | |
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY):two randomised trials. Lancet. 2011;377:1760–1769. | |
Bajwah S, Ross JR, Peacock JL, et al. Interventions to improve symptoms and quality of life of patients with fibrotic interstitial lung disease: a systematic review of the literature. Thorax. 2013;68(9):867–879. | |
Spagnolo P, Del Giovane C, Luppi F, et al. Non-steroid agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2010;8(9):CD003134. | |
Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(5):538–542. | |
Zappala CJ, Latsi PI, Nicholson AG, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:830–835. | |
Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med. 2012;185(10):1044–1048. | |
du Bois RM, Nathan SD, Richeldi L, Schwarz MI, Noble PW. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med. 2012;186(8):712–715. | |
Wells AU, Behr J, Costabel U, Cottin V, Poletti V, Richeldi L; for European IPF Consensus Group. Hot of the breath: mortality as a primary end-point in IPF treatment trials: the best is the enemy of the good. Thorax. 2012;67(11):938–940. | |
King TE, Albera C, Bradford WZ, et al. All-cause mortality (ACM) rate in patients with idiopathic pulmonary fibrosis (IPF):implications for the design and execution of mortality trials. ATS. 2013;A2356. | |
Vancheri C, du Bois RM. A progression-free end-point for idiopathic pulmonary fibrosis trials: lessons from cancer. Eur Respir J. 2013;41(2):262–269. | |
Azuma A, Taguchi Y, Ogura T, et al. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation of patients with idiopathic pulmonary fibrosis as benefiting from treatment. Respir Res. 2011;12:143. | |
Taniguchi H, Kondoh Y, Ebina M, et al. The clinical significance of 5% change in vital capacity in patients with idiopathic pulmonary fibrosis: extended analysis of the pirfenidone trial. Respir Res. 2011;12:93. | |
Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40(3):519–521. | |
Okuda R, Hagiwara E, Baba T, Kitamura H, Kato T, Ogura T. Safety and efficacy of pirfenidone in idiopathic pulmonary fibrosis in clinical practice. Respir Med. 2013;107(9):1431–1437. | |
Shionogi & Co, Ltd. [Immunosuppressive effect of pirfenidone]. Internal Data Reference No 200801771, Shionogi & Co, Ltd; 2008. Japanese. | |
Iwasawa T, Ogura T, Sakai F, et al. CT analysis of the effect of pirfenidone in patients with idiopathic pulmonary fibrosis. Eur J Radiol. 2014;83(1):32–38. | |
Ito M, Niimi Y, Nakamura A, et al. Post-marketing surveillance of pirfenidone for idiopathic pulmonary fibrosis in Japan: Interim analysis of 973 patients. ERS Annual Congress; September 1–5, 2012; Vienna, Austria: P3158. | |
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | |
Behr J. Evidence-based treatment strategies in idiopathic pulmonary fibrosis. Eur Respir Rev. 2013;22(128):163–168. | |
Xaubet A, Ancochea J, Bollo E, et al. Guidelines for the diagnosis and treatment of idiopathic pulmonary fibrosis. Arch Bronconeumol. 2013;49(8):343–353. | |
Homma S, Azuma A, Taniguchi H, et al. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology. 2012;17(3):467–477. | |
Roth GJ, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J Med Chem. 2009;52(14):4466–4480. | |
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.