")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Efficacy and safety of once-daily inhaled umeclidinium/vilanterol in Asian patients with COPD: results from a randomized, placebo-controlled study
Authors Zheng J , Zhong N, Newlands AH, Church A, Goh AH
Received 17 January 2015
Accepted for publication 11 May 2015
Published 2 September 2015 Volume 2015:10(1) Pages 1753—1767
DOI https://doi.org/10.2147/COPD.S81053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Jinping Zheng,1 Nanshan Zhong,1 Amy Newlands,2 Alison Church,3 Aik H Goh4
1State Key Lab of Respiratory Disease, National Clinical Research Centre of Respiratory Disease, First Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China; 2GlaxoSmithKline, London, UK; 3GlaxoSmithKline, Research Triangle Park, NC, USA; 4GlaxoSmithKline, Shanghai, People’s Republic of China
Background: Combination of the inhaled long-acting muscarinic antagonist umeclidinium (UMEC; GSK573719) with the long-acting β2-agonist vilanterol (VI) is an approved maintenance treatment for COPD in the US and EU. We compared the efficacy and safety of UMEC/VI with placebo in patients with COPD of Asian ancestry.
Patients and methods: In this 24-week, Phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study, patients were randomized 1:1:1 to UMEC/VI 125/25 µg, UMEC/VI 62.5/25 µg, or placebo. The primary efficacy end point was trough forced expiratory volume in 1 second (FEV1) on day 169; secondary end points were Transition Dyspnea Index (TDI) focal score at week 24 and weighted mean (WM) FEV1 over 0–6 hours postdose on day 1. Additional end points and safety were also assessed.
Results: Both UMEC/VI 125/25 µg and UMEC/VI 62.5/25 mg statistically significantly improved trough FEV1 at day 169 versus placebo (UMEC/VI 125/25 µg, 0.216 L, [95% confidence interval [CI] 0.175–0.257]; UMEC/VI 62.5/25 µg, 0.151 L, 95% CI 0.110–0.191; both P<0.001). Statistically significant improvements in TDI score were observed for both UMEC/VI groups versus placebo (UMEC/VI 125/25 µg, 0.9, 95% CI 0.3–1.4, P=0.002; UMEC/VI 62.5/25 µg, 0.7, 95% CI 0.1–1.2, P=0.016). On day 1, both UMEC/VI groups improved 0–6-hour WM FEV1 versus placebo (UMEC/VI 125/25 µg, 0.182 L 95% CI 0.161–0.203; UMEC/VI 62.5/25 µg, 0.160 L, 95% CI 0.139–0.181; both P<0.001). Statistically significant improvements for UMEC/VI groups versus placebo were observed for rescue albuterol use at weeks 1–24 (puffs/day, both P<0.001). The incidence of adverse events was similar across groups.
Conclusion: In Asian patients with COPD, once-daily UMEC/VI 125/25 µg and UMEC 62.5/25 µg resulted in clinically meaningful and statistically significant improvements in lung-function end points versus placebo. Symptomatic and quality of life measures also improved. The safety profile of UMEC/VI was consistent with previous studies.
Keywords: chronic obstructive pulmonary disease, umeclidinium, vilanterol, Asian
Introduction
What is known?
Previous studies have shown that combination treatment with umeclidinium (UMEC)/vilanterol (VI) improves lung function compared with monotherapies, and the tolerability and safety of UMEC/VI has also been studied. However, few patients in these studies were Asian, and specific subanalyses of these populations were not carried out.
What is new?
This is the first study to comprehensively investigate long-acting muscarinic antagonist (LAMA)/long-acting β2-agonist (LABA) combination dual-bronchodilator therapy in Asian patients with COPD. In this population, once-daily UMEC/VI 125/25 μg and UMEC 62.5/25 μg resulted in clinically meaningful and statistically significant improvements in lung-function end points compared with placebo, which is consistent with previous studies.
COPD is characterized by persistent airflow obstruction, and is a major health burden worldwide.1 It has been estimated that 65 million people worldwide have moderate–severe COPD.2 The prevalence of COPD in Asian countries has been reported to be between 5.4% and 13.4%, with figures expected to rise.3–5 Pharmacological management of stable COPD is predominantly focused on improvement of lung function, symptom reduction, decreasing COPD exacerbations, and improving quality of life (QoL) and exercise tolerance. Inhaled bronchodilators, such as LABAs and LAMAs, are central to the pharmacological management of COPD.6 LABAs exert their bronchodilatory effect through stimulation of β2-adrenergic receptors, subsequently increasing cyclic adenosine monophosphate and causing relaxation of airway smooth muscle.1 LAMAs inhibit muscarinic receptors in the airways and block cholinergic bronchoconstriction, thereby decreasing airway smooth-muscle contraction.6 The complementary mechanisms of bronchodilatory action of coadministered LAMAs and LABAs leads to improvements in lung function in patients with COPD compared with LAMA or LABA monotherapy.6–9 LAMA/LABA combination therapy may also decrease the risk of side effects when compared with dose escalation of LAMA and LABA monotherapies,1 and has the potential to offer improved convenience over monotherapies and subsequently improve treatment compliance.
The combination of the inhaled LAMA UMEC with the LABA VI is an approved maintenance treatment for COPD in the US and EU.9 Previous studies in predominantly Western populations have provided evidence for the efficacy of UMEC/VI 125/25 μg and 62.5/25 μg as maintenance therapy in the treatment of moderate–very severe COPD.6,9,10 As interethnic differences are known to exist for some drug classes, the characterization of the efficacy and safety profile of inhaled UMEC/VI in patients with COPD of Asian ancestry is warranted. This study evaluated the efficacy and safety of UMEC/VI 125/25 μg and 62.5/25 μg administered once daily over 24 weeks in patients of Asian ancestry with COPD.
Patients and methods
Study design
This was a 24-week, Phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study (ClinicalTrials.gov identifier NCT01636713, GSK study DB2114634) of once-daily UMEC/VI 125/25 μg and 62.5/25 μg conducted in mainland People’s Republic of China, Philippines, South Korea, Taiwan, and Thailand.
Patients
Eligible patients were male or female, ≥40 years of age at screening, with an established clinical history of COPD, as defined by the American Thoracic Society/European Respiratory Society criteria.11 Patients were also current or former smokers with a smoking history ≥10 pack-years; had a postalbuterol forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio of <0.70 and a postalbuterol FEV1 ≤70% of predicted normal values (based on National Health and Nutrition Examination Survey III reference equations at visit 1),12,13 and a dyspnea score of ≥2 on the modified Medical Research Council Dyspnea Scale at screening. Patients were excluded if they had a current diagnosis of asthma or any other known respiratory disorder, including α1-antitrypsin deficiency or active lung infection, eg, tuberculosis, lung cancer, clinically significant bronchiectasis, pulmonary hypertension, sarcoidosis, or interstitial lung disease. Patients with a previous history or current evidence of clinically significant or uncontrolled cardiovascular, neurological, psychiatric, renal, hepatic, immunological, endocrine, or hematological abnormalities were also excluded. A full list of inclusion and exclusion criteria is detailed in the Supplementary materials.
Written informed consent was obtained from each patient prior to the performance of any study-specific procedure. This study was conducted in accordance with International Conference on Harmonisation Good Clinical Practice and all applicable subject privacy requirements, and the ethical principles that are outlined in the Declaration of Helsinki 2008. This study was approved by the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University and other local ethics committees.
Treatments
Eligible patients were randomized to UMEC/VI 125/25 μg (delivering 113 μg UMEC and 22 μg VI), UMEC/VI 62.5/25 μg (delivering 55 μg UMEC and 22 μg VI), and placebo treatment groups in a 1:1:1 ratio in accordance with the randomization schedule, following a 7- to 14-day run-in period. The randomization schedule was generated by GlaxoSmithKline using the validated computerized system RandAll version 2.5. All randomized study medication was delivered via dry-powder inhalers each morning.
All patients received supplemental albuterol (metered-dose inhaler and/or nebules) to be used as rescue medication throughout the study. The use of inhaled corticosteroid was permitted provided the dose did not exceed 1,000 μg of fluticasone propionate or equivalent per day, and the inhaled corticosteroid was not initiated or discontinued within 30 days prior to study entry.
Investigational product taken during the 24-week treatment period was administered in a double-blind fashion. Neither the subject nor the study physician knew which study medication the subject was receiving.
Study outcomes
Lung-function end points
The primary efficacy end point was trough FEV1 on day 169 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the dose administered on day 168). The secondary lung-function end point was weighted mean (WM) FEV1 over 0–6 hours after dosing on day 1. Other lung-function end points included trough FEV1 at other time points; serial FEV1 over 0–6 hours postdose at day 1; the proportion of patients achieving an increase in FEV1 of ≥12% and ≥0.200 L above baseline at any time 0–6 hours postdose on day 1; the proportion of patients achieving an increase of ≥0.100 L above baseline in trough FEV1; and trough and serial FVC and time to onset (defined as an increase of 0.100 L in FEV1 above baseline) 0–6 hours postdose at day 1.
Symptomatic end points
Symptomatic end points included Transition Dyspnea Index (TDI) focal score at week 24 (which was a secondary end point in this study), TDI focal score recorded at other time points, and proportion of TDI responders (a responder to TDI was defined as a patient who reported a TDI score of ≥1 unit). Additional symptomatic end points were rescue-albuterol use (percentage of rescue-free days and puffs/day) and time to first COPD exacerbation (defined as an acute worsening of symptoms of COPD requiring the use of rescue albuterol or any treatment beyond study medication).
Health-related QoL assessments
Health-related QoL (HRQoL) was measured by the St George’s Respiratory Questionnaire (SGRQ), COPD assessment test (CAT), and a COPD-related health care resource-utilization assessment. Further information on HRQoL assessments is detailed in the Supplementary materials.
Safety assessments
Safety assessments included the incidence of adverse events (AEs) and serious AEs (SAEs), vital signs (including pulse rate and systolic and diastolic blood pressure), 12-lead electrocardiogram (ECG) parameters, and clinical chemistry and hematology parameters (including routine urinalysis). AEs and SAEs were recorded from visit 2 (study-treatment start) until visit 9 (follow-up). Any SAEs assessed as related to study participation (eg, study treatment, protocol-mandated procedures, invasive tests, or change in existing therapy) or related to a GlaxoSmithKline concomitant medication, were recorded from the time informed consent was given up to and including any follow-up contact.
Statistical analysis
Sample size was calculated based on the primary end point of trough FEV1 on day 169 and assumed 90% power, and a two-sided 5% significance level. Furthermore, an estimate of residual standard deviation (SD) of 0.240 L was calculated, in addition to a treatment difference from any UMEC/VI group and placebo of 0.100 L. Based upon these assumptions, 123 evaluable subjects were required for each treatment arm. However, this was increased to 191 evaluable subjects per arm, in order to meet individual country and regional requirements and account for an estimated 21% withdrawal rate over the 24-week study period. Primary analyses were performed on the intent-to-treat (ITT) population, defined as all patients randomized to treatment who had received at least one dose of randomized study medication during the treatment period. For the primary end point of trough FEV1 on day 169, mixed model for repeated measures (MMRM) analysis was performed with all available postbaseline assessments for subjects in the ITT population, including the following covariates: baseline FEV1 (mean of the two assessments made 30 and 5 minutes predose on day 1), smoking status, day, country/region, treatment, day-by-baseline interaction and day-by-treatment interaction, where “day” was nominal. Analysis of mean TDI focal score on days 28, 84, and 168 used MMRM analysis with Baseline Dyspnea Index score in place of the baseline FEV1. WM clinic visit FEV1 over 0–6 hours postdose was analyzed using analysis of covariance with baseline FEV1 (mean of the two assessments made 30 and 5 minutes predose on day 1), treatment, smoking status, and country/region as covariates. Time to onset of bronchodilation and time to first COPD exacerbation were analyzed using Cox’s proportional hazard model with covariates of treatment, smoking status, and country/region.
A step-down testing procedure was employed to adjust for multiplicity on the primary efficacy end point, whereby interpretation of the comparisons between UMEC/VI 62.5/25 μg versus placebo were only carried out if the UMEC/VI 125/25 μg versus placebo comparison was shown to be statistically significant.
Results
Study population
Of the 739 patients screened, a total of 580 patients were enrolled from July 16, 2012 to October 25, 2013 and included in the ITT population. The majority of patients (n=497 [86%]) completed the study (Figure 1). Patients were enrolled across 44 centers in the People’s Republic of China (n=385 [66%]), Taiwan (n=82 [14%]), South Korea (n=51 [9%]), the Philippines (n=33 [6%]), and Thailand (n=29 [5%]). Patient baseline demographics and clinical characteristics were mainly similar between treatment groups; however, there was a higher percentage of subjects in GOLD (Global initiative for chronic Obstructive Lung Disease) stage IV in the UMEC/VI 62.5/25 μg treatment group (18%) compared with placebo (13%) and UMEC 125/25 μg treatment groups (11%). The majority of patients were male, of East Asian heritage and had moderate–severe impairment in airflow obstruction (Table 1). A total of 83 (14%) patients withdrew from the study, and withdrawal rates were similar across all treatment groups (UMEC/VI 125/25 μg, 12%; UMEC/VI 62.5/25 μg, 14%; placebo, 17%).
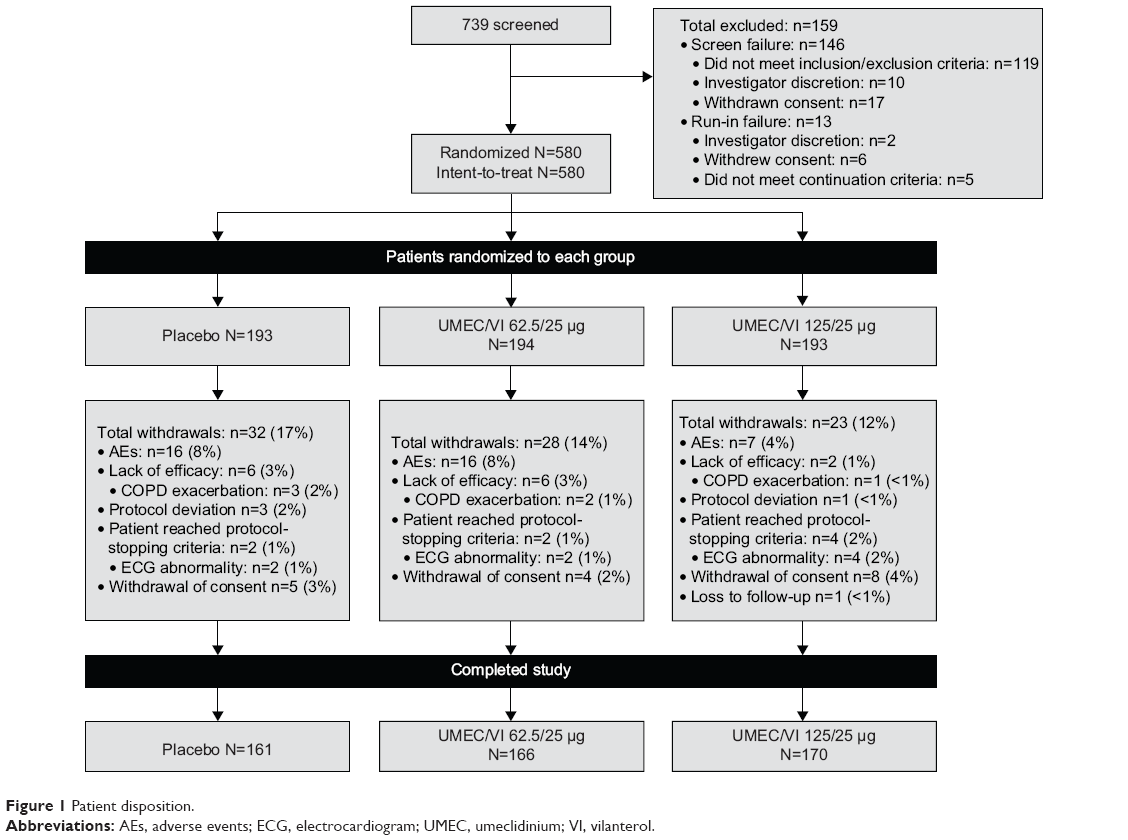 | Figure 1 Patient disposition. |
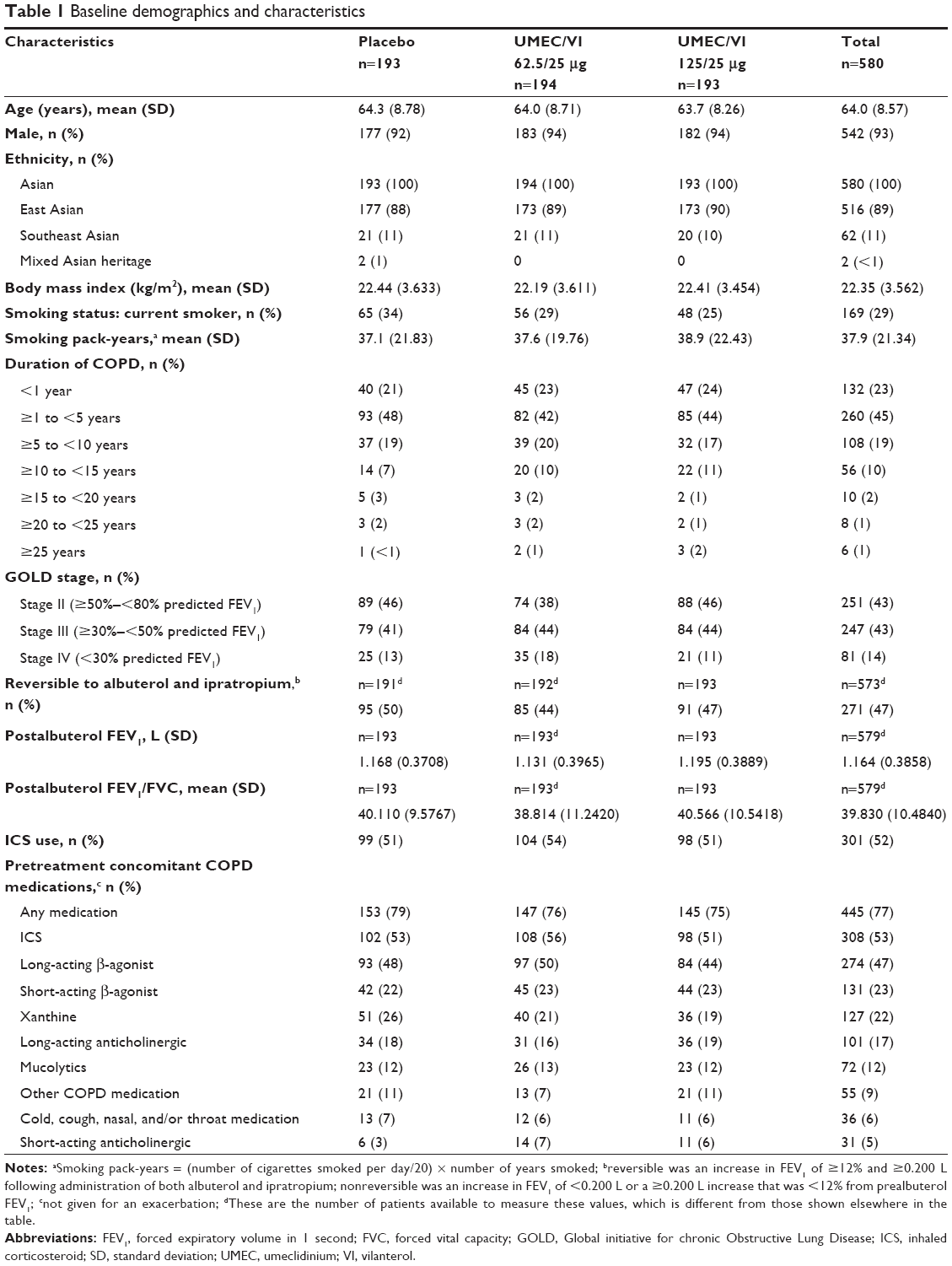 | Table 1 Baseline demographics and characteristics |
Efficacy – lung function
Both UMEC/VI 125/25 μg and UMEC/VI 62.5/25 μg demonstrated a statistically significantly greater change from baseline in trough FEV1 at day 169 compared with placebo (UMEC/VI 125/25 μg, 0.216 L, 95% confidence interval [CI] 0.175–0.257; UMEC/VI 62.5/25 μg, 0.151 L, 95% CI 0.110–0.191; both P<0.001) (Table 2). The treatment differences relative to placebo were also statistically significant versus placebo at all other time points assessed (Figure 2). On day 1, statistically significantly greater changes from baseline in 0- to 6-hour WM FEV1 were observed for both the UMEC/VI 125/25 μg and UMEC/VI 62.5/25 μg treatment groups compared with placebo (UMEC/VI 125/25 μg, 0.182 L, 95% CI 0.161–0.203; UMEC/VI 62.5/25 μg, 0.160 L, 95% CI 0.139–0.181; both P<0.001) (Table 2).
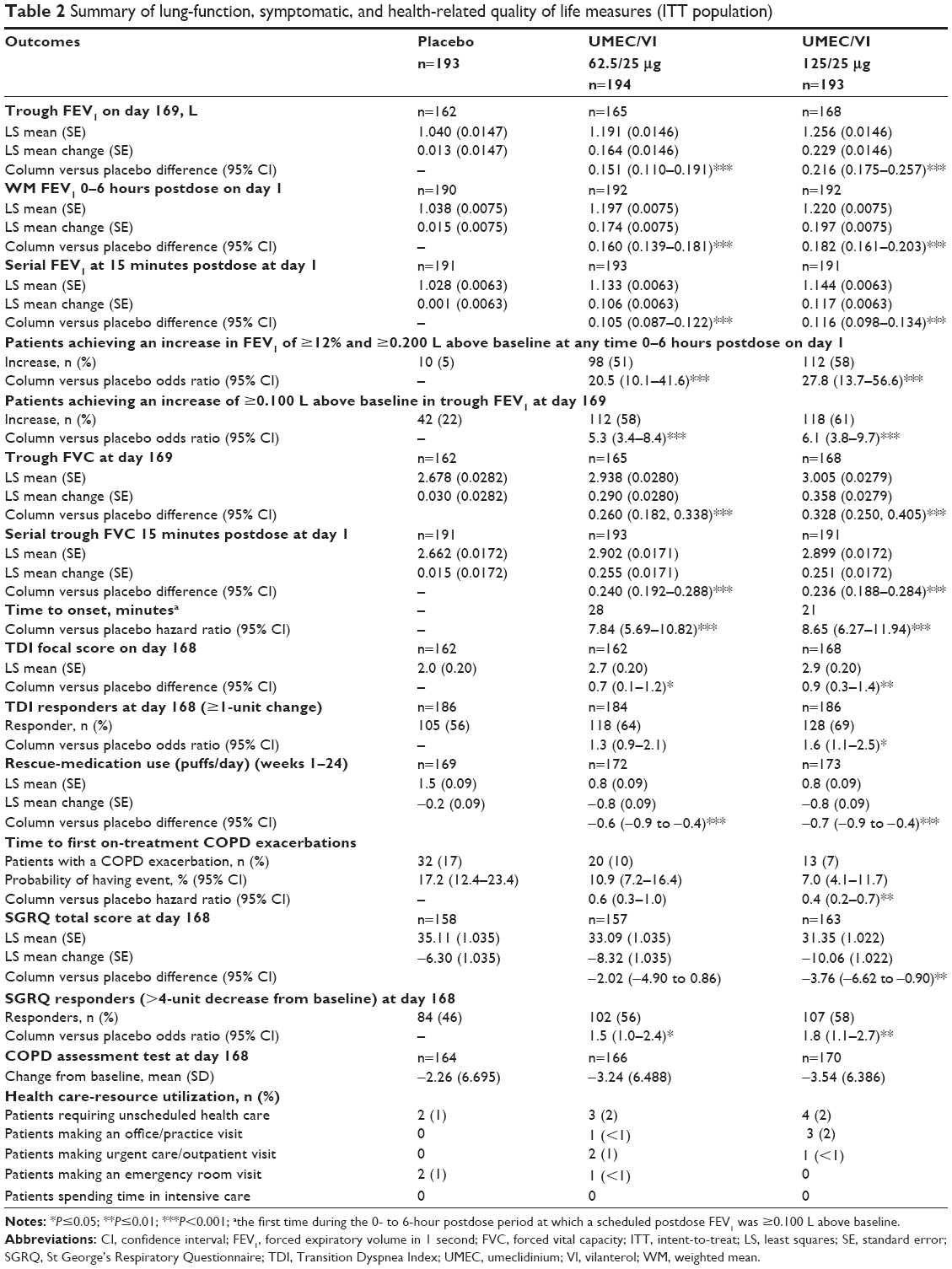 | Table 2 Summary of lung-function, symptomatic, and health-related quality of life measures (ITT population) |
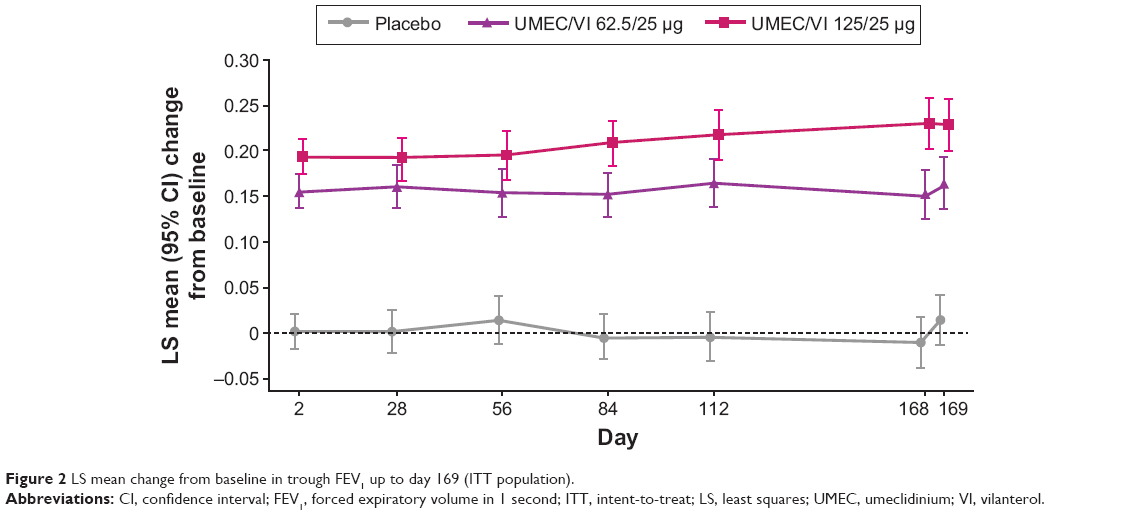 | Figure 2 LS mean change from baseline in trough FEV1 up to day 169 (ITT population). |
On day 1, serial FEV1 for both UMEC/VI 125/25 μg and UMEC/VI 62.5/25 μg demonstrated statistically significantly greater changes compared with placebo at 15 minutes postdose (UMEC/VI 125/25 μg, 0.116 L, 95% CI 0.098–0.134; UMEC/VI 62.5/25 μg, 0.105 L, 95% CI 0.087–0.122; both P<0.001), which were sustained at each time point up to 24 hours postdose (Figure 3). Furthermore, patients had statistically significantly higher odds of achieving an increase in FEV1 of ≥12% and ≥0.200 L above baseline at day 1 compared with placebo (UMEC/VI 125/25 μg, odds ratio [OR] 27.8, 95% CI 13.7–56.6; UMEC/VI 62.5/25 μg, OR 20.5, 95% CI 10.1–41.6; both P<0.001) and also a statistically significantly higher OR of achieving an increase in trough FEV1 of ≥0.100 L above baseline at day 169 compared with placebo (UMEC/VI 125/25 μg, OR 6.1, 95% CI 3.8–9.7; UMEC/VI 62.5/25 μg, OR 5.3, 95% CI 3.4–8.4; both P<0.001).
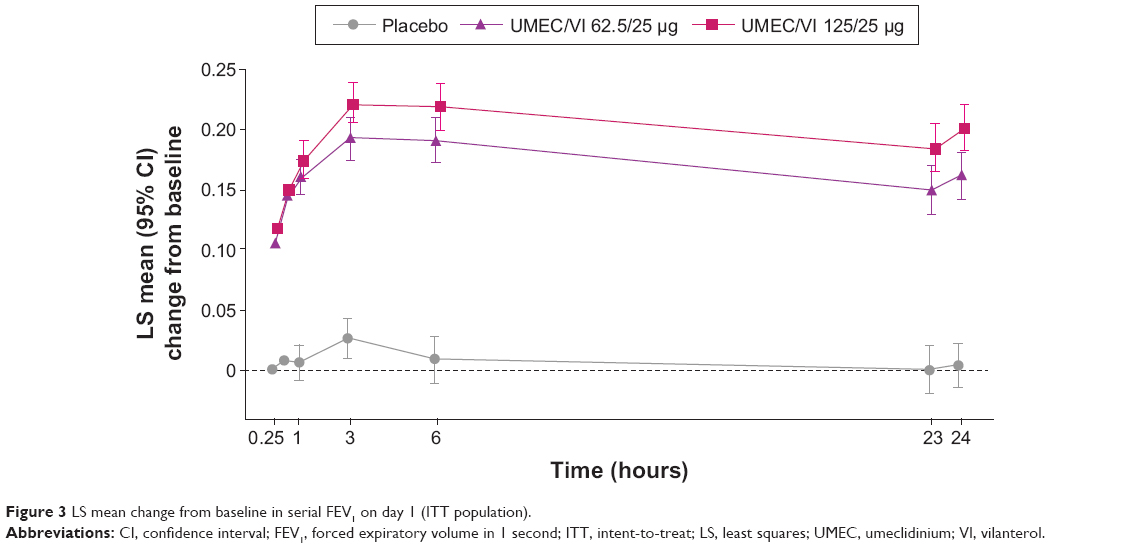 | Figure 3 LS mean change from baseline in serial FEV1 on day 1 (ITT population). |
Treatment differences for trough FVC were statistically significantly greater for patients receiving UMEC/VI compared with patients receiving placebo at day 169 and all other time points assessed (day 169: UMEC/VI 125/25 μg, 0.328, 95% CI 0.250–0.405; UMEC/VI 62.5/25 μg, 0.260, 95% CI 0.182–0.338; both P<0.001). Serial trough FVC measurements for both UMEC/VI treatment groups demonstrated improvements in least-squares mean change from baseline at 15 minutes postdose compared with placebo (UMEC/VI 125/25 μg, 0.236, 95% CI 0.188–0.284; UMEC/VI 62.5/25 μg, 0.240, 95% CI 0.192–0.288; both P<0.001), which were sustained up to 24 hours postdose on day 1.
Efficacy – symptoms
Clinically meaningful improvements in TDI score (defined as a TDI score of ≥1 unit) were observed for both UMEC/VI doses at day 168 and both other days assessed (day 28 and day 84). Furthermore, TDI scores for the UMEC/VI treatment groups were statistically significantly greater compared with placebo at day 168 (UMEC/VI 125/25 μg, 0.9, 95% CI 0.3–1.4, P=0.002; UMEC/VI 62.5/25 μg, 0.7, 95% CI 0.1–1.2, P=0.016) (Table 2). The treatment differences for both UMEC/VI 62.5/25 and 125/25 μg versus placebo were also statistically significant at days 28 and 84 (P<0.001 for both treatments (Figure 4).
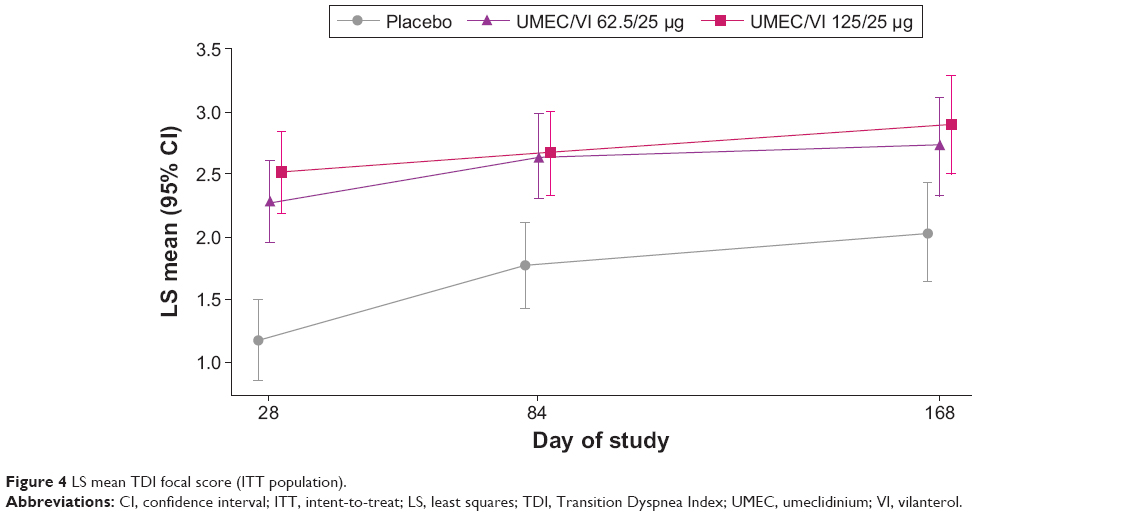 | Figure 4 LS mean TDI focal score (ITT population). |
Patients receiving UMEC/VI also had statistically significantly higher odds of being a TDI responder compared with placebo at all time points assessed, except for day 168 with UMEC/VI 62.5/25 μg (day 168: OR UMEC/VI 125/25 μg, 1.6, 95% CI 1.1–2.5, P=0.022; UMEC/VI 62.5/25 μg, 1.3, 95% CI 0.9–2.1, P=0.163) (Table 2). Rescue-medication use (puffs/day) was statistically significantly reduced with UMEC/VI compared with placebo (UMEC/VI 125/25 μg, −0.7, 95% CI −0.9 to −0.4; UMEC/VI 62.5/25 μg, −0.6, 95% CI −0.9 to −0.4; P<0.001 for both), while the percentage of rescue-free days over weeks 1–24 was greater for UMEC 125/25 μg (67.8%) and UMEC 62.5/25 μg (64.0%) compared with placebo (48.6%).
On-treatment COPD exacerbations were reported by more patients receiving placebo (17%) compared with UMEC/VI 125/25 μg and 62.5/25 μg (7% and 10%, respectively). Analysis of time to first COPD exacerbation found that UMEC/VI 125/25 μg also reduced the risk of COPD exacerbation compared with placebo, but not for UMEC/VI 62.5/25 μg (UMEC/VI 125/25 μg, hazard ratio 0.4, 95% CI 0.2–0.7], P=0.004; UMEC/VI 62.5/25 μg, hazard ratio 0.6, 95% CI 0.3–1.0, P=0.069).
Health-related QoL
UMEC/VI was associated with statistically significant reductions in SGRQ total score that were approximate to the minimal clinically important reduction of 4 units14 at all time points for UMEC/VI 125/25 μg and at day 84 for UMEC/VI 62.5/25 μg compared with placebo (see Table S2 for data). Furthermore, patients treated with UMEC/VI had higher odds of being an SGRQ responder (defined as a >4-unit decrease from baseline) at day 168 relative to placebo (UMEC/VI 125/25 μg, OR 1.8, 95% CI 1.1–2.7, P=0.010; UMEC/VI 62.5/25 μg, OR 1.5, 95% CI 1.0–2.4, P=0.045) (Table 2). Clinically meaningful improvements from baseline in mean CAT scores were observed for both UMEC/VI treatment groups at day 168 (UMEC/VI 125/25 μg, −3.54, minimum–maximum −27.0 to 17.0, SD =6.386; UMEC/VI 62.5/25 μg, −3.24, minimum–maximum −21.0 to 16.0, SD =6.488). The proportion of patients who reported unscheduled health care utilization was low (1%–2% across treatment groups).
Safety
The incidence of AEs was similar across treatment groups (placebo, 39%; UMEC/VI 125/25 μg, 34%; UMEC/VI 62.5/25 μg, 34%); nasopharyngitis and upper respiratory tract infection were the most common AEs across the treatment groups (Table 3). AEs associated with antimuscarinic effects, such as dry mouth and urinary retention, were low and similar across UMEC/VI treatment groups and placebo. Overall, the incidence of drug-related AEs was low across both UMEC/VI treatment groups (UMEC/VI 125/25 μg, 5%; UMEC/VI 62.5/25 μg, 4%) and similar to placebo (5%). There was a low incidence of cardiovascular AEs of special interest (2%–6%) and pneumonia and lower respiratory tract infections (as an AE of special interest, 2%–3%) across treatment groups.
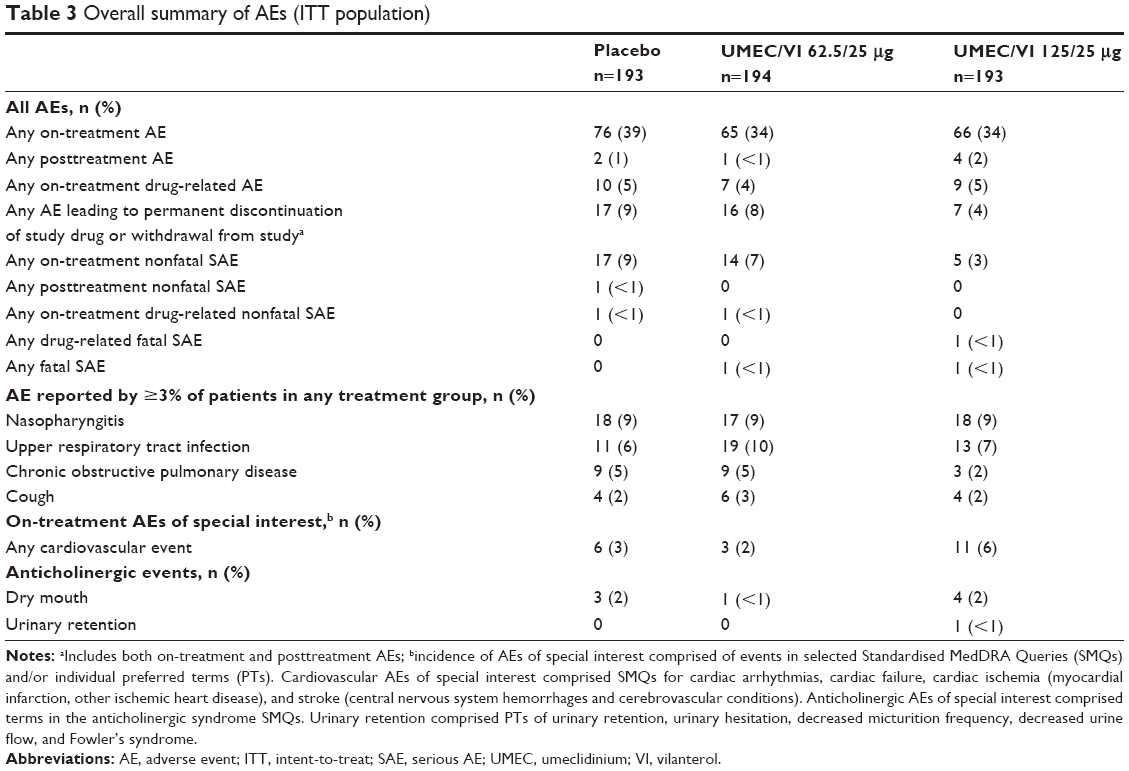 | Table 3 Overall summary of AEs (ITT population) |
On-treatment nonfatal SAEs reported by >1% of patients in any treatment group were COPD (placebo, 5%; UMEC/VI 125/25 μg, 1%; UMEC/VI 62.5/25 μg, 5%) and pneumonia (placebo, 1%; UMEC/VI 125/25 μg, <1%; UMEC/VI 62.5/25 μg, 1%). Two deaths occurred during the study. One patient in the UMEC/VI 125/25 μg group had acute respiratory failure, COPD, pneumonia, and septic shock, which was considered to be treatment-related by the study investigator. One patient in the UMEC/VI 62.5/25 μg group died of drowning, which was not considered to be treatment-related. There were no clinically-meaningful effects on vital signs, ECGs, or clinical chemistry and hematology parameters.
Discussion
This study aimed to examine the efficacy and safety of once-daily inhaled UMEC/VI 125/25 μg and 62.5/25 μg in patients of Asian ancestry with COPD over a 24-week period. In this previously unstudied population, treatment with once-daily UMEC/VI 125/25 μg and UMEC/VI 62.5/25 μg resulted in clinically meaningful improvements in lung-function measurements compared with placebo over a 24-week period.
In addition to improvements in lung function, there was evidence of symptomatic improvement with both UMEC/VI treatment doses, with statistically significant improvements in focal TDI score and a suggested benefit on the frequency of COPD exacerbations. The improvements in lung function and COPD symptoms had additional benefits on QoL parameters, with improvements observed in SGRQ and CAT scores.
These observations are generally consistent with the results of previous studies in predominantly Caucasian patients with COPD, which also reported lung-function improvements, symptom improvements, and improvements in HRQoL with once-daily UMEC/VI.6–10,15 In one such study, trough FEV1 (primary end point) was statistically significantly improved at day 169 for UMEC/VI 62.5/25 μg compared with placebo (difference of 0.167, 95% CI 0.128–0.207 L).6 Similarly, improvements in trough FEV1 from baseline of 0.205–0.211 L for UMEC/VI 62.5/25 μg8,15 and 0.207–0.223 L for UMEC/VI 125/25 μg7,8 have been reported. Greater improvements in 0–6 hour WM FEV1 (secondary end point) at day 168 were reported for UMEC/VI 62.5/25 μg compared with placebo (difference of 0.242 L, 95% CI 0.202–0.282).6 Improvements from baseline in 0–6 hour WM FEV1 of 0.254–0.276 L and 0.263–0.282 L have also been reported for UMEC/VI 62.5/25 μg8,15 and UMEC/VI 125/25 μg, respectively.7,8 TDI score at week 24, reported in several studies, ranged from 2.3 to 2.4 for UMEC/VI 62.5/25 μg6,8 and from 1.8 to 2.9 for UMEC 125/25 μg.7,8 Change from baseline in rescue use (puffs/day) and SGRQ total score ranged from −1.3 to −2.7 and −7.27 to −9.95, respectively, with UMEC 62.5/25 μg,6,8,15 and from −2.2 to −2.7 and −3.6 to −9.95, respectively, with UMEC 125/25 μg.7,8
The safety profile of UMEC/VI was similar to that previously reported in Caucasian patients with COPD, with headache, nasopharyngitis, extrasystoles, upper respiratory tract infection, cough, and back pain being the most commonly reported AEs.6–8,10,15 Both doses of UMEC/VI appeared to be well tolerated, with no notable treatment-related changes in vital signs, ECGs, or clinical laboratory parameters, and no new safety concerns were raised.
The efficacy and safety profile of UMEC/VI in Asian patients with COPD therefore appears to be consistent with previous studies in predominantly Western COPD patient populations,6,8,10 suggesting that ethnicity is not a factor of variance in response to UMEC/VI treatment.
Studies of other LABA/LAMA dual bronchodilators, such as QVA149 (indacaterol and glycopyrronium), have also reported benefits in patients with COPD; however, comparisons with such studies cannot be made, due to differences in study populations and design. In addition, these studies were also carried out in predominantly Caucasian populations (approximately 20%–30% of patients were Asian), and specific subanalyses for race were not reported.16,17 Similarly, a combination of olodaterol and tiotropium has been shown to provide additional improvements in lung function compared with tiotropium alone, but specific Asian subanalyses have not been reported.18
A limitation of the present study was that although the baseline characteristics were generally balanced across the treatment groups, there was a higher percentage of patients with GOLD stage IV COPD in patients receiving UMEC/VI 62.5/25 μg (18%) compared with placebo (13%) and UMEC/VI 125/25 μg (11%). This could explain why there were greater differences in the lung-function response observed between the two UMEC/VI doses than was observed in other studies.8 A large placebo response was also observed in the TDI responder analysis. The reason for this is unknown, but a similar placebo effect has been observed in a previous placebo-controlled UMEC/VI study6 and another COPD trial conducted with patients of Asian heritage.19
Findings from the present study suggested a beneficial effect on the frequency of COPD exacerbations; however, it should be noted that this study was not specifically designed to evaluate the effects of UMEC/VI on COPD exacerbations, thereby limiting the interpretation of these data. Patients in this study were not required to have a history of COPD exacerbations to enroll in the study, and thus only 24%–25% of patients had an exacerbation requiring oral/systemic steroids or antibiotics. Furthermore, patients were required to be withdrawn if they experienced a COPD exacerbation; therefore, no effect on rate of exacerbations could be determined.
Conclusion
The results of the present study indicate that UMEC/VI 125/25 μg and UMEC/VI 62.5/25 μg are beneficial for the once-daily, maintenance bronchodilator treatment of airflow obstruction in Asian patients with moderate – very severe COPD, consistent with studies in predominantly Western populations. The safety profile for UMEC/VI was also comparable to previous studies, and no new safety concerns were identified in this patient population.
Acknowledgments
The authors would like to thank all investigators in the study. GlaxoSmithKline funded this study, and was involved in the study design, the conduct of the study, and the collection, analysis, and interpretation of data. Joanne Ashworth of Fishawack Indicia Ltd provided editorial and formatting assistance in the preparation of the manuscript, which was funded by GlaxoSmithKline.
The authors acknowledge the investigators and participant sites for their great contributions to this study. From the People’s Republic of China: Ping Chen and MingXiao Hou, General Hospital of Shenyang Military Command; Ping Chen and Lianyue Yang, The 2nd Xiangya Hospital, Central South University; Shuliang Guo and Tianyou Luo, The First Affiliated Hospital, Chongqing Medical University; Yong He and Shizhi Fang, Daping Hospital, Third Military Medical University; Xiaoyun Hu and Sijin Li, The First Hospital of Shanxi Medical University; Fuxin Hui and Qing He, Wuxi People’s Hospital; Jian Kang and Xiaosong Yu, The First Hospital of China Medical University; Jianguo Li and Li Yan, Sun Yat-sen Memorial Hospital; Zhikui Li and Mingquan Li, Xijing Hospital; Yuejian L and Shengxi H, Sichuan Provincial People’s Hospital; Shengua Sun and Jiexiang L, The 3rd Xiangya Hospital, Central South University; Huaping Tang and Huamin Ding, Qingdao Municipal Hospital; Changzheng Wang and Jiancheng X, Xinqiao Hospital; Haoyun Wang and Mei Wei, Beijing Friendship Hospital; Zhing-Guang Wen and Jiaqi Lu, Beijing 304 Military Hospital; Bin Wu and Can Chen, Affiliated Hospital of Guangdong Medical College; Lan Yang and Dong Yallin, 1st Affiliated Hospital, Xian Jiaotong University; Jie Zhang and Xiujuan Fu, The Second Hospital of Jilin University; Zhang Wei and Zeqi Zheng, The First Affiliated Hospital of Nanchang University; Li Zhao and Danan Wang, Shengjing Hospital of China Medical University; Rongchang Chen, Guangzhou Institute of Respiratory Disease; Xiaoning Zhong and Mujun Li, 1st Affiliated Hospital of Guangxi Medical University; Xiangdong Zhou and Jun Wu, Southwest Hospital, Third Military Medical University; Xin Zhou and Guohua Liu, Shanghai 1st People’s Hospital. From the Republic of Korea: Ki-Suck Jung and Sang Ho Yoo, Hallym University Sacred Heart Hospital; Cheong Kim and Kwang-Moon Kim, National Health Insurance Corporation Ilsan Hospital, Division of Pulmonary; Yong Chul. Lee and Sung Kwang Park, Respiratory Medicine and Allergy Division; Yong-Ho Roh and Sung Ki Park, Veterans Hospital Services Medical Center; Jee-Hong Yoo and Hyung In Yang, Department of Pulmonary and Critical Care Medicine, Kyung Hee University Hospital at Gangdong. From the Republic of the Philippines: Tito Atziena, Veterans Memorial Medical Center; Marie Isidro and Doris A Mendoza, West Visayas State University Medical Center; Joel Santiaguel and Evelyn Reside, Quirino Memorial Medical Center. From Taiwan: Shih-Lung Cheng, Far-Eastern Memorial Hospital; Jeng-Yuan Hsu and Yen-Chuan Ou, Taichung Veterans General Hospital, Department of Internal Medicine; Wu-Huei H and Martin MT Fuh, China Medical University Hospital, Internal Medicine; Han-Pin Kuo and Tsang-Tang Hsieh, Chang Gung Memorial Hospital-Linkou; Ching-Hsiung Lin and Kun-Tu Yeh, Changhua Christian Hospital; Yu-Chih Liu and Tsang-Tang Hsieh, Chang Gung Memorial Hospital-Keelong; Wei-Juin Su and Shung-Tai Ho, Taipei Veterans General Hospital; Shih-Ming Tsao and Chih-Ping Han, Chung Shan Medical University Hospital; Chin-Chou Wang and Tsang-Tang Hsieh, Chang Gung Memorial Hospital- Kaohsiung Medical Center. From Thailand: Watchara Boonsawat and Suchat Areemit, Srinagarind Hospital, Department of Pulmonary Medicine, Khon Kaen University; Petchara Boonyongsunchai and Suchat Areemit, Srinagarind Hospital, Department of Pulmonary Medicine, Khon Kaen University; Petchara Boonyongsunchai and Yawana Tanapat, Phramongkutklao Hospital, Department of Internal Medicine; Kanok Pipatvech, Nan Hospital; and Yawana Tanapat, Ministry of Public Health.
Author contributions
JPZ, NSZ, AN, AC, and AHG all made a substantial contribution to the conception and design (eg, protocol development and/or design advice), acquisition of data (eg, study investigation), and data analysis and interpretation. All authors contributed toward drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
JPZ has received lecture fees from GlaxoSmithKline. AN, AC, and AHG are employees of GlaxoSmithKline and hold stocks and shares in the company. NSZ reports no conflicts of interest in this work.
References
GOLD (Global initiative for chronic Obstructive Lung Disease). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. Bethesda (MD): GOLD; 2015. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2015_Apr2.pdf. Accessed May 15, 2015. | ||
World Health Organization. Chronic respiratory diseases: burden of COPD. Available from: http://www.who.int/respiratory/copd/burden/en. Accessed May 15, 2015. | ||
Kim C, Yoo KH, Rhee CK, et al. Health care use and economic burden of patients with diagnosed chronic obstructive pulmonary disease in Korea. Int J Tuberc Lung Dis. 2014;18:737–743. | ||
Tan WC, Ng TP. COPD in Asia: where East meets West. Chest. 2008;133:517–527. | ||
Zhong N, Wang C, Yao W, et al. Prevalence of chronic obstructive pulmonary disease in China: a large, population-based survey. Am J Respir Crit Care Med. 2007;176:753–760. | ||
Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107:1538–1546. | ||
Celli B, Crater G, Kilbride S, et al. Once-daily umeclidinium/vilanterol 125/25 μg in COPD: a randomized, controlled study. Chest. 2014;145:981–991. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2:472–486. | ||
Malerba M, Morjaria JB, Radaeli A. Differential pharmacology and clinical utility of emerging combination treatments in the management of COPD − role of umeclidinium/vilanterol. Int J Chron Obstruct Pulmon Dis. 2014;9:687–695. | ||
Donohue JF, Niewoehner D, Brooks J, O’Dell D, Church A. Safety and tolerability of once-daily umeclidinium/vilanterol 125/25 mcg and umeclidinium 125 mcg in patients with chronic obstructive pulmonary disease: results from a 52-week, randomized, double-blind, placebo-controlled study. Respir Res. 2014;15:78. | ||
Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. | ||
Hankinson JL, Kawut SM, Shahar E, Smith LJ, Stukovsky KH, Barr RG. Performance of American Thoracic Society-recommended spirometry reference values in a multiethnic sample of adults: the multi-ethnic study of atherosclerosis (MESA) lung study. Chest. 2010;137:138–145. | ||
Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187. | ||
Jones PW. St George’s Respiratory Questionnaire: MCID. COPD. 2005;2:75–79. | ||
Maleki-Yazdi MR, Kaelin T, Richard N, Zvarich M, Church A. Efficacy and safety of umeclidinium/vilanterol 62.5/25 mcg and tiotropium 18 mcg in chronic obstructive pulmonary disease: results of a 24-week, randomized, controlled trial. Respir Med. 2014;108:1752–1760. | ||
Bateman ED, Ferguson GT, Barnes N, et al. Dual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. Eur Respir J. 2013;42:1484–1494. | ||
Dahl R, Chapman KR, Rudolf M, et al. Safety and efficacy of dual bronchodilation with QVA149 in COPD patients: the ENLIGHTEN study. Respir Med. 2013;107:1558–1567. | ||
ZuWallack R, Allen L, Hernandez G, Ting N, Abrahams R. Efficacy and safety of combining olodaterol Respimat® and tiotropium HandiHaler® in patients with COPD: results of two randomized, double-blind, active-controlled studies. Int J Chron Obstruct Pulmon Dis. 2014;9:1133–1144. | ||
Zheng JP, Yang L, Wu YM, et al. The efficacy and safety of combination salmeterol (50 μg)/fluticasone propionate (500 μg) inhalation twice daily via accuhaler in Chinese patients with COPD. Chest. 2007;132:1756–1763. |
Supplementary materials
Full inclusion/exclusion criteria
Inclusion criteria
- Type of subject: Outpatient.
- Informed consent: Subjects gave their signed and dated written informed consent prior to study participation.
- Age: 40 years of age or older at Screening (Visit 1).
- Sex: Male or female subjects. A female was eligible if she was of non-childbearing potential or was using acceptable contraceptive methods.
- COPD history: Established clinical history of COPD in accordance with the definition by the American Thoracic Society/European Respiratory Society.1
- Tobacco use/smoking history: Current/former cigarette smokers with a history of cigarette smoking of ≥10 pack-years (number of pack-years = [number of cigarettes per day/20] x number of years smoked [eg, 20 cigarettes per day for 10 years, or 10 cigarettes per day for 20 years both equal 10 pack-years]). Former smokers were defined as those who had stopped smoking ≥6 months prior to Visit 1. Note: Pipe and/or cigar use could not be used to calculate pack-year history.
- Severity of disease: A post-albuterol FEV1/FVC ratio of <0.70 and a post-albuterol FEV1 of ≤70% of predicted normal values calculated using National Health and Nutrition Examination Survey III reference equations at Visit 1.2,3
- Dyspnea: A score of ≥2 on the Modified Medical Research Council Dyspnea Scale at Screening (Visit 2).
Exclusion criteria
- Pregnancy: Women who were pregnant or lactating or are planning on becoming pregnant during the study.
- Asthma: A current diagnosis of asthma.
- Other respiratory disorders: Known α-1 antitrypsin deficiency, active lung infections (eg, tuberculosis) and lung cancer were absolute exclusionary conditions. A subject who had any other significant respiratory conditions in addition to COPD were excluded. Eg, clinically significant, bronchiectasis, pulmonary hypertension, sarcoidosis, or interstitial lung disease.
- Other diseases/abnormalities: Subjects with historical or current evidence of clinically significant cardiovascular, neurological, psychiatric, renal, hepatic, immunological, endocrine (including uncontrolled diabetes or thyroid disease) or hematological abnormalities that were uncontrolled and/or a previous history of cancer in remission for <5 years prior to Visit 1 (localized carcinoma of the skin that has been resected for cure was not exclusionary). Significant was defined as any disease that, in the opinion of the investigator, would put the safety of the subject at risk through participation, or which would have affected efficacy or safety analysis if the disease/condition exacerbated during the study.
- Chest X-ray: A chest X-ray or computed tomography (CT) scan that revealed evidence of clinically significant abnormalities not believed to be due to the presence of COPD. A chest X-ray must have been taken at Visit 1 if a chest X-ray or CT scan was not available within 6 months prior to Visit 1.
- Contraindications: history of allergy or hypersensitivity to any anticholinergic/muscarinic receptor antagonist, β2-agonist, lactose/milk protein or magnesium stearate or a medical condition such as of narrow-angle glaucoma, prostatic hypertrophy, or bladder neck obstruction that contraindicated study participation or use of an inhaled anticholinergic.
- Hospitalization: Hospitalization for COPD or pneumonia within 12 weeks prior to Visit 1.
- Lung resection: Subjects with lung volume reduction surgery within the 12 months prior to Visit 1.
- 12-lead ECG: An abnormal and significant ECG finding from the 12-lead ECG conducted at Visit 1, including the presence of a paced rhythm on a 12-lead ECG which caused the underlying rhythm and ECG to be obscured. Investigators were provided with ECG reviews conducted by a centralized independent cardiologist to assist in evaluation of subject eligibility.
- Screening labs: Significantly abnormal finding from clinical chemistry and hematology tests at Visit 1.
- Medication prior to spirometry: Unable to withhold albuterol for the 4-hour period required prior to spirometry testing at each study visit.
- Medications prior to Screening: Use of the medications according to the defined time intervals prior to Visit 1 as shown in Table S1.
Health-related quality of life assessments
St George’s Respiratory Questionnaire (SGRQ)
The SGRQ4 is a disease-specific questionnaire designed to measure the impact of respiratory disease and its treatment on a subject’s health related quality of life. As well as producing an overall summary score, it is also possible to calculate scores for the individual domains of symptoms, activity, and impacts. It has been widely used in studies of COPD and has been translated and validated for use in most major languages. Research has demonstrated that it is sensitive to change and interpretation of the results has been enhanced by determination of the score change necessary to achieve a clinically-meaningful improvement in quality of life.5
The SGRQ contains 76 items grouped into three domains (symptoms, activity, and impacts). The domain score is calculated as the sum of the weighted scores for the non-missing items within each domain, divided by the maximum possible score for those non-missing items and multiplied by 100. The SGRQ total score is calculated as the sum of the weighted scores from all 76 items, divided by the maximum possible score for the SGRQ, multiplied by 100. A lower SGRQ score indicates better health status. The minimum clinically-important difference for the SGRQ is a -4-unit difference.5 The SGRQ was self-completed by subjects prior to spirometry at Visits 2, 4, 6, and 8.
COPD assessment test (CAT)
The CAT is a subject-completed instrument designed to provide a simple and reliable measure of health status in COPD. The CAT was designed to measure overall COPD-related health status for the assessment and long-term follow-up of individual subjects. The instrument consists of 8 items, each formatted as a semantic 6-point differential scale.6
The CAT was completed independently and without supervision by all subjects at Visits 2, 4, 6, and 8. The CAT was administered before any other study procedures were performed (including concurrent medication assessment or AE assessment, etc).
COPD-related healthcare resource utilization assessment
All unscheduled COPD-related visits to a physician’s office, urgent care facility, or emergency department, and COPD-related hospitalizations were recorded on the COPD-related healthcare resource use assessment worksheet within the subject’s diary, by the subject. At Visits 2 through 9 or at the Early Withdrawal Visit, the resource utilization worksheet completed by the subject to record all health care contacts since the last visit, was reviewed by the investigator (or designee). The investigator (or designee) asked the subject if any of the health care contacts recorded on the worksheets were due to COPD exacerbation. The investigator could refer to his/her records to verify or supplement information given by the subject if necessary. If any unscheduled health care contact was due to a COPD exacerbation, then the COPD exacerbation section of the electronic case report form was completed.
References
Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. | ||
Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187. | ||
Hankinson JL, Kawut SM, Shahar E, Smith LJ, Stukovsky KH, Barr RG. Performance of American Thoracic Society-recommended spirometry reference values in a multiethnic sample of adults: the multi-ethnic study of atherosclerosis (MESA) lung study. Chest. 2010;137:138–145. | ||
Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory Questionnaire. Am Rev Respir Dis. 1992;145:1321–1327. | ||
Jones PW. St George’s Respiratory Questionnaire: MCID. COPD. 2005;2:75–79. | ||
Jones PW, Harding G, Berry P, Wiklund I, Chen WH, Kline Leidy N. Development and first validation of the COPD Assessment Test. Eur Respir J. 2009;34:648–654. |
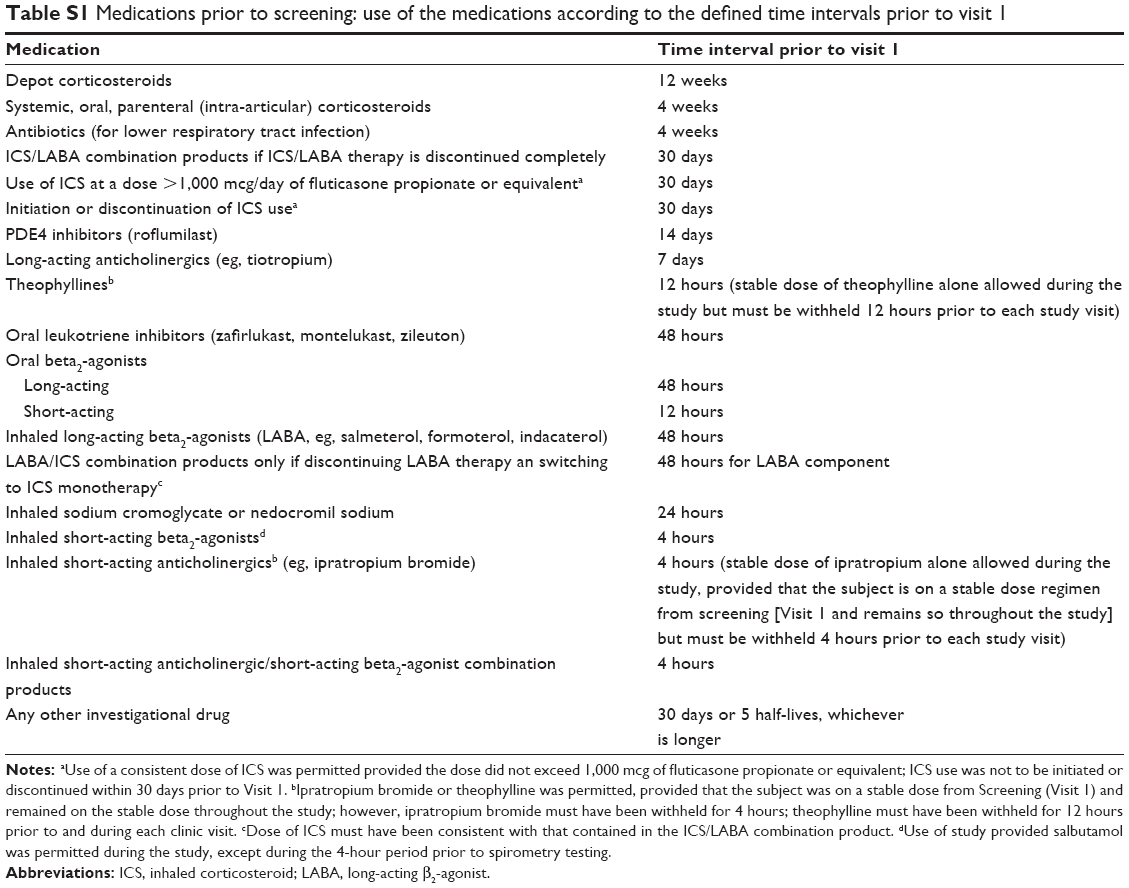 | Table S1 Medications prior to screening: use of the medications according to the defined time intervals prior to visit 1 |
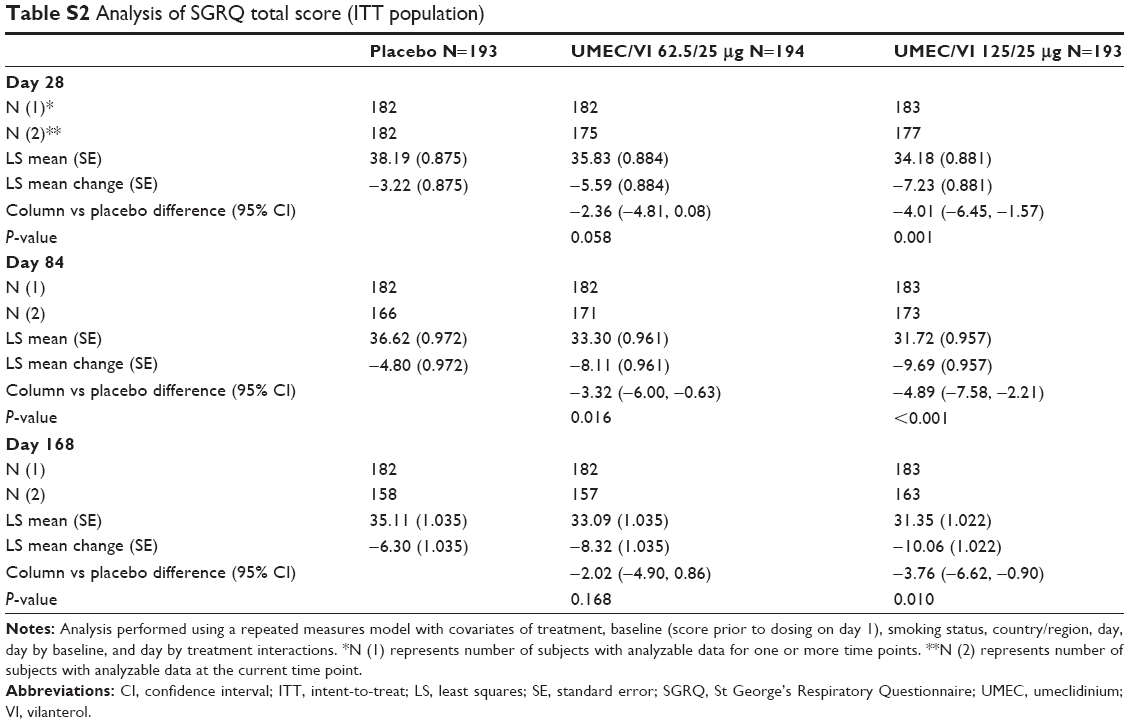 | Table S2 Analysis of SGRQ total score (ITT population) |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.