")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Efficacy and safety of four doses of glycopyrrolate/formoterol fumarate delivered via a metered dose inhaler compared with the monocomponents in patients with moderate-to-severe COPD
Authors Reisner C , Pearle J, Kerwin EM , St Rose E, Darken P
Received 24 February 2018
Accepted for publication 16 May 2018
Published 19 June 2018 Volume 2018:13 Pages 1965—1977
DOI https://doi.org/10.2147/COPD.S166455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Colin Reisner,1,2 James Pearle,3 Edward M Kerwin,4 Earl St Rose,1 Patrick Darken1
1Pearl – a member of the AstraZeneca Group, Morristown, NJ, USA; 2AstraZeneca, Gaithersburg, MD, USA; 3California Research Medical Group, Inc., Fullerton, CA, USA; 4Clinical Research Institute of Southern Oregon, Medford, OR, USA
Purpose: To determine the efficacy and safety of glycopyrrolate/formoterol fumarate metered dose inhaler (GFF MDI 36/9.6, 36/7.2, 18/9.6, 9/9.6 μg) using innovative co‑suspension delivery technology, compared with glycopyrrolate (GP) MDI 36 μg and formoterol fumarate (FF) MDI 9.6 μg, in patients with moderate-to-severe COPD.
Methods: In this Phase IIb, randomized, double-blind, balanced incomplete-block, two-period, cross-over study (NCT01349816), patients received treatment twice-daily for 7 days. The primary efficacy endpoint was forced expiratory volume in 1 second (FEV1) area under the curve from 0 to 12 hours (AUC0–12) on Day 7. Secondary efficacy endpoints were peak change from baseline in FEV1 through 2 hours; time to onset of action (≥10% improvement in mean FEV1); proportion of patients achieving ≥12% improvement in FEV1 on Day 1; peak change from baseline in inspiratory capacity (IC) on Days 1 and 7; change from baseline in morning pre-dose FEV1; peak change from baseline in FEV1 through 6 hours; and change from baseline in mean evening 12-hour post-dose trough FEV1 on Day 7. Safety was assessed.
Results: All 185 randomized patients received treatment. All doses of GFF MDI significantly improved the primary endpoint compared with GP MDI 36 μg (all P≤0.0137). For peak change in FEV1 and IC and time to onset of action secondary endpoints, ≥2 doses of GFF MDI demonstrated superiority to GP MDI 36 μg. No significant differences were observed between GFF MDI and FF MDI 9.6 μg for primary and secondary endpoints. The incidence of adverse events was similar between treatments.
Conclusion: While all doses of GFF MDI were superior to GP MDI 36 μg for the primary endpoint, in this study neither superiority of GFF MDI to FF MDI 9.6 μg nor a clear dose-response was observed. All treatments were well tolerated with no unexpected safety findings.
Keywords: bronchodilator, chronic obstructive pulmonary disease, co-suspension delivery technology, fixed-dose combination, LAMA/LABA, long-acting β2-agonist, long-acting muscarinic antagonist
Introduction
Chronic obstructive pulmonary disease was responsible for 3.2 million deaths worldwide in 2015,1 and COPD is projected to be the fourth leading cause of death by 2030.2 Pharmacologic treatment of COPD, which centers around long-acting muscarinic antagonist (LAMA) and long-acting β2-agonist (LABA) bronchodilators, has been shown to decrease both symptoms and exacerbations, and to improve patients’ health status.3 Stepping up treatment to combined LAMA/LABA therapy is recommended for patients who have persistent symptoms or experience further exacerbations after treatment with LAMA or LABA monotherapy.3
Glycopyrrolate/formoterol fumarate (GFF) is a LAMA/LABA fixed-dose combination therapy delivered via metered dose inhaler (MDI) using innovative co-suspension delivery technology. Co-suspension delivery technology allows the uniform delivery of both treatments in the same inhaler,4,5 and GFF MDI (Bevespi Aerosphere®, AstraZeneca, Wilmington, DE, USA) is deposited in the lungs with high efficiency.6 GFF MDI 18/9.6 μg (equivalent to glycopyrronium/formoterol fumarate dihydrate 14.4/10 μg) has been approved in the USA for the long-term maintenance treatment of airflow obstruction in patients with COPD.7
This Phase IIb study was part of a wider clinical trial program for GFF MDI in COPD, to allow the identification of the optimal dose of GFF MDI for use in Phase III studies.8,9 Findings from other dose-ranging Phase II studies have been published previously, and supported the selection of 18 μg glycopyrrolate (GP) and 9.6 μg foromoterol fumarate (FF) in the GFF MDI fixed-dose combination.10,11 Here we investigated the efficacy and safety of four doses of GFF MDI (36/9.6 μg, 36/7.2 μg, 18/9.6 μg, and 9/9.6 μg) compared with the monocomponents, GP MDI 36 μg and FF MDI 9.6 μg, in patients with moderate-to-severe COPD.
Methods
Study design
This was a Phase IIb, randomized, double-blind, 7-day dosing, two-period, six-treatment, balanced incomplete-block, cross-over study in patients with moderate-to-severe COPD, which was conducted across 14 sites in the USA between 06 July 2011 and 19 November 2011 (Figure 1). Patients were randomized using an interactive web response system to one of 30 treatment sequences, each of which included two of the six treatments: GFF MDI 36/9.6 μg, 36/7.2 μg, 18/9.6 μg, 9/9.6 μg, GP MDI 36 μg, and FF MDI 9.6 μg (all twice-daily).
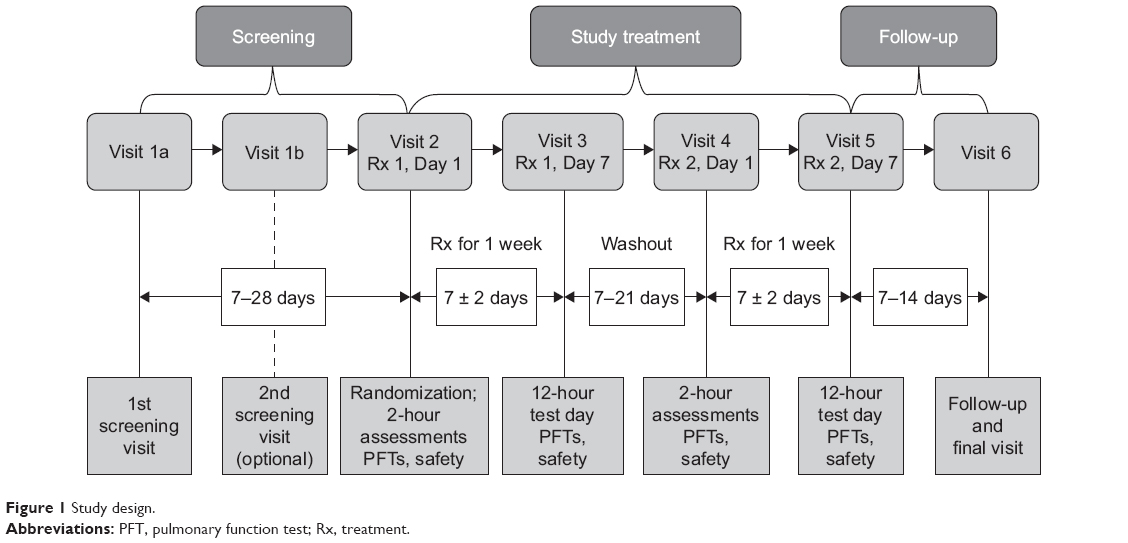 | Figure 1 Study design. |
Each treatment period lasted 7 ± 2 days. Patients reported to the clinic on Days 1 and 7 of each treatment period and remained until all scheduled assessments were completed. Patients underwent a washout period of 7-28 days prior to the first treatment period, and 7-21 days between treatment periods. Seven to 14 days after the end of the second treatment period, patients returned to the clinic for a follow-up visit (Figure 1).
During the screening and washout periods, patients were permitted to use ipratropium MDI, albuterol/ipratropium MDI combination, or sponsor-provided albuterol MDI, at the Investigator’s discretion. During the treatment periods, patients were permitted to use sponsor-provided albuterol MDI as a rescue medication on an as-needed basis. Patients receiving an inhaled corticosteroid (ICS) as a maintenance therapy were allowed to continue the ICS if they had been on a stable dose for 4 weeks or longer. If the ICS was taken as part of a fixed-dose combination with a LABA, patients were instead provided with the corresponding ICS dose as a single therapy, and were permitted to use albuterol MDI, ipratropium MDI, or albuterol/ipratropium MDI combination at the Investigator’s discretion. Use of any COPD medications, including ICS, was prohibited for at least 6 hours before each clinic visit. Patients were prohibited from smoking for at least 4 hours before, and during, each clinic visit, were not allowed to consume grapefruit or grapefruit juice during the study, and were prohibited from ingesting xanthine-containing foods or beverages (such as coffee, tea, chocolate and cola) for at least 6 hours before, and during, each clinic visit. Patients were reminded of these criteria at each visit and the study visit was rescheduled if a patient failed to meet any of the criteria.
The study was conducted in accordance with Good Clinical Practice, including the International Conference on Harmonization, the US Code of Federal Regulations and the Declaration of Helsinki. The protocol and informed consent form were reviewed and approved by a central (Independent Investigational Review Board, Inc., Plantation, FL, USA) and local (Western Institutional Review Board, Olympia, WA, USA) institutional review board. Written informed consent was obtained from each patient prior to screening. This study was registered on the US National Institutes of Health ClinicalTrials.gov website (NCT01349816).
Study population
Inclusion criteria
Male and female patients 40-80 years of age, who were current or former smokers with a history of ≥10 pack-years were eligible for inclusion. Patients were required to have an established clinical history of COPD as defined by the American Thoracic Society/European Respiratory Society,12 with a pre- and postbronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) <0.70, a postbronchodilator FEV1 ≥30% and <80% of predicted normal value and ≥750 mL at screening, and a prebronchodilator FEV1 <80% of predicted normal value at baseline (predicted normal values were calculated using the Third National Health and Nutrition Examination Survey reference equations). In addition, patients had to be willing and able, in the opinion of the Investigator, to change current COPD therapy. Clinical laboratory tests and an electrocardiogram (ECG) conducted at screening and a chest X-ray or CT scan taken within 6 months of screening had to be deemed acceptable by the Investigator (based on his/her clinical expertise) for inclusion in the study. Female patients must have agreed to take acceptable contraceptive precautions during the study, where appropriate.
Exclusion criteria
Patients with a primary diagnosis of asthma, α-1 antitrypsin deficiency, other respiratory disorders that in the opinion of the Investigator would have affected the study, or any other clinically significant medical conditions were excluded from this study. A patient with a previous diagnosis of asthma was eligible if COPD was his/her primary diagnosis at screening. Patients who had undergone prior lung volume reduction surgery, had poorly controlled COPD (defined as acute worsening of COPD requiring corticosteroid or antibiotic treatment 6 weeks before screening or between screening and randomization) or had been hospitalized due to poorly controlled COPD in the 3 months prior to screening, were receiving long-term oxygen therapy for >12 hours per day, had lower respiratory tract infections that required treatment with antibiotics in the 6 weeks before screening, or could not perform spirometry that was deemed acceptable (≥3 acceptable flow-volume curves with ≥2 meeting American Thoracic Society reproducibility criteria) were excluded from this study. Patients who for medical reasons could not withhold their short-acting bronchodilators for 6 hours before spirometry assessments were performed, and those who had poor hand-to-breath coordination with an MDI, and therefore required a spacer device were also excluded. Additionally, patients who had previously experienced hypersensitivity to any β2-agonists or muscarinic antagonists, or any component of the MDI, and patients with a history of substance abuse within 2 years prior to screening were not eligible for this study.
Efficacy endpoints
The primary efficacy endpoint of this study was FEV1 area under the curve from 0 to 12 hours (AUC0–12) relative to baseline and normalized by the nominal total period of evaluation (12 hours) on Day 7. The secondary efficacy endpoints evaluated on Day 1 were peak change from baseline in FEV1 through 2 hours; time to onset of action (≥10% improvement in mean FEV1); proportion of patients achieving ≥12% improvement in FEV1; and peak change from baseline in inspiratory capacity (IC, mean of 1 and 2 hours postdose). Secondary efficacy endpoints evaluated on Day 7 were change from baseline in morning predose FEV1 (average of 1 hour and 30 minutes predose); peak change from baseline in FEV1 through 6 hours; peak change from baseline in IC; and change from baseline in mean evening 12-hour postdose trough FEV1 (mean of 11.5 and 12 hours postdose). Baseline FEV1 and IC were the average of the two predose assessments (30 minutes and 1 hour) on Day 1 of both treatment periods.
Efficacy assessments
FEV1, FVC, and IC pulmonary function tests (PFTs) were carried out in accordance with American Thoracic Society criteria.13 All sites were provided with identical spirometry systems (KoKo® Spirometer, nSpire Health, Inc., Longmont, CO, USA), and all study staff who performed PFTs were experienced in conducting such tests and received standardized training at Investigator meetings. They were required to demonstrate the ability to perform technically acceptable tests based on American Thoracic Society criteria before testing patients in this study. Throughout the study, all spirometry data were independently reviewed against American Thoracic Society criteria using a centralized quality assurance process. On Day 1 of each treatment period, spirometry assessments were performed 30 minutes and 1 hour predose, and 15 minutes, 30 minutes, 1 hour, and 2 hours postdose. IC was performed 30 minutes and 1 hour predose, and 1 and 2 hours postdose. On Day 7 of each treatment period, spirometry assessments were performed 30 minutes and 1 hour predose, and 15 minutes, 30 minutes, and 1, 2, 4, 6, 8, 10, 11.5, and 12 hours postdose. IC was evaluated 30 minutes and 1 hour predose, and 1, 2, 11.5, and 12 hours postdose. Patients were provided with diaries at screening and on Day 1 of each treatment period to record the actual time of dosing every day.
Safety evaluations
ECGs and vital signs (including systolic and diastolic blood pressure and heart rate) were assessed 30 minutes and 1 hour predose on Days 1 and 7, up to 2 hours postdose on Day 1, and up to 12 hours postdose on Day 7 of each treatment period. The following abnormal clinical findings may have led to discontinuation from the study: QT interval prolongation (Fridericia’s corrected QT interval [QTcF]>500 msec any time after treatment, or a >60 msec increase from test day baseline); increased heart rate (>120 bpm within the 12 hours after treatment, or a >40 bpm increase from test day baseline) or increased systolic blood pressure (>180 mmHg within the 12 hours after treatment, or a >40 mmHg increase from test day baseline). Clinical laboratory tests (including hematology and clinical blood chemistry) were assessed 1 hour predose on Days 1 and 7, and up to 2 hours postdose on Days 1 and 7 of each treatment period. Adverse events (AEs), including serious AEs, were recorded on Days 1 and 7 of each treatment period, and at the follow-up visit of 7-14 days after the end of the last treatment period. Paradoxical bronchospasm, dry mouth, and tremor were AEs of interest.
Statistical analyses
The safety population included all patients who were randomized, received any study treatment, and had a postbaseline safety assessment for that treatment. The intent-to-treat (ITT) population included all patients who were randomized, received any study treatment, and had both baseline and postbaseline efficacy data for that study treatment. Patients who took less than one full dose of a study treatment were eligible for inclusion in the safety and ITT populations. The modified ITT (mITT) population included all patients who have completed the two treatment periods with predose data on Day 1 for both treatment periods, had at least one predose assessment on Day 7 for both treatment periods, and had no protocol deviations believed to have a potential impact on efficacy results.
The analysis for the primary efficacy endpoint (FEV1 AUC0–12 on Day 7 relative to baseline and normalized by the nominal total period of evaluation [12 hours]) involved eight predefined a priori treatment comparisons for superiority comprising each of the four GFF MDI doses compared with (i) GP MDI 36 μg and (ii) FF MDI 9.6 μg. In addition, a comparison of GP MDI 36 μg versus FF MDI 9.6 μg was also reported. Strong control of the family-wise Type I error was achieved by hierarchical testing.14 Assessment was based on two independent sets of testing hierarchies, one for the comparison of the GFF MDI doses versus GP MDI 36 μg and one for the comparison of the GFF MDI doses versus FF MDI 9.6 μg. These comparisons were made using a linear mixed effects model. This model with FEV1 AUC0–12 as the dependent variable included the following factors: baseline FEV1 (a covariate), patient (sequence) (a random factor), period (a fixed factor), treatment (a fixed factor), and sequence (a fixed factor). For this model, potential additional correlation for values within-patient, beyond treating patient as a random effect, was fit as unstructured.
Preliminary data summaries were prepared to determine whether a transformation of the data was required to satisfy the necessary distributional assumptions underlying the statistical methodology to be employed. The assumption of normality in the data was checked by visually inspecting the distribution of the residuals. The assumption of homogeneity of variance was also verified by inspection of scatterplots or box plots of residuals by treatment group. If the assumption of homogeneity of treatment variance did not hold, the mixed model above was to be used for analysis but it was to be run allowing for unequal treatment variances. In the event of nonnormality, a log transformation was to be performed.
Secondary efficacy analysis involved the same comparisons as described for the primary efficacy endpoint on the secondary efficacy endpoints. It was analyzed using the same mixed model and the same algorithms, with the exception of the proportion of patients achieving ≥10% or ≥12% improvement in FEV1 which were analyzed using McNemar’s test, and time to onset of action which was analyzed using Murray’s method for weighted Kaplan–Meier statistics for paired data.15
Approximately 175 patients were planned for recruitment, to ensure approximately 150 completed patients at study termination, to achieve 91% power to demonstrate differences of 100 mL in FEV1 AUC0–12 on Day 7 (assuming a significance test at the 5% level, with no multiplicity adjustment and within- and between-patient variance components standard deviations of 130 mL).
Results
Study population
A total of 301 patients were screened and 185 were randomized and treated (Figure 2), and were included in the ITT and safety populations. Of the 155 patients (83.8%) who completed this study, 146 (94.2%) were included in the mITT population. A total of 30 patients withdrew early from the study for the reasons detailed in Figure 2.
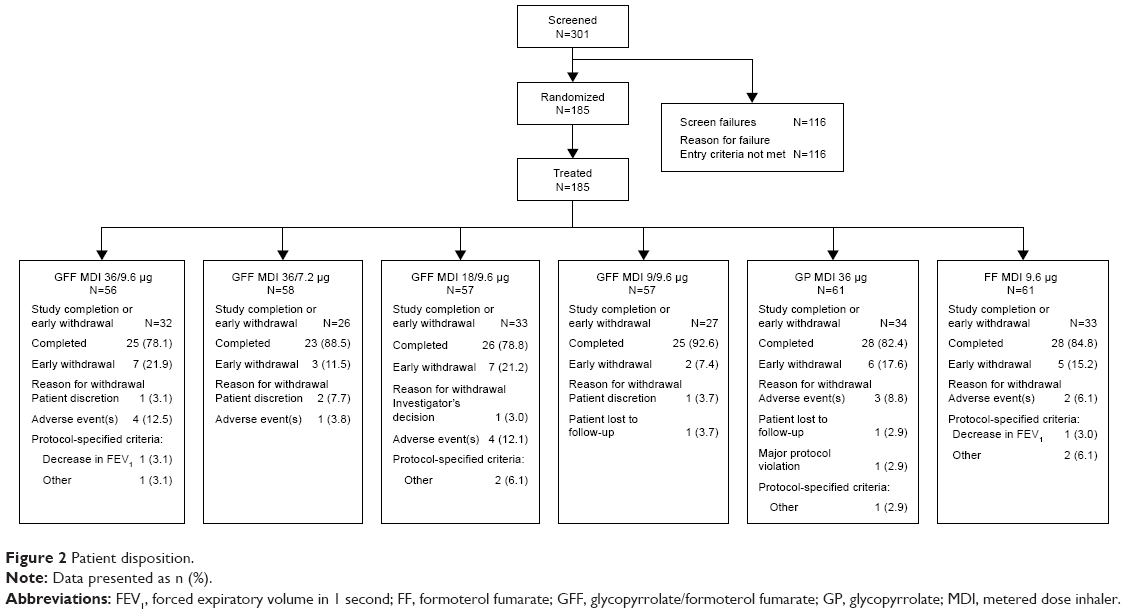 | Figure 2 Patient disposition. |
In the safety/ITT population, the mean age was 62.1 years, and the majority of patients were male (57.8%) and Caucasian (91.9%; Table 1). There were no clinically relevant differences in demographic characteristics, smoking status, and disease duration between treatments, although the percentage of male patients was somewhat higher in patients treated with GFF MDI 36/7.2 μg (67.2%) and lower in patients treated with GFF MDI 9/9.6 μg (49.1%; Table 1).
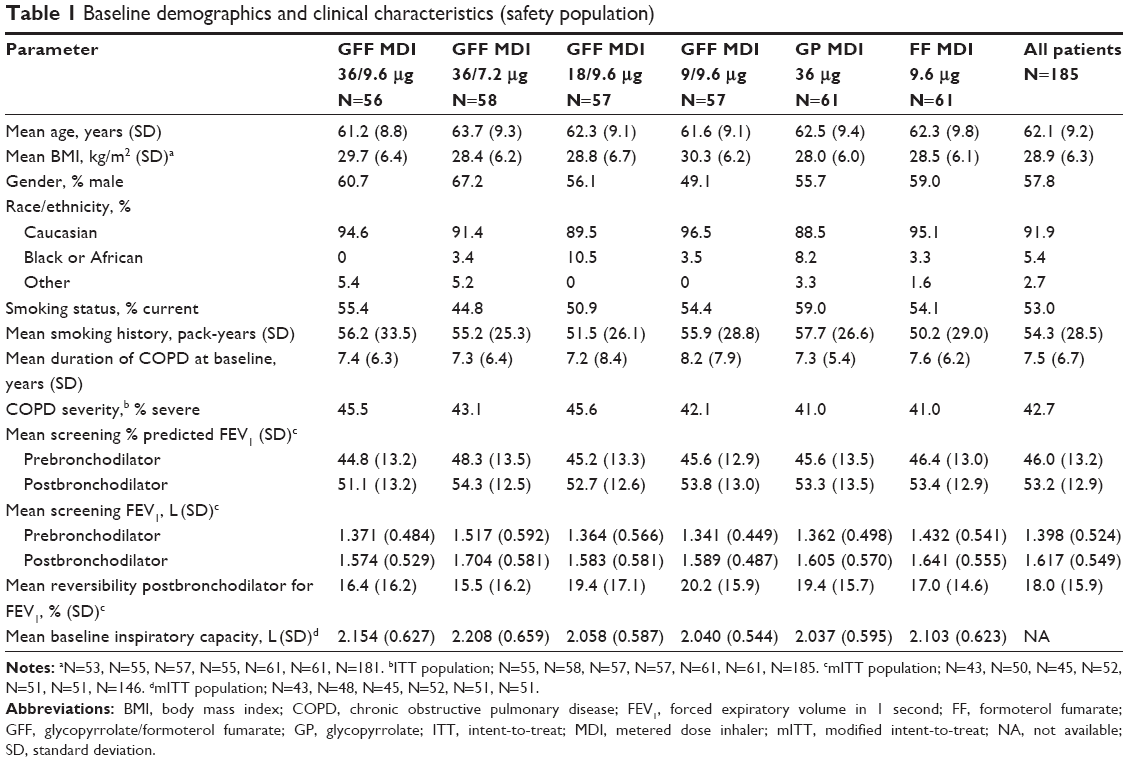 | Table 1 Baseline demographics and clinical characteristics (safety population) |
Use of permitted short-acting bronchodilators was similar between treatment groups, ranging from 1.8%–4.9% for ipratropium MDI (1–3 patients per group), 5.3%–14.8% for albuterol/ipratropium MDI combination (3–9 patients), and 48.2%–62.3% (27–38 patients) for albuterol MDI.
Primary efficacy endpoint
FEV1 AUC0–12 on Day 7
As the data for the primary efficacy endpoint were not normally distributed, analysis was carried out on log-transformed data, and the differences between treatments were reported as geometric mean ratios. All doses of GFF MDI resulted in significant improvements versus GP MDI 36 μg in FEV1 AUC0–12 on Day 7 (Geometric mean ratios: 1.045–1.072; P≤0.0137; Figure 3A). For the nonlog transformed data, the estimated least squares mean (LSM) differences for all doses of GFF MDI compared with GP MDI 36 μg were 51–86 mL. When all doses of GFF MDI were compared with FF MDI 9.6 μg, no significant differences were observed (Geometric mean ratios: 0.998–1.023; Figure 3A). For the nonlog-transformed data, the estimated LSM differences for all doses of GFF MDI compared with FF MDI 9.6 μg were 1–36 mL. In addition, no pairwise comparisons among the GFF MDI doses were significant.
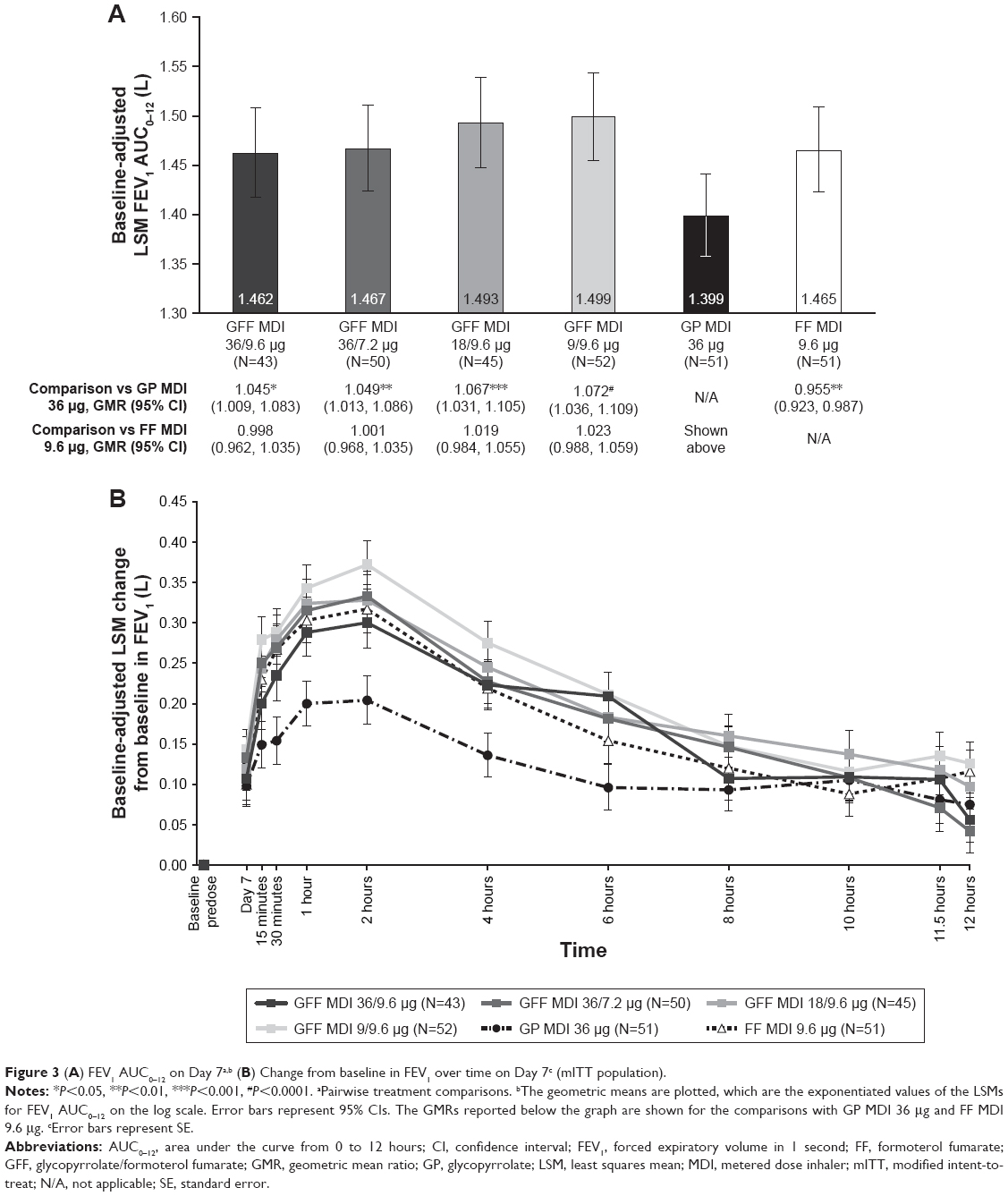 | Figure 3 (A) FEV1 AUC0–12 on Day 7a,b (B) Change from baseline in FEV1 over time on Day 7c (mITT population). |
All doses of GFF MDI, GP MDI 36 μg, and FF MDI 9.6 μg showed a similar profile for FEV1 over time, with an early onset of action that peaked at 2 hours postdose and then gradually decreased over the 12-hour period. GP MDI 36 μg led to a robust increase in FEV1 of 204 mL compared with baseline at 2 hours postdose, with sustained improvements of 75 mL compared with baseline at 12 hours postdose. All doses of GFF MDI led to further improvements in FEV1 compared with GP MDI 36 μg up to 10 hours post-dose, confirming the benefits of adding formoterol to GP in GFF MDI (Figure 3B).
Secondary efficacy endpoints
Secondary efficacy endpoints on Day 1
All doses of GFF MDI demonstrated superiority to GP MDI 36 μg for peak change in FEV1 through 2 hours (LSM differences: 64–102 mL; P≤0.0392), with the exception of GFF MDI 36/9.6 μg (LSM difference: 60 mL; P=0.0556; Figure 4A). The onset of action of GFF MDI 36/7.2 and 9/9.6 μg was faster than GP MDI 36 μg (mean differences: −0.30 and −0.27 hours, respectively; P≤0.0088; Table 2). For the proportion of patients achieving ≥12% improvement in FEV1, none of the pairwise comparisons between any of the GFF MDI doses and GP MDI 36 μg were nominally significant (Table 2). GFF MDI 18/9.6 and 9/9.6 μg were superior to GP MDI 36 μg for peak change from baseline in IC (LSM differences: 111 and 98 mL, respectively; P≤0.0395; Figure 4B). No pairwise comparisons between GFF MDI doses and FF MDI 9.6 μg or among the GFF MDI doses were nominally significant for any of the secondary endpoints on Day 1 (Figure 4, Table 2).
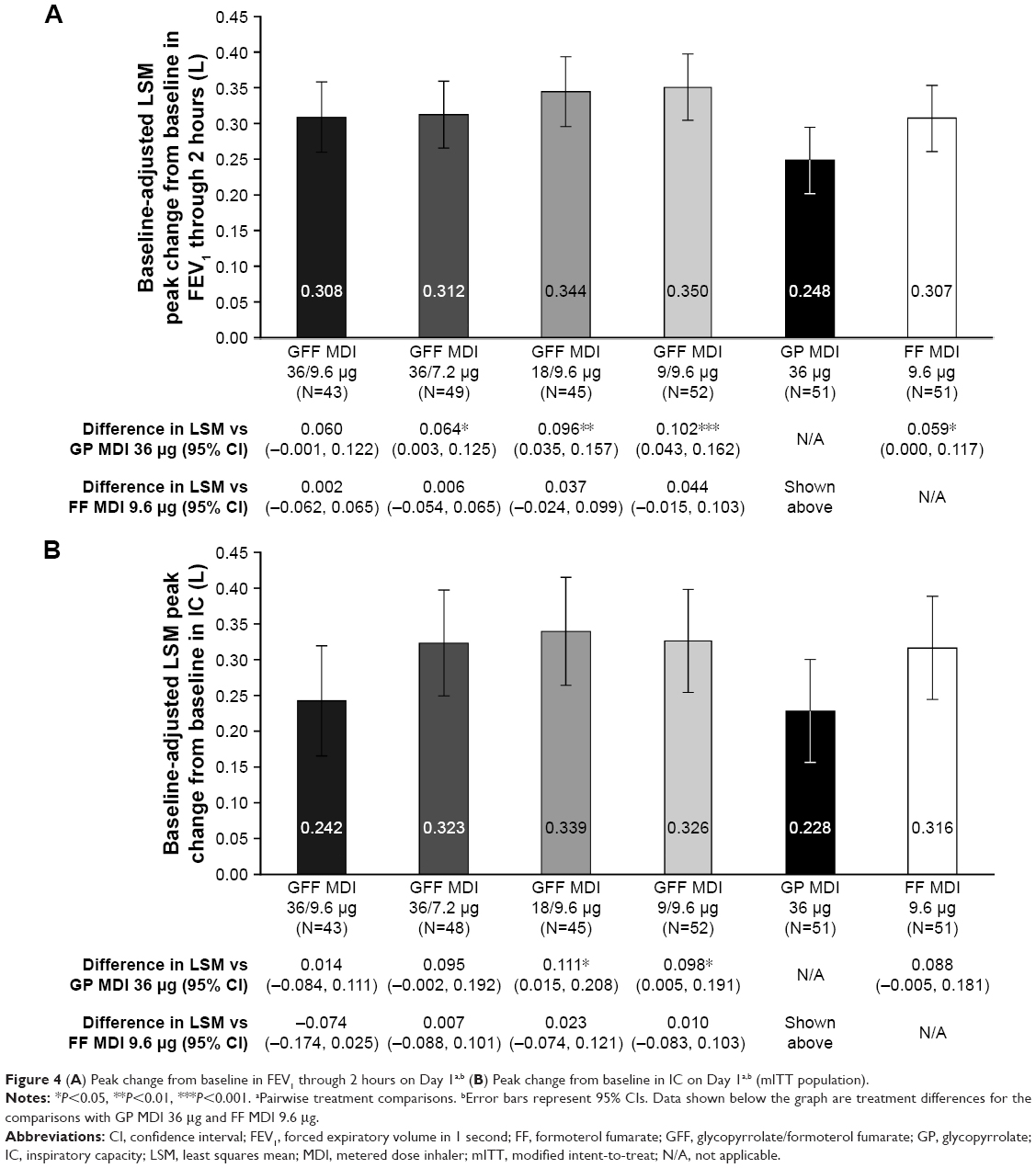 | Figure 4 (A) Peak change from baseline in FEV1 through 2 hours on Day 1a,b (B) Peak change from baseline in IC on Day 1a,b (mITT population). |
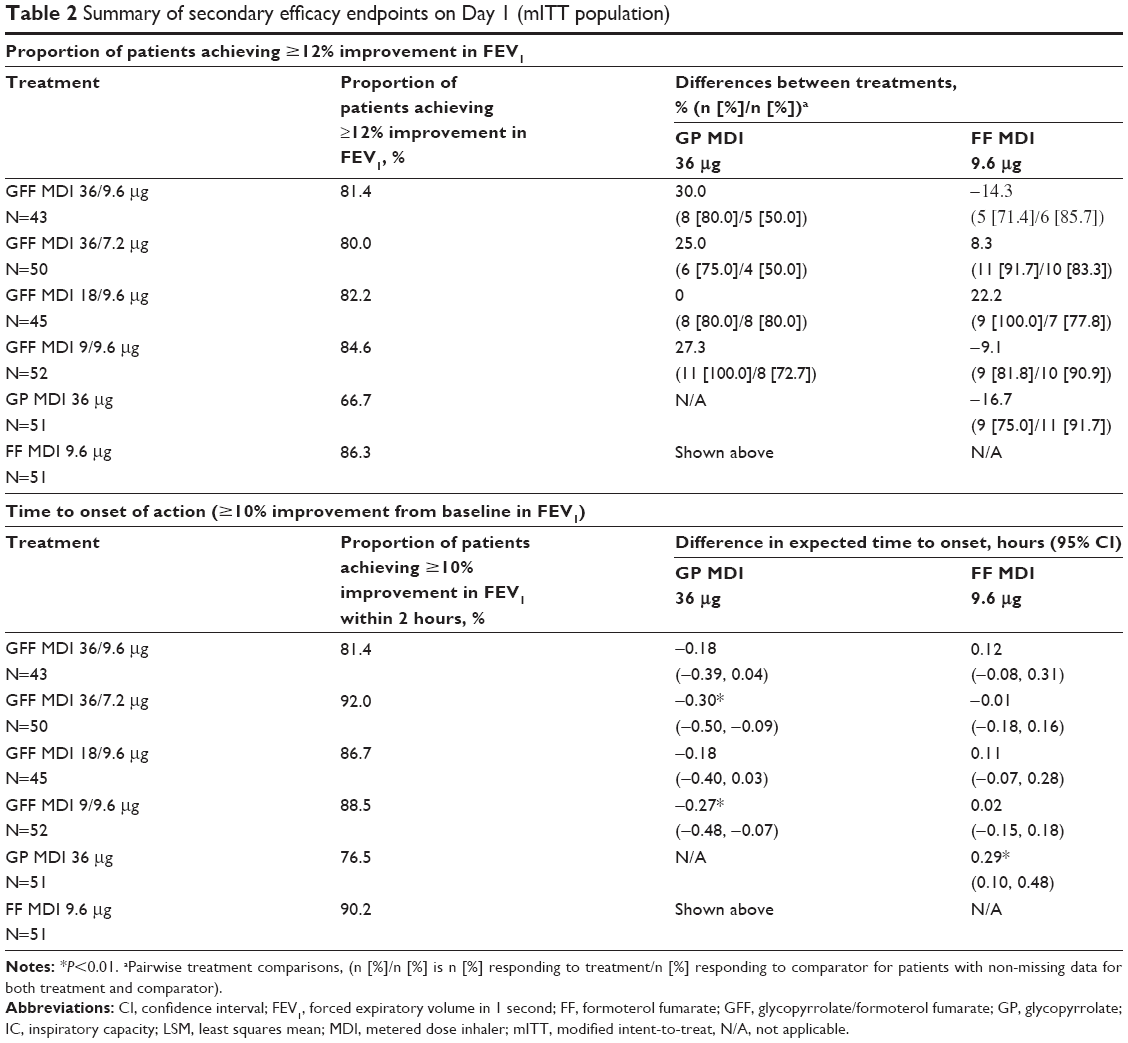 | Table 2 Summary of secondary efficacy endpoints on Day 1 (mITT population) |
Secondary efficacy endpoints on Day 7
For change from baseline in morning predose FEV1 and change from baseline in mean evening 12-hour postdose trough FEV1, no pairwise comparisons between any GFF MDI doses and GP MDI 36 μg were nominally significant (Table 3). All doses of GFF MDI were superior to GP MDI 36 μg for peak change from baseline in FEV1 through 6 hours (LSM differences: 72–131 mL; P≤0.0348; Figure 5A). Only GFF MDI 18/9.6 and 9/9.6 μg demonstrated superiority to GP MDI 36 μg for peak change from baseline in IC (LSM differences: 112 and 124 mL, respectively; P≤0.0419; Figure 5B). No pairwise comparisons between GFF MDI doses and FF MDI 9.6 μg or among the GFF MDI doses were nominally significant for any of the secondary endpoints on Day 7 (Figure 5, Table 3).
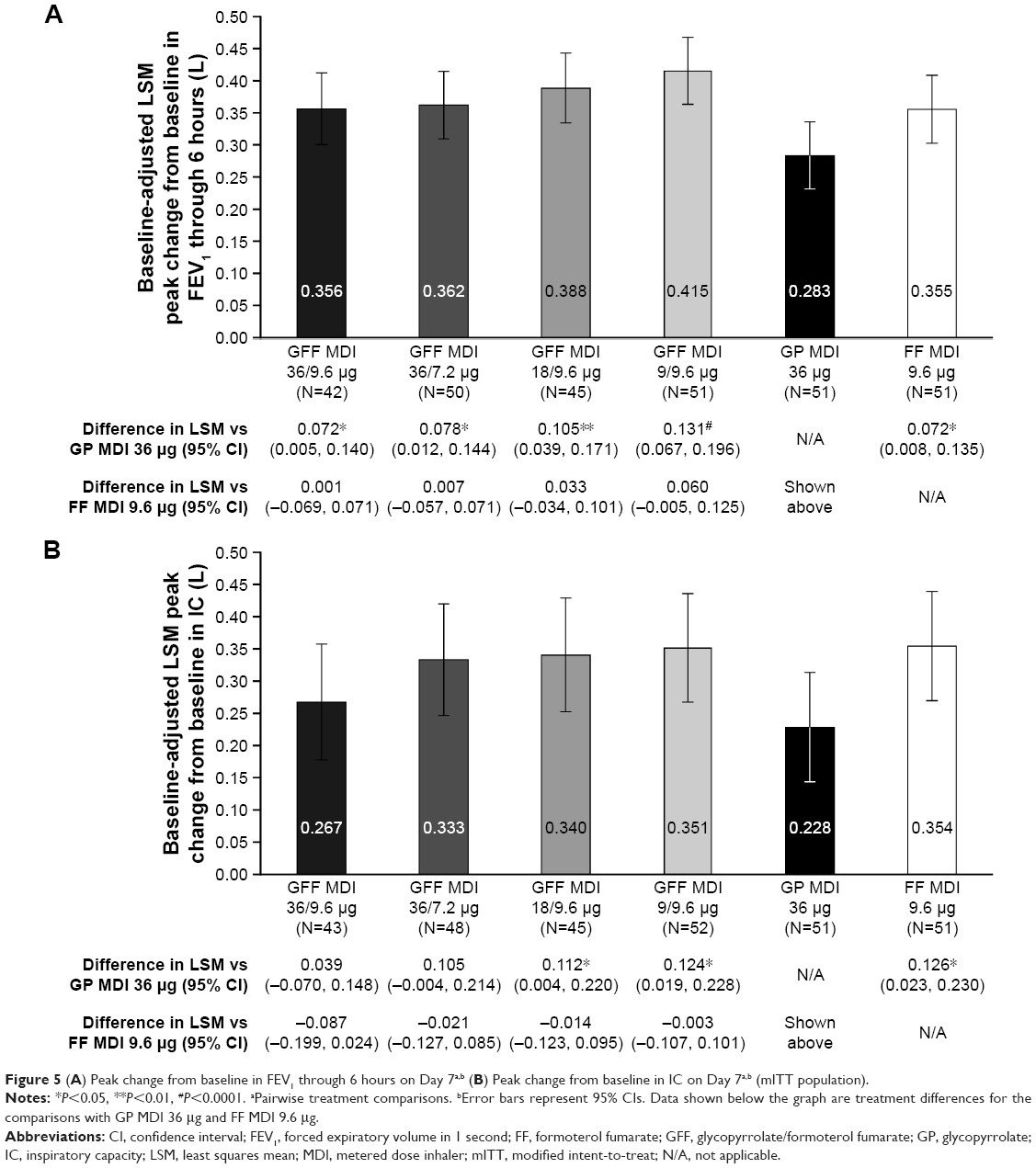 | Figure 5 (A) Peak change from baseline in FEV1 through 6 hours on Day 7a,b (B) Peak change from baseline in IC on Day 7a,b (mITT population). |
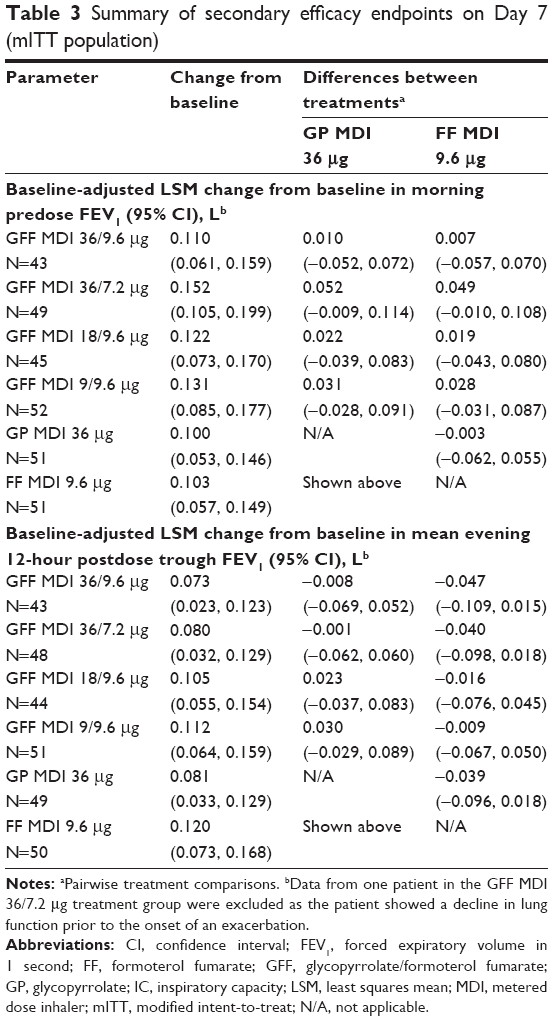 | Table 3 Summary of secondary efficacy endpoints on Day 7 (mITT population) |
Safety
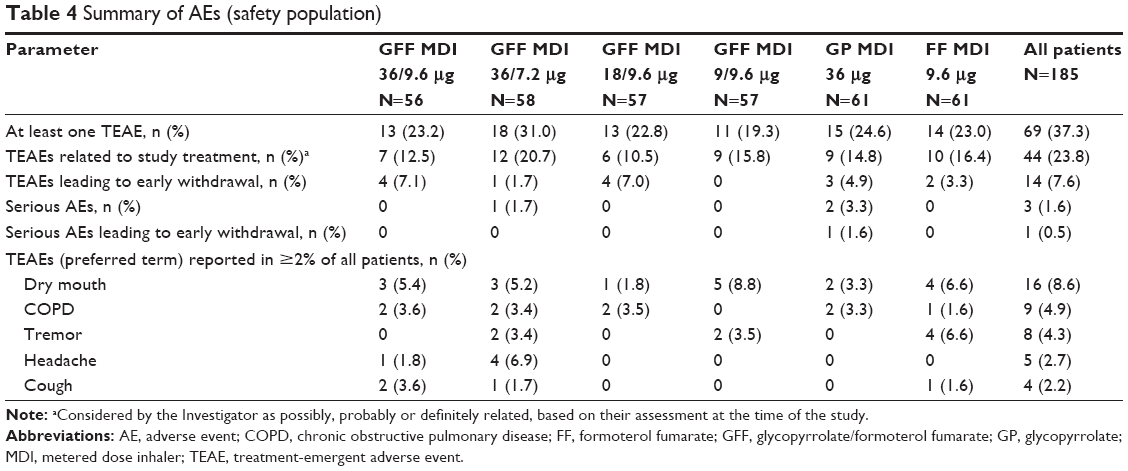
Overall, 69 patients (37.3%) reported at least one treatment-emergent adverse event (TEAE) at any time during the study, and 44 patients (23.8%) reported TEAEs that in the opinion of the investigator were possibly, probably or definitely related to the study treatment (Table 4). Three patients (1.6%) reported serious AEs. One patient who received GFF MDI 36/7.2 μg experienced a serious AE of hypokalemia that was considered to be possibly related to study treatment. The patient withdrew from the study, with the reason recorded as “patient discretion”. The serious AEs of pyelonephritis and acute renal failure, which both occurred in patients receiving GP MDI 36 μg, were not considered to be related to treatment. No deaths were reported.
 | Table 4 Summary of AEs (safety population) |
Fourteen patients (7.6%) reported TEAEs that led to early withdrawal from the study (Table 4), including one serious AE of acute renal failure (further described below). Seven patients withdrew due to COPD exacerbations (two each with GFF MDI 36/9.6 μg, GFF MDI 18/9.6 μg, and GP MDI 36 μg; one with FF MDI 9.6 μg). This was only considered possibly related to study treatment in the two patients treated with GP MDI 36 μg. Three patients withdrew due to worsening hypertension (two with GFF MDI 36/9.6 μg and one with GFF MDI 36/7.2 μg). This was considered possibly related to study treatment in the two patients treated with GFF 36/9.6 μg. One patient treated with GFF MDI 18/9.6 μg withdrew due to dyspnea, and one patient treated with FF MDI 9.6 μg withdrew due to dry mouth; both were considered possibly related to study treatment. One patient treated with GFF MDI 18/9.6 μg withdrew due to a herniated disc and one patient treated with GP MDI 36 μg withdrew due to a serious AE of acute renal failure; both were considered not related to study treatment. The patient who withdrew due to acute renal failure was admitted to hospital 7 days after beginning treatment with GP MDI 36 μg with symptoms of nausea, vomiting, and diarrhea and was initially diagnosed with hypokalemia, hyponatremia, and mild renal insufficiency. After treatment with IV fluids and potassium supplements, his condition resolved and he was discharged the following day with a discharge diagnosis of volume depletion with hypokalemia and hyponatremia, and acute renal failure.
There were no clinically relevant differences in the occurrence of TEAEs among treatments. TEAEs that occurred in ≥2% of all patients were dry mouth (8.6%), worsening of COPD (4.9%), tremor (4.3%), headache (2.7%), and cough (2.2%; Table 4). No incidences of paradoxical bronchospasm were reported. In addition, no important trends were observed in clinical laboratory results, vital signs, and ECGs among the treatments.
Discussion
This Phase IIb study assessed the efficacy and safety of four doses of GFF MDI (36/9.6 μg, 36/7.2 μg, 18/9.6 μg, and 9/9.6 μg), compared with GP MDI 36 μg and FF MDI 9.6 μg, all twice-daily, in patients with moderate-to-severe COPD.
All doses of GFF MDI demonstrated statistically significant increase in the primary efficacy endpoint FEV1 AUC0–12 on Day 7 compared with GP MDI 36 μg. However, when all doses of GFF MDI were compared with FF MDI 9.6 μg, only small numerical differences were observed for the primary endpoint. The comparison between GFF MDI and GP MDI 36 μg confirmed the findings for FEV1 AUC0–12 from other studies with GFF MDI, although, in these studies GFF MDI also led to significant improvements in FEV1 AUC0–12 versus FF MDI 9.6 μg.9–11 The two-period, six-treatment, balanced incomplete-block study design may have contributed to the lack of statistical significance for the comparisons between GFF MDI and FF MDI. Other cross-over studies had a four-period, eight-treatment incomplete-block design,10,11 whereas a 12-hour PFT sub-study of a parallel-group study enrolled a much larger number of patients.9 Inclusion of ≥4 treatment periods may have allowed better crossover controlled data.
Among the secondary efficacy endpoints, two or more of the four GFF MDI doses demonstrated superiority to GP MDI 36 μg for peak change in FEV1 through 2 hours, time to onset of action (≥10% improvement in mean FEV1), and peak change from baseline in IC on Day 1, and for peak change from baseline in FEV1 through 6 hours, and peak change from baseline in IC on Day 7. However, as for the primary endpoint, there were no nominally significant pairwise comparisons between any of the GFF MDI doses and FF MDI 9.6 μg for the secondary efficacy measures. The findings for the comparisons with GP MDI are in line with a study by Tashkin and colleagues, in which all GFF MDI doses between 1.2/9.6 μg and 18/9.6 μg led to significant improvements compared with GP MDI 18 μg for peak change in FEV1 through 2 hours and peak change from baseline in IC on Day 1, and for peak change in FEV1 through 6 hours, and peak change from baseline in IC on Day 7.11 Additionally, with the exception of peak change in FEV1 through 6 hours on Day 7, none of the doses of GFF MDI showed significant improvements for any of these endpoints compared with FF MDI 9.6 μg.11 However, in the study by Reisner and colleagues, GFF MDI 36/9.6 μg showed superiority to FF MDI 9.6 μg for all secondary endpoints investigated on Day 7, with the exception of peak change from baseline in IC. A higher dose of GFF MDI (72/9.6 μg), but not GFF MDI 36/9.6 μg, also showed superiority to FF MDI 9.6 μg for peak change from baseline in FEV1 and IC on Day 1.10 Furthermore, in the Phase III PINNACLE-1 and -2 studies, GFF MDI 18/9.6 μg showed statistically significant improvements compared with GP MDI 18 μg and FF MDI 9.6 μg for the secondary lung function endpoints change from baseline in morning predose trough FEV1 over 24 weeks and peak change from baseline in FEV1 within 2 hours postdose at Week 24.9
Efficacy results were similar across the GFF MDI doses with no nominally significant pairwise comparisons between the GFF MDI doses across the primary and secondary efficacy endpoints. A clear dose-response was not observed. When GFF MDI doses between 1.2/9.6 μg and 18/9.6 μg were evaluated in the study by Tashkin and colleagues, the highest dose of GFF MDI consistently showed the greatest improvement over the monocomponent MDIs (GP MDI 18 μg and FF MDI 9.6 μg) for FEV1 AUC0–12, change from baseline in morning predose trough FEV1, peak change from baseline in FEV1 through 6 hours and change from baseline in evening 12-hour postdose trough FEV1 on Day 7 compared with the lower GFF MDI doses.11
All treatments were well tolerated. Throughout the study, patients were monitored for the occurrence of paradoxical bronchospasm (no reports), dry mouth (8.6%), and tremor (4.3%). Dry mouth and tremor are well known side effects after LAMA and LABA bronchodilator therapy, respectively.16–18 In general, changes from baseline in clinical laboratory results, vital signs, and ECGs were small and not clinically significant, and no safety signals were detected among the treatments. The safety findings in this study were generally comparable with findings in other Phase II studies with GFF MDI.10,11
Conclusion
In conclusion, GFF MDI 36/9.6, 36/7.2, 18/9.6, and 9/9.6 μg twice-daily demonstrated statistically significant increase in FEV1 AUC0–12 on Day 7 (primary efficacy endpoint) compared with GP MDI 36 μg in patients with moderate-to-severe COPD. However, a clear dose-response or superiority to FF MDI 9.6 μg was not observed among the GFF MDI doses in this study. All treatments were well tolerated with no unexpected safety signals observed.
Acknowledgments
The authors would like to thank all of the patients and their families, the team of investigators, research nurses, and operations staff involved in this study. Medical writing support, under the direction of the authors, was provided by Pauline Craig, PhD, of CMC CONNECT, a division of Complete Medical Communications Ltd, Glasgow, UK, which was funded by AstraZeneca, Cambridge, UK in accordance with Good Publication Practice (GPP3) guidelines.19
This study was supported by Pearl – a member of the AstraZeneca Group. Employees of the sponsor (CR, ESR, and PD), were involved in various aspects of the conception and design of the study, acquisition of data and analysis and interpretation of data, and input into manuscript development. The sponsor did not place any restriction on authors about the statements made in the final article.
Author contributions
All authors contributed toward conception and design, data acquisition, or data analysis and interpretation, critically revising and providing final approval of the manuscript, and agree to be accountable for all aspects of the work.
Disclosure
CR is Chief Executive Officer of Pearl – a member of the AstraZeneca Group, and an employee of AstraZeneca. JP has received speaking fees from AstraZeneca, Genentech, Pfizer, and others, and has conducted trials on behalf of Boehringer Ingelheim, Forest, GlaxoSmithKline, Merck, Novartis, Sunovion, Teva, and others. EMK has served on advisory boards, speaker panels, or received travel reimbursement from Amphastar, AstraZeneca, Forest, Mylan, Novartis, Oriel, Pearl – a member of the AstraZeneca Group, Sunovion, Teva, and Theravance. He has conducted multicenter clinical trials for ~40 pharmaceutical companies. ESR and PD are employees of Pearl – a member of the AstraZeneca Group. The authors report no other conflicts of interest in this work.
References
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. | ||
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. | ||
Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD. 2018. Available from: http://www.goldcopd.org. Accessed May 5, 2018. | ||
Vehring R, Lechuga-Ballesteros D, Joshi V, Noga B, Dwivedi SK. Cosuspensions of microcrystals and engineered microparticles for uniform and efficient delivery of respiratory therapeutics from pressurized metered dose inhalers. Langmuir. 2012;28(42):15015–15023. | ||
Doty A, Schroeder J, Vang K, et al. Drug delivery from an innovative LAMA/LABA co-suspension delivery technology fixed-dose combination MDI: evidence of consistency, robustness, and reliability. AAPS PharmSciTech. 2018;19(2):837–844. | ||
Taylor G, Warren S, Dwivedi S, et al. Gamma scintigraphic pulmonary deposition study of glycopyrronium/formoterol metered dose inhaler formulated using co-suspension delivery technology. Eur J Pharm Sci. 2018;111:450–457. | ||
AstraZeneca Pharmaceuticals LP. Bevespi Aerosphere™ Prescribing Information. 2017. Available from: http://www.azpicentral.com/bevespi/bevespi_pi.pdf. Accessed May 5, 2018. | ||
Hanania NA, Tashkin DP, Kerwin EM, et al. Long-term safety and efficacy of glycopyrrolate/formoterol metered dose inhaler using novel Co-Suspension™ Delivery Technology in patients with chronic obstructive pulmonary disease. Respir Med. 2017;126:105–115. | ||
Martinez FJ, Rabe KF, Ferguson GT, et al. Efficacy and safety of glycopyrrolate/formoterol metered dose inhaler formulated using co-suspension delivery technology in patients with COPD. Chest. 2017;151(2):340–357. | ||
Reisner C, Fabbri LM, Kerwin EM, et al. A randomized, seven-day study to assess the efficacy and safety of a glycopyrrolate/formoterol fumarate fixed-dose combination metered dose inhaler using novel Co-Suspension™ Delivery Technology in patients with moderate-to-very severe chronic obstructive pulmonary disease. Respir Res. 2017;18(1):8. | ||
Tashkin DP, Martinez FJ, Rodriguez-Roisin R, et al. A multicenter, randomized, double-blind dose-ranging study of glycopyrrolate/formoterol fumarate fixed-dose combination metered dose inhaler compared to the monocomponents and open-label tiotropium dry powder inhaler in patients with moderate-to-severe COPD. Respir Med. 2016;120:16–24. | ||
Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Bauer P, Röhmel J, Maurer W, Hothorn L. Testing strategies in multi-dose experiments including active control. Stat Med. 1998;17(18):2133–2146. | ||
Murray S. Using weighted Kaplan-Meier statistics in nonparametric comparisons of paired censored survival outcomes. Biometrics. 2001;57(2):361–368. | ||
Alagha K, Palot A, Sofalvi T, et al. Long-acting muscarinic receptor antagonists for the treatment of chronic airway diseases. Ther Adv Chronic Dis. 2014;5(2):85–98. | ||
Sharafkhaneh A, Majid H, Gross NJ. Safety and tolerability of inhalational anticholinergics in COPD. Drug Healthc Patient Saf. 2013;5:49–55. | ||
Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res. 2010;11:149. | ||
Battisti WP, Wager E, Baltzer L, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461–464. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.