")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Efficacy and safety of fluticasone furoate/vilanterol or tiotropium in subjects with COPD at cardiovascular risk
Authors Covelli H, Pek B, Schenkenberger I, Scott-Wilson C, Emmett A, Crim C
Received 30 June 2015
Accepted for publication 7 September 2015
Published 18 December 2015 Volume 2016:11(1) Pages 1—12
DOI https://doi.org/10.2147/COPD.S91407
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Henry Covelli,1 Bonavuth Pek,2 Isabelle Schenkenberger,3 Catherine Scott-Wilson,4 Amanda Emmett,5 Courtney Crim4
1Kootenai Health, Coeur d’Alene, ID, USA; 2Clinique de Pneumologie et de Sommeil de Lanaudière, Quebec, Canada; 3Klinische Forschung, Berlin, Germany; 4GlaxoSmithKline Inc., Research Triangle Park, 5PAREXEL International, Durham, NC, USA
Background: Fluticasone furoate/vilanterol (FF/VI) is a novel, once-daily, inhaled corticosteroid/long-acting β2-agonist combination approved for the treatment of COPD and asthma. We compared the safety and efficacy of FF/VI and tiotropium (TIO) in subjects with moderate-to-severe COPD with greater risk for comorbid cardiovascular disease (CVD).
Methods: This randomized, blinded, double-dummy, parallel-group study compared a once-daily morning dose of FF/VI 100/25 mcg delivered via ELLIPTA™ with TIO 18 mcg via HandiHaler® for 12 weeks in subjects with diagnosed COPD, forced expiratory volume in 1 second (FEV1) 30%–70% predicted, and CVD or CVD risk. The primary endpoint was change from baseline in 24-hour weighted mean FEV1 on Day 84. Other efficacy endpoints included time to onset of bronchodilation, trough FEV1, other spirometry measures, rescue medication use, symptoms, quality of life (St George’s Respiratory Questionnaire-COPD [SGRQ-C]), and health status (COPD Assessment Tests [CAT]) measures. Safety endpoints included cardiovascular monitoring, cortisol excretion, COPD exacerbations, and adverse events, including prespecified drug effects.
Results: Both FF/VI and TIO improved the 24-hour weighted mean FEV1 from baseline after 12 weeks with no significant difference between treatments. Other endpoints favored FF/VI for time to onset of bronchodilation, rescue medication use, dyspnea, SGRQ-C and CAT scores, or favored TIO for change from baseline in forced vital capacity and inspiratory capacity. Pneumonia occurred more frequently in the FF/VI group, and two TIO-treated subjects died following cardiovascular events. Other safety measures were similar between groups, and cardiovascular monitoring did not reveal increased CVD risk.
Conclusion: Both FF/VI and TIO were efficacious in improving lung function in subjects with COPD and comorbid CVD or CVD risk factors, with minor differences in efficacy and safety profiles.
Keywords: fluticasone furoate/vilanterol, tiotropium, COPD, cardiovascular disease, ICS/LABA, anticholinergic
Introduction
Successful management of moderate-to-severe COPD can be accomplished with combination therapy that delivers a concurrent dose of inhaled corticosteroid and a long-acting β2-agonist (ICS/LABA) or with tiotropium (TIO), an inhaled long-acting anticholinergic bronchodilator.1 Both twice-daily ICS/LABA and once-daily TIO treatment for COPD have been demonstrated to improve lung function and health status as well as reduce COPD exacerbations.1–6 Head-to-head comparisons between ICS/LABA drugs and TIO, however, are limited and the efficacy results inconclusive,7,8 leading researchers to call for further large, long-term randomized controlled trials to establish relative efficacy and safety of these treatments, as well as compare newly developed alternative ICS/LABAs with TIO.8 A newly introduced ICS/LABA treatment for COPD in a novel inhaler device combines fluticasone furoate (FF), an ICS with enhanced selectivity, retention, and affinity for the glucocorticoid receptor,9 and vilanterol (VI), a LABA with prolonged bronchodilating properties.10 FF/VI is the only once-daily ICS/LABA treatment and is well tolerated, improves lung function, and sustains bronchodilation after 24 weeks and 52 weeks of treatment for COPD.11–13 When once-daily treatment with FF/VI for 12 weeks was compared with twice-daily fluticasone propionate/salmeterol 250/50 (FP/SALM) for COPD, there was evidence of numerically greater effect on the forced expiratory volume in 1 second (FEV1) and faster onset for FF/VI compared with FP/SALM with similar safety profiles for the two drugs.14
COPD is not only a disease of the airways and lung parenchyma, but there are several extrapulmonary comorbidities associated with COPD,15 of which cardiovascular disease (CVD) is arguably the most significant. CVD is a comorbidity of COPD,16–18 and the risk of cardiovascular (CV) events rises in subjects with COPD with a history of myocardial infarction.19 Furthermore, concerns regarding CV safety and COPD medications have been raised for both TIO5 and LABA drugs.20 Therefore, we conducted the study in a population of subjects with or at risk for CVD. The objective of this study was to compare the efficacy and safety of FF/VI and TIO, both delivered once daily, for COPD treatment over a 12-week period in a randomized, controlled trial. An additional objective was to investigate whether the efficacy and/or safety profile of FF/VI or TIO would differ in subjects with CVD or in subjects at risk for CVD.
Methods
The study (Protocol HZC115805, NCT01627327) was approved by an institutional review board or independent ethics committee at each site and conducted in accordance with good clinical practice guidelines and the Declaration of Helsinki. All subjects provided written informed consent. The study was conducted between April and December 2012 at 56 centers in six countries (Canada, Czech Republic, Germany, Poland, Romania, and the USA).
Subjects
Men and women aged ≥40 years with a clinical diagnosis of COPD were recruited. All subjects were current or former smokers (≥10 pack years) and had a postbronchodilator FEV1 ≥30% to ≤70% of predicted normal (NHANES III) and an FEV1/forced vital capacity (FVC) ratio ≤70% at screening. Subjects also had a history of CVD/a CVD event or, in addition to being a current/former smoker, had at least one current CV risk factor (hypertension, hypercholesterolemia, or treated diabetes). Female subjects were either postmenopausal or using effective contraception.
Subjects were excluded if they had asthma or respiratory disorders other than COPD; recent (≤12 months) lung resection; clinically significant abnormal chest X-ray, laboratory, Holter, or electrocardiogram (ECG) finding at screening; recent (≤12 weeks) hospitalization for COPD; recent (≤6 weeks) acute worsening of COPD requiring treatment with corticosteroids or antibiotics or lower respiratory tract infection requiring antibiotics; noncompliance, COPD exacerbation, or lower respiratory tract infection during the run-in period; cancer not in complete remission for ≥5 years; long-term or nocturnal oxygen therapy (>12 h/day); or other diseases or contraindications that would put the subject at risk.
Study design and treatment
This study was a 12-week, randomized (1:1), blinded, stratified, double-dummy, parallel-group, active comparator study. After providing written informed consent and prior to screening, subjects discontinued COPD medications (including maintenance medications; excluded medications included systemic/oral/parenteral corticosteroids [within 6 weeks prior to screening and thereafter], ICS or ICS/LABA combinations [within 4 weeks prior to screening and thereafter], anticholinergics [long acting, 1 week prior to screening and thereafter; short acting, 4 hours prior to screening and thereafter], oral phosphodiesterase type 4 inhibitors [PDE-4] inhibitors [1 week prior to screening and thereafter], and LABAs [within 48 hours prior to screening and thereafter]); exceptions were study-supplied rescue medication (albuterol) and mucolytics at a constant dosage. After the screening visit, eligible subjects entered a 2-week single-blind placebo run-in period to obtain baseline rescue medication use and COPD symptom (scores recorded on diary cards) and to evaluate subjects’ compliance and disease stability. At the end of the run-in period, eligible subjects were randomly assigned to once-daily morning treatment with either FF/VI 100/25 mcg (BREO™ [RELVAR™, REVINTY™], GlaxoSmithKline plc [GSK], London, UK) delivered via the ELLIPTA™ inhaler (GSK, London, UK) or TIO 18 mcg (Spiriva®, Boehringer Ingelheim, Ingelheim, Germany) delivered from blister capsules by the HandiHaler® (Spiriva, Boehringer Ingelheim). When used, the novel ELLIPTA dry powder inhaler simultaneously delivers one breath-actuated dose of both FF 100 mcg and VI 25 mcg from their individual blister strips.
A central randomization schedule was generated using a validated computerized system (RandAll; GSK) and communicated with a validated computerized voice response system, the Registration and Medication Ordering System (RAMOS; GSK), when the investigative site called to randomize a subject. To provide a double-dummy design, subjects were given two inhalers, one containing active drug and the other placebo, and they and their investigators were blinded to assigned treatment. TIO and placebo capsules were closely matched in color, but TIO capsules had trade markings that were not present on the placebo capsules. Whether subjects would notice and correctly or incorrectly interpret this difference is unclear. Capsule type was consistent for each subject for the duration of the study. Both the TIO and placebo blister packages were covered with opaque overlabels that hide the information on the TIO packaging. HandiHalers were covered with labels to mask identifying marks. Staff involved with safety and efficacy assessments were not present during dosing in the clinic so that they would not identify the capsules removed from the blisters.
Every morning, subjects in the FF/VI group inhaled once from the ELLIPTA inhaler containing active drug followed by two inhalations from the HandiHaler containing placebo. Subjects in the TIO group inhaled once from the ELLIPTA inhaler containing placebo followed by two inhalations from the HandiHaler containing TIO. Compliance with the study drug was assessed at each visit by reviewing the dose counter on the ELLIPTA or counting capsules from the HandiHaler. Subjects were also provided with albuterol (metered dose inhaler [MDI] and/or nebules) to use as needed for symptomatic relief of COPD during the run-in and treatment periods, except for the 4-hour period prior to spirometry and during each study visit.
Subjects were stratified at randomization for COPD exacerbation history (either did or did not have ≥1 COPD exacerbation requiring oral corticosteroid and/or antibiotic treatment and/or hospitalization in the 3 years prior to screening) and for reversibility status (either reversible [defined as an increase in FEV1 of ≥12% and ≥200 mL] or nonreversible [defined as an increase in FEV1 <200 mL or ≥200 mL and <12% after albuterol administration at screening]). Subjects revisited the clinic on treatment days 28, 56, and 84. A follow-up safety telephone contact was performed 7 days after the last treatment visit. Subjects who experienced a COPD exacerbation or pneumonia were withdrawn; however, only exacerbations that resulted in hospitalization (ie, severe exacerbations) were captured as an adverse event (AE).
Study assessments
Standardized equipment (spirometry: microQuark, 12-lead ECG: Universal ECG, and 24-hour Holter monitor: Mortara Instrument), study-customized software, and training were provided, and data were transmitted and overread by independent centralized certified staff at CompleWare.
At the screening visit, pre- and postbronchodilator spirometry, medical history, physical examination, 12-lead ECG, 24-hour Holter monitoring (subset of subjects), chest X-ray, vital signs, serum pregnancy tests, clinical laboratory tests, oropharyngeal examination, and 24-hour urine cortisol collection (subset of subjects) were conducted, and diary cards were dispensed for recording of medical problems, concomitant medications, rescue medication use, and symptoms (dyspnea, cough, sputum production). At all subsequent visits, eligible randomized subjects completed the St George’s Respiratory Questionnaire-COPD (SGRQ-C) and COPD Assessment Tests (CAT) prior to other assessments, and safety monitoring was conducted, including oropharyngeal examination, AE reporting, exacerbation and pneumonia assessment, and diary card review. At the randomization Visit 2, subjects also underwent vital sign measurement and blood draws for biomarkers (subset of subjects). At Visit 5, which was an overnight stay, 24-hour spirometry, ECGs, 24-hour Holter monitoring (subset of subjects), 24-hour urinary cortisol collection (subset of subjects), physical examinations, vital sign measurement, blood draws for clinical laboratory tests and biomarkers, and pregnancy tests were conducted. Each Holter was reviewed by a centralized overreader at CompleWare. Serum was collected for seven known inflammatory biomarkers at baseline and Week 12 and analyzed as exploratory endpoints: high-sensitivity C-reactive protein (hsCRP), pulmonary and activation-regulated chemokine (more currently referred to as chemokine [C-C motif] ligand 18 [CCL-18]), fibrinogen, interleukin-6 (IL-6), surfactant protein D (SP-D), Clara cell secretory protein-16 (CC-16), and brain natriuretic peptide (BNP).
Spirometry was performed after withholding rescue medication, study medication, and caffeinated drinks and after abstinence from smoking and physical exertion. Inspiratory capacity (IC) assessments were performed prior to FEV1 assessments. At the randomization Visit 2, after predose spirometry and the first study dose, subjects underwent postdose serial spirometry at 5 minutes, 15 minutes, and 30 minutes and at 1 hour, 2 hours, and 4 hours. At Visit 5 on Day 84 after predose spirometry and after the last study dose, subjects underwent postdose serial spirometry at 5 minutes, 15 minutes, and 30 minutes and at 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 13 hours, 14 hours, 16 hours, 20 hours, and 24 hours.
Statistical analysis
Assuming a standard deviation of 215 mL and significance at the two-sided 5% level, a sample size of 271 subjects per treatment arm was required for 90% power to detect a 60 mL difference between FF/VI and TIO for 24-hour weighted-mean FEV1 at Week 12, an estimate based on historical data. To allow for a 10% withdrawal rate, at least 300 subjects were to be randomized per treatment arm. Primary analyses were performed on the intent-to-treat population, which comprised all subjects randomized to treatment and who had received at least one dose of study medication.
The primary endpoint, change from baseline in 24-hour weighted mean FEV1 at Week 12, was analyzed using analysis of covariance with covariates of baseline FEV1, randomization stratum (COPD exacerbation history and reversibility status), smoking status at screening, and country. For calculation of 24-hour weighted mean FEV1, missing serial spirometry FEV1 values were linearly interpolated between two nonmissing values; however, the 0-hour and 24-hour values, as well as additional values between 0 hour and 6 hours and between 12 hours and 24 hours, must not have been missing.
The secondary endpoints (time to onset [increase of 100 mL above baseline in FEV1] 0–4 hours postdose on Day 1 and change from baseline in trough FEV1 on Day 84) were nested under the primary endpoint. To account for multiplicity across primary and secondary efficacy endpoints, a P-value <0.05 was required for the primary endpoint to allow statistical significance to be inferred for secondary endpoints. Time to onset was analyzed using a log-rank test, stratified by exacerbation history and reversibility stratum. Change from baseline in trough (predose) FEV1 was analyzed using the same methodology as the primary endpoint.
Daily diary measures were averaged weekly and over the entire 12-week treatment period for analysis. Change from baseline in SGRQ-C and CAT scores was analyzed using mixed model repeated measures with covariates of baseline score, exacerbation history and reversibility stratum, smoking status, country, treatment group, day, day by baseline, and day by treatment group. A responder analysis was also performed for the SGRQ-C. Subjects were classified as a responder at a given visit if they had a change from baseline at that visit of ≥4 points improvement, the minimal clinically important difference (MCID).21 Otherwise, the subject was classified as a nonresponder. A logistic regression model with terms for treatment group, exacerbation history and reversibility stratum, and baseline score was used to compare the treatment groups. Other efficacy endpoints, as well as safety measures of pulse rate and log-transformed urinary cortisol, were analyzed using analysis of covariance.
Results
Subject characteristics
At screening, 890 subjects were evaluated for eligibility (Figure 1). Of those, 267 (30%) subjects were not randomized mainly because they failed the strict enrollment criteria, for example, FEV1 30%–70% predicted (n=112), and/or had an abnormal Holter (n=75) and/or ECG (n=55) finding. Following the run-in period, 310 and 313 subjects were randomized to treatment with FF/VI and TIO, respectively, and constituted the intent-to-treat population. Of those, 291 (94%) and 274 (88%) subjects, respectively, completed the study. More subjects were withdrawn from treatment with TIO, mainly due to AEs and lack of efficacy/COPD exacerbations, than with FF/VI. Three percent of TIO-treated and 1% of FF/VI-treated subjects withdrew from the study due to protocol deviations.
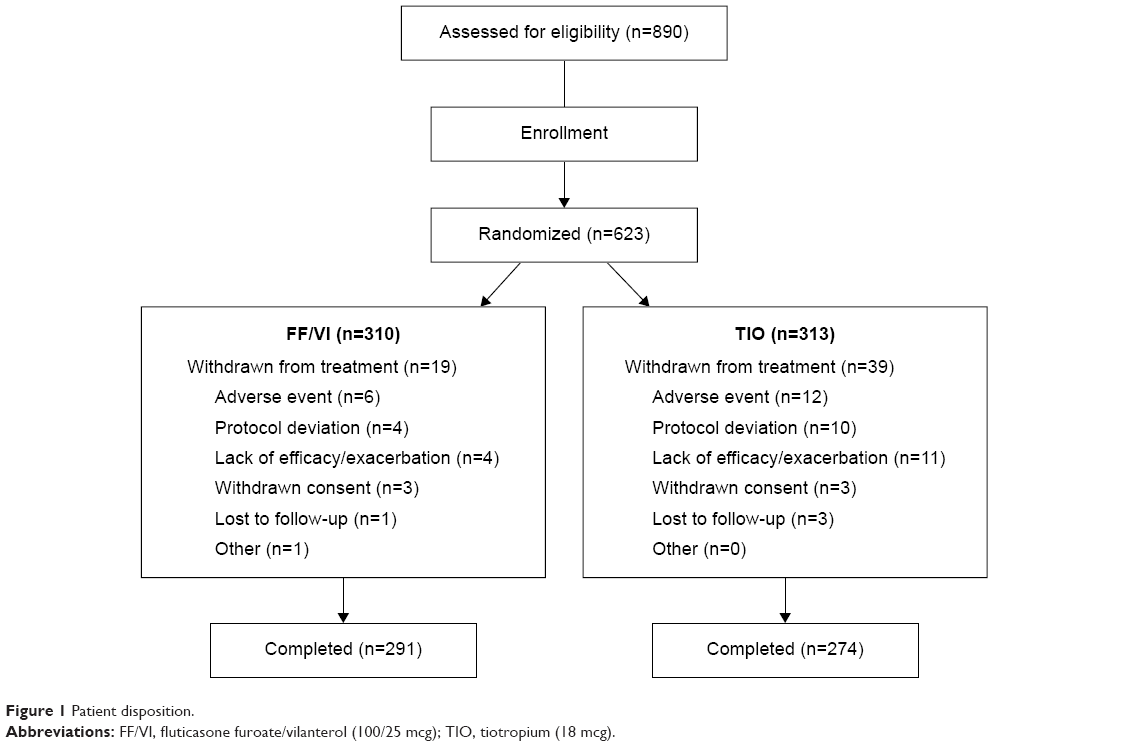 | Figure 1 Patient disposition. |
Subjects’ demographic and baseline characteristics were similar between treatment groups (Table 1), with the majority of subjects being white and male. In this population of subjects with a history of CVD/CV event or risk factors, the most common baseline conditions were hypertension and hypercholesterolemia. Slightly under half of the subjects had not had a COPD exacerbation in the last 3 years. Baseline lung function results indicated that the study population had moderate-to-severe airflow limitation (Table 1), and approximately one-third of the subjects demonstrated acute bronchodilator reversibility. Baseline health-related quality of life (QoL) and health status, indicated by SGRQ-C and CAT scores, respectively, suggested impairment and a high level of COPD symptoms.
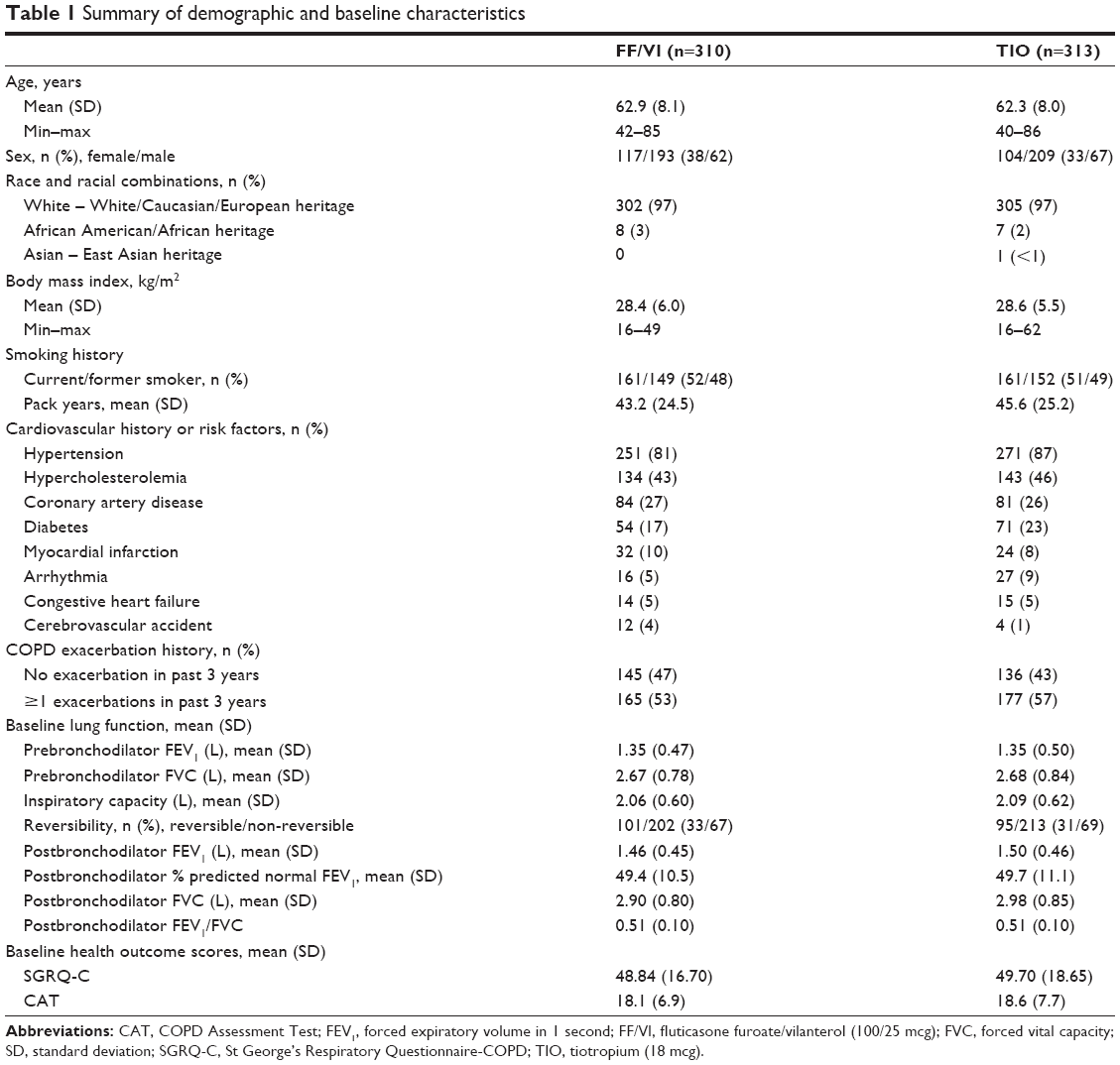 | Table 1 Summary of demographic and baseline characteristics |
Efficacy
Results of the primary efficacy measure, change from baseline trough in 24-hour weighted mean FEV1 after 12 weeks’ treatment, were similar between the treatment groups (Figure 2), with a least squares (LS) mean change from baseline of 117 mL and 95 mL in the FF/VI and TIO groups, respectively, with a difference of 0.022 L (95% confidence interval [CI] −0.012 to 0.055) (P=0.201). In the reversible subgroup, however, the mean change was greater in the FF/VI group compared with the TIO group (181 mL and 110 mL, respectively), with a difference of 0.071 L (95% CI 0.012–0.129). Other stratification subgroups (ie, nonreversible, ≥1 COPD exacerbation in last 3 years, no COPD exacerbation in last 3 years) demonstrated small or no treatment group differences.
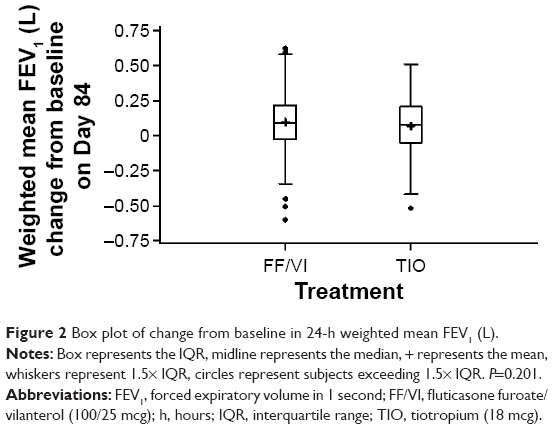 | Figure 2 Box plot of change from baseline in 24-h weighted mean FEV1 (L). |
The secondary endpoints were nested under the primary endpoint. Because the primary endpoint difference did not reach statistical significance, all differences reported for the secondary efficacy endpoints are presented descriptively only as statistical inference cannot be made.
Table 2 shows that 79% and 82% of subjects receiving FF/VI or TIO, respectively, achieved an increase in FEV1 of ≥100 mL from baseline within 4 hours postdose on treatment Day 1, with a somewhat faster onset in the FF/VI group, with the median time to onset of 17.0 minutes compared with the median time of 20.5 minutes in the TIO group. More subjects in the FF/VI group than the TIO group demonstrated an onset of effect within the first 5 minutes of dosing (Table 2).
 | Table 2 Time to onset of action (postdose change from baseline of ≥100 mL) on treatment day 1 |
Other efficacy endpoints are summarized in Table 3. FF/VI and TIO improved trough FEV1 on Day 84 to a similar degree. Both change from baseline in trough IC and change from baseline in trough FVC favored TIO treatment.
 | Table 3 Other efficacy endpoints |
Subjects reported their COPD symptoms daily on diary cards. Changes from baseline in the individual symptoms of cough and sputum production were similar between groups (Table 3). However, reductions in dyspnea over the 12-week period favored FF/VI.
Treatment with FF/VI reduced subjects’ use of rescue medication during Weeks 1–12, with an LS mean change difference of −0.37 inhalations (95% CI −0.55 to −0.19) and during each of the week-long treatment periods (Figure 3A). Likewise, the percentage of rescue-free 24-hour periods was increased by FF/VI treatment compared with TIO during Weeks 1–12, with an LS mean change difference of 9.1% (95% CI 4.0–14.2) and during each week (Figure 3B).
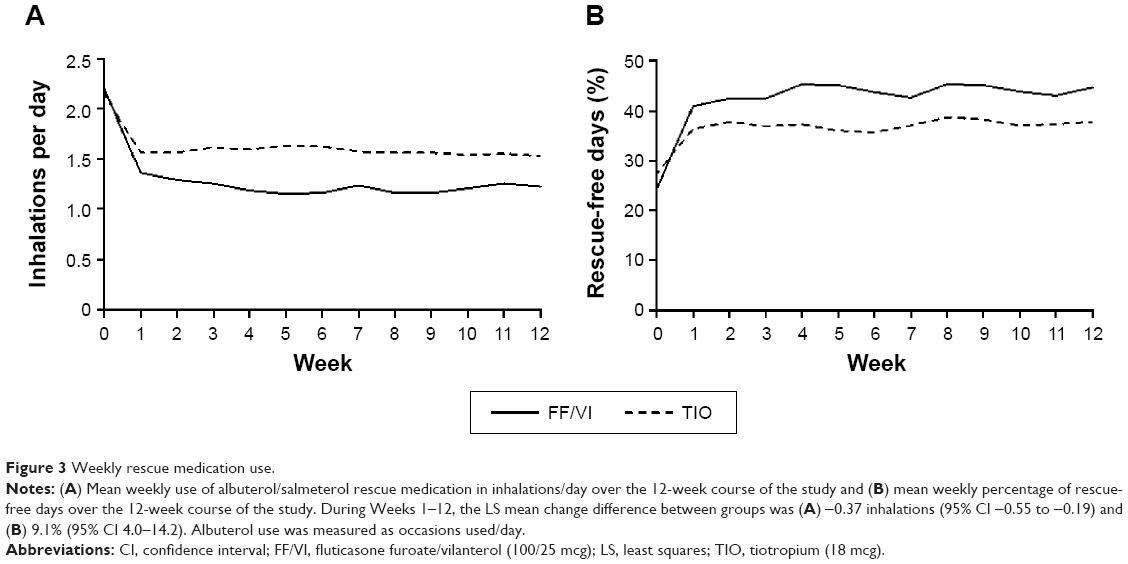 | Figure 3 Weekly rescue medication use. |
Health-related QoL was measured at baseline and at each posttreatment visit. While SGRQ-C baseline scores were similar between treatment groups (Table 1) and both groups improved with treatment (Figure 4A), FF/VI demonstrated a consistently greater numerical improvement in total score than TIO treatment, and only FF/VI treatment reached the clinically significant MCID change score of −4 (Week 8); achievement of the total score MCID was not persistent at Week 12. With respect to individual SGRQ-C components (symptoms, impacts, activity), FF/VI consistently reduced each component score more than TIO, and only the FF/VI group achieved the MCID for symptoms across all time points. A responder analysis for total SGRQ-C score revealed that a higher percentage of FF/VI-treated than TIO-treated subjects, respectively, achieved the MCID after 4 weeks (46% vs 34%), 8 weeks (49% vs 36%), and 12 weeks (48% vs 42%) of treatment.
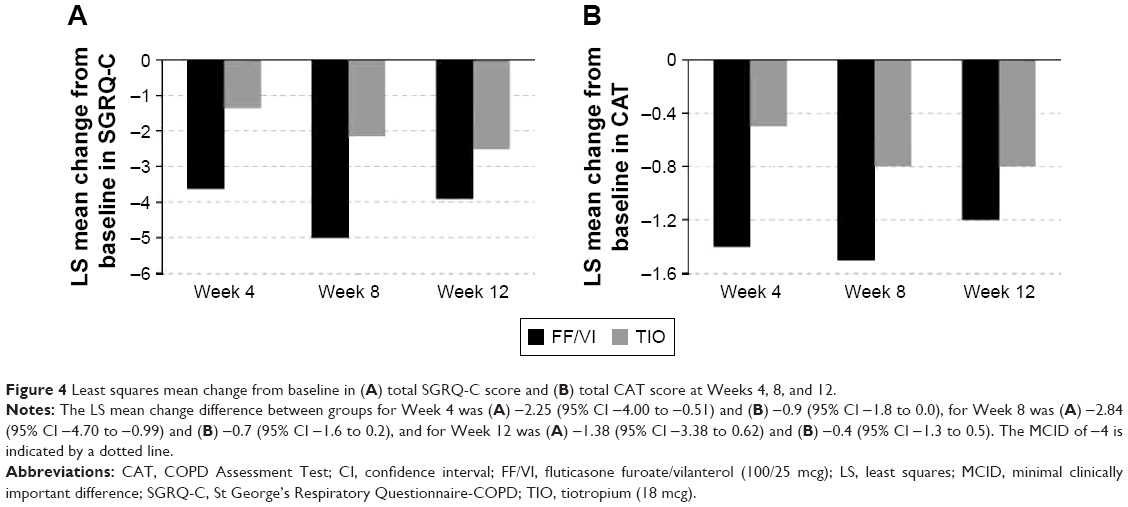 | Figure 4 Least squares mean change from baseline in (A) total SGRQ-C score and (B) total CAT score at Weeks 4, 8, and 12. |
CAT scores at baseline were high (≥10) and similar between groups (Table 1). The FF/VI group consistently demonstrated a numerically greater improvement in CAT score over the treatment period (Figure 4B).
Biomarkers
Seven biomarkers (hsCRP, CCL-18, SP-D, fibrinogen, IL-6, CC-16, and BNP) were measured in a subset of subjects (165 FF/VI, 169 TIO) at baseline and Week 12. There were no significant changes from baseline in BNP, fibrinogen, or IL-6 with either FF/VI or TIO. For both the treatment groups, there were decreases in CCL-18 and CC-16 and an increase in SP-D; hsCRP only increased at Week 12 in the TIO group. However, for all seven biomarkers, the change from baseline was not significantly different between the two treatment groups (data not shown).
Safety
Over one-third of the subjects experienced an AE during treatment (Table 4), the most frequent of which were headache, nasopharyngitis, back pain, and oral candidiasis. More TIO-treated than FF/VI-treated subjects were withdrawn from the study due to AEs. Two TIO-treated subjects died (one due to cardiopulmonary arrest and the other due to cardiorespiratory arrest and cardiac failure). Both subjects had hypertension; one smoked and had a family history of CVD. More FF/VI than TIO-treated subjects experienced an AE considered drug related by the investigator.
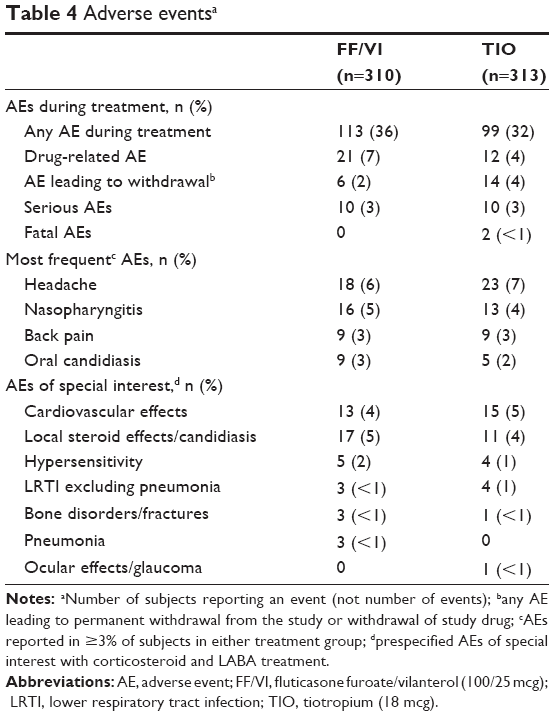 | Table 4 Adverse eventsa |
Pneumonia was reported in three subjects during treatment with FF/VI, all of which were radiologically confirmed and treated with antibiotics. Two of the three subjects were hospitalized. The third subject experienced a COPD exacerbation 4 days after developing pneumonia. All three subjects were withdrawn from the study. One FF/VI-treated subject developed pneumonia 19 days after study medication (FF/VI) was finished. None of the pneumonias were fatal.
AEs of special interest based on the known effects of corticosteroid and LABA treatment were prespecified. The most commonly reported were CV effects and local steroid effects (Table 4). The number of other events was low and similar between groups.
Fewer subjects in the FF/VI group (7/310; 2%) experienced a COPD exacerbation than in the TIO group (11/313; 4%), and all were withdrawn from the study. Although the protocol required subjects to be withdrawn upon experiencing an exacerbation, one subject in the FF/VI group was not withdrawn until after having a second event. Of those withdrawn, five FF/VI-treated and nine TIO-treated subjects had a history of COPD exacerbation. None of the COPD exacerbations were fatal. Three of eight exacerbations in the FF/VI group required hospitalization compared with one of eleven in the TIO group; per protocol, only the hospitalized events were captured as adverse events. Compared with the FF/VI group, more subjects in the TIO group who had an exacerbation were treated with antibiotics alone or in conjunction with oral corticosteroids.
There were no clinically significant differences between groups in mean changes from baseline for pulse rate, heart rate, or QTc intervals. At Week 12, few subjects (2% in each group) had a QTc(F) >450, and few (≤1% in each group) had a change from baseline in QTc(F) ≥60 ms.
The percentage of subjects with ECG findings defined a priori as abnormalities of potential clinical importance was similar at baseline (9% FF/VI group, 13% TIO group) and at the end of treatment (8% FF/VI group, 7% TIO group). Most abnormalities were categorized as depolarization abnormalities or prior myocardial infarction.
Holter monitoring was conducted in a subset of subjects (158 FF/VI, 153 TIO), with no significant difference between FF/VI and TIO groups in abnormalities of potential clinical significance, including mean heart rate, cardiac rhythm abnormalities (ventricular ectopic events), or other prespecified abnormalities of potential clinical importance. Most of the abnormalities were nonsustained ventricular tachycardias (defined as >100 beats/min, 3–30 beats) or right bundle branch blocks. There was a higher incidence of nonsustained ventricular tachycardia in the FF/VI group (9%) than the TIO group (4%) and of right bundle branch block in the TIO group (5%) than the FF/VI group (2%). There were no reports of sustained ventricular tachycardia or sustained supraventricular tachycardia (defined as >100 beats/min, >30 beats) at any time postrandomization in either group. Abnormalities of potential clinical importance were reported in more subjects at the end of treatment (14% FF/VI, 12% TIO) than at screening (4% FF/VI, 5% TIO). More subjects in both groups had ventricular ectopics with ≥1 ventricular run at the end of treatment (11% FF/VI, 6% TIO) than at screening (<1% FF/VI, 2% TIO).
Clinical chemistry and hematology results were generally similar between groups, and mean changes from baseline were minimal. No subject had laboratory abnormalities of potential clinical concern. Cortisol excretion was measured by 24-hour urine collection in a subset of subjects (104 on FF/VI, 103 on TIO). There were no clinically significant differences in LS mean urinary cortisol ratio of Week 12 to baseline between groups (FF/VI [1.06] vs TIO [1.02] ratio of 1.038; 95% CI 0.86–1.26).
Discussion
Our results in this head-to-head comparator trial between FF/VI and TIO in subjects with COPD who had or were at risk of CVD showed that both treatments resulted in lung function improvement >12 weeks and were consistent with previous reports of FF/VI treatment effects from randomized controlled trials.11–13,22–24 The change from baseline in 24-hour weighted mean FEV1 after 12-week treatment (117 mL for FF/VI and 95 mL for TIO) exceeded a proposed MCID for weighted mean FEV125 and was within the putative MCID range of 100–140 mL for trough FF/VI;26 however, the treatment difference between the two therapies was not statistically significant.
In our study, secondary endpoints were nested under the primary endpoint; since the primary endpoint difference did not reach statistical significance, all differences reported for the secondary efficacy endpoints must be regarded as descriptive only. Nonetheless, numerical differences were observed between the treatment groups in some other lung function endpoints, symptoms, use of rescue medication, and QoL. Rescue medication use and dyspnea were consistently reduced over the 12 weeks of treatment with FF/VI compared with TIO. QoL (measured by mean SGRQ-C total and component scores and by the percentage of SGRQ-C responders) and health status (measured by mean CAT scores) improved with both treatments, but the improvement was greater with FF/VI treatment compared with TIO. The total SGRQ-C improvement with FF/VI was −5.0 at 8 weeks and −3.9 at 12 weeks, similar to the result of −4.3 at 12 weeks in another study.25 A previous comparison of another ICS/LABA with TIO provided similar findings of a superior effect of ICS/LABA on QoL.7 In our study, mean SGRQ-C improvements reached or approached the MCID after treatment with FF/VI, but not TIO. In contrast, CAT improvement scores were more modest after FF/VI treatment and did not approach the MCID. Although the most reliable estimate of MCID may be two points,27 the CAT only allows integer scores, and the bar may be set too high for population-based comparisons.28
As COPD progresses, patients experience increasing air trapping and hyperinflation reflected by IC declines.29 In our study, 12 weeks’ TIO treatment compared with FF/VI provided improvements in trough IC and FVC. Improvements in IC have been associated with decreased exertional but not resting dyspnea.30,31 In this regard, it is noteworthy that although treatment with TIO was associated with a greater improvement in IC, dyspnea improved more so with FF/VI.
The safety profiles of FF/VI and TIO in this study were similar, with a couple of exceptions. Pneumonia was reported more frequently in FF/VI-treated subjects, all radiographically confirmed, consistent with previous reports of a higher incidence with FF/VI treatment13 or other ICS treatment alone or in combination with a LABA.32,33
More subjects failed to complete the study while receiving TIO (12% vs 6% for FF/VI), including those who experienced COPD exacerbations (4% vs 2%, respectively). In 1-year studies of FF/VI versus VI,13 treatment with FF/VI reduced exacerbation rates compared with treatment with VI alone. Likewise, in a 2-year study comparing TIO with FP/SALM, more TIO-treated subjects withdrew, but the rates of COPD exacerbation did not differ between groups.7
In our population of patients with comorbid CVD or increased risk of CVD, there were no apparent effects of FF/VI or TIO treatment on the CV safety profile or differences between treatments, with the exception of two deaths from CV events in the TIO-treated subjects. We paid particular attention to measuring CV safety, employing 24-hour Holter monitoring in a subset of subjects, performing frequent ECGs, and monitoring CV AEs because this was an at-risk population and due to previous reports of TIO and LABA CV safety. While some earlier analyses suggested that the risk of CVD or CV mortality was increased in COPD patients using the TIO Respimat® (Boehringer Ingelheim) inhaler,5,34,35 this was not confirmed in a subsequent trial,35 and the TIO HandiHaler has not been associated with an increased risk compared with placebo in recent studies.6,36–39
In our study, the rate of ventricular tachycardias was somewhat higher than anticipated. However, the definition can vary from that used here (>100 beats/min) to that of >120 beats/min.40–41 Also, subjects in both groups experienced a greater incidence of ventricular ectopic beats at the end of the study than at screening, but the circumstances of those Holter measurements differed, as the end-of-study visit was conducted with a 24-hour in-patient stay.
The CV risk profile of FF/VI versus TIO has also been examined in a recent clinical study of FF/VI effects on arterial stiffness, a marker that is elevated in COPD and is a predictor of CV events and mortality.42,43 Arterial stiffness, as measured by aortic pulse wave velocity, was lowered by both FF/VI and TIO in subjects with moderate-to-very severe COPD and baseline aortic pulse wave velocity ≥11.0 m/s.44 These subjects also had a high incidence of baseline CVD, but the number of CV events over the 12-week treatment period was low and similar between treatments.44 Results of the ongoing SUMMIT trial in 16,000 subjects with comorbid CVD risk and COPD will further determine the impact of FF/VI on CVD and mortality.45
The major limitations of our study were its lack of placebo arm, relatively small sample size, and 12-week duration, making the study incapable of establishing effects on pneumonia incidence, CVD, or mortality.
However, this randomized study had several strengths. First, it provided for prospective collection of COPD exacerbation and pneumonia data. Second, it applied rigorous CV monitoring and compared ICS/LABA and TIO treatment in a population with comorbid CVD. Most importantly, the study is the first to compare the efficacy of a once-daily ICS/LABA with TIO. Patients with COPD demonstrate low adherence to study medication,46 which is significantly associated with increased risk of death and COPD exacerbations.47 Once-daily medications such as TIO have been shown to improve adherence in medication use over medications with more frequent dosing regimens,48–50 and it is expected that FF/VI will have similar benefits, especially in certain patient populations who prefer once-daily medications.51 Adherence to study medication in the current study was high, with no difference between the FF/VI and TIO groups.
Conclusion
In conclusion, treatment with FF/VI or TIO for 12 weeks improved lung function in COPD subjects who have or are at risk for comorbid CVD, but there was no significant difference between the treatment groups. Reduction in rescue medication use and dyspnea and improving SGRQ-C-indicated QoL favored FF/VI, whereas improvements in FVC and IC favored TIO. Other than more reports of pneumonia in FF/VI-treated subjects and CV AEs in TIO-treated subjects, the safety profiles of the treatments were similar. The risk of pneumonia should be weighed against the benefits of FF/VI treatment, such as the reduction in COPD exacerbations, increased QoL, and medication adherence, when clinicians consider the potential use of FF/VI for patients with COPD. The results of an ongoing larger FF/VI trial in subjects with COPD with CVD (SUMMIT)45 are anticipated to provide additional evidence regarding efficacy and safety in this population.
Acknowledgments
Editorial support in the development of the first draft of this manuscript under the guidance of the authors, collation of author comments, editorial suggestions to drafts of this manuscript, preparation of tables and figures, referencing, and copyediting was provided by Elizabeth Field, PhD, of Field Advantage Medical Communications, LLC. Editorial support, in the form of preparation of the manuscript for submission, was provided by Rhiannon Owen, BSc, at Gardiner-Caldwell Communications (Macclesfield, UK). This support was funded by GSK.
Disclosure
GSK sponsored this study and provided support for study design and data collection, analysis, and interpretation. The sponsor had no part in the decision of the authors to submit this manuscript for publication. HC has received support for conducting clinical research for GSK, Boehringer Ingelheim, Forest Labs, Novartis, Pearl Therapeutics, and Intermune. BP and IS received support from GSK to conduct clinical research. CS-W and CC are employees of GSK and hold shares/share options in the company. AE is an employee of PAREXEL International (and was previously employed by GSK; work performed on this publication was done while employed by GSK) and holds shares/share options in GSK. The authors report no other conflicts of interest in this work.
References
goldcopd.org [homepage on the Internet]. Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD. 2015. Available from: http://www.goldcopd.org/:2015. Accessed June 24, 2015. | ||
Nannini LJ, Poole P, Milan SJ, Holmes R, Normansell R. Combined corticosteroid and long-acting beta2-agonist in one inhaler versus placebo for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2013;11:CD003794. | ||
Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;256:775–789. | ||
Celli B, Ferguson G, Anderson J, et al. Salmeterol/fluticasone propionate improves lung function and reduces the rate of decline over three years in the TORCH survival study. Am J Respir Crit Care Med. 2008;178:332–338. | ||
Karner C, Chong J, Poole P. Tiotropium versus placebo for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;7:CD009285. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359:1543–1554. | ||
Wedzicha JA, Calverley PM, Seemungal TA, et al; INSPIRE Investigators. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177:19–26. | ||
Welsh EJ, Cates CJ, Poole P. Combination inhaled steroid and long-acting beta2-agonist versus tiotropium for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2013;5:CD007891. | ||
Valotis A, Hogger P. Human receptor kinetics and lung tissue retention of the enhanced-affinity glucocorticoid fluticasone furoate. Respir Res. 2007;8:54. | ||
Kempsford R, Norris V, Siederer S. Vilanterol trifenatate, a novel inhaled long-acting beta2 adrenoceptor agonist, is well tolerated in healthy subjects and demonstrates prolonged bronchodilation in subjects with asthma and COPD. Pulm Pharmacol Ther. 2013;26:256–264. | ||
Kerwin EM, Scott-Wilson C, Sanford L, et al. A randomized trial of fluticasone furoate/vilanterol (50/25 μg; 100/25 μg) on lung function in COPD. Respir Med. 2013;107:560–569. | ||
Martinez FJ, Boscia J, Feldman G, et al. Fluticasone furoate/vilanterol (100/25; 200/25 μg) improves lung function in COPD: a randomized trial. Respir Med. 2013;107:550–559. | ||
Dransfield MT, Bourbeau J, Jones PW, et al. Once-daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbations of COPD: two replicate double-blind, parallel-group, randomized controlled trials. Lancet Respir Med. 2013;1:210–223. | ||
Dransfield MT, Feldman G, Korenblat P, et al. Efficacy and safety of once-daily fluticasone furoate/vilanterol (100/25 mcg) versus twice-daily fluticasone propionate/salmeterol (250/50 mcg) in COPD patients. Respir Med. 2014;108:1171–1179. | ||
Agusti AGN. Systemic effects of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:367–370. | ||
Jones PW, Wedzicha JA, Agusti A, et al. Baseline demographic and clinical profile of patients with and without cardiovascular diseases in the Assessment of Comorbidities in COPD in European Symptomatic Subjects from primary care (ACCESS) study. Eur Respir J. 2013;42(suppl 57):1067s. | ||
Curkendall SM, DeLuise C, Jones JK, et al. Cardiovascular disease in patients with chronic obstructive pulmonary disease, Saskatchewan Canada cardiovascular disease in COPD patients. Ann Epidemiol. 2006;16:63–70. | ||
Finkelstein J, Cha E, Scharf SM. Chronic obstructive pulmonary disease as an independent risk factor for cardiovascular morbidity. Int J Chron Obstruct Pulmon Dis. 2009;4:337–349. | ||
Calverley PM, Anderson JA, Celli B, et al; TORCH Investigators. Cardiovascular events in patients with COPD: TORCH study results. Thorax. 2010;65:719–725. | ||
Cazzola M, Matera MG, Donner CF. Inhaled β2-adrenoceptor agonists. Drugs. 2005;65:1595–1610. | ||
Jones PW. Health status measurement in chronic obstructive pulmonary disease. Thorax. 2001;56:880–887. | ||
Lotvall J, Bakke PS, Bjermer L, et al. Efficacy and safety of 4 weeks’ treatment with combined fluticasone furoate/vilanterol in a single inhaler given once daily in COPD: a placebo-controlled randomized trial. BMJ Open. 2012;2:e000370. | ||
Boscia JA, Pudi KK, Zvarich MT, Sanford L, Siederer K, Crim C. Effect of once-daily fluticasone furoate/vilanterol on 24-hour pulmonary function in patients with chronic obstructive pulmonary disease: a randomized, three-way, incomplete block, crossover study. Clin Ther. 2012;34:1655–1666. | ||
Bollmeier SG, Prosser TR. Combination of fluticasone furoate and vilanterol for the treatment of chronic obstructive pulmonary disease. Ann Pharmacother. 2014;48:250–257. | ||
Agustí A, de Teresa L, De Backer W, et al. A comparison of the efficacy and safety of once-daily fluticasone furoate/vilanterol with twice-daily fluticasone propionate/salmeterol in moderate to very severe COPD. Eur Respir J. 2014;43:763–772. | ||
Donohue J. Minimal clinically important differences in COPD lung function. COPD. 2005;21:111–124. | ||
Kon SS, Canavan JL, Jones SE, et al. Minimum clinically important difference for the COPD assessment test: a prospective analysis. Lancet Respir Med. 2014;2:195–203. | ||
Jones PW. Estimation and application of the minimum clinically important difference in COPD. Lancet Respir Med. 2014;2:167–169. | ||
Celli BR, Decramer M, Lystig T, Kesten S, Tashkin DP. Longitudinal inspiratory capacity changes in chronic obstructive pulmonary disease. Respir Res. 2012;13:66. | ||
Taube C, Lehnigk B, Paasch K, Kirsten DK, Jörres RA, Magnussen H. Factor analysis of changes in dyspnea and lung function parameters after bronchodilation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:216–220. | ||
O’Donnell DE, Flüge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnea and exercise tolerance in COPD. Eur Respir J. 2004;117(suppl 12A):49S–59S. | ||
Kew KM, Seniukovich A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;3:CD010115. | ||
Crim C, Dransfield MT, Bourbeau J, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol compared with vilanterol alone in patients with COPD. Ann Am Thorac Soc. 2015;12:27–34. | ||
Singh S, Loke YK, Enright P, Furberg CD. Mortality associated with tiotropium mist inhaler in patients with chronic obstructive pulmonary disease: systematic review and meta-analysis of randomized controlled trials. BMJ. 2011;342:d3215. | ||
Dong Y-H, Lin H-H, Shau W-Y, Wu Y-C, Chang C-H, Lai M-S. Comparative safety of inhaled medications in patients with chronic obstructive pulmonary: systematic review and mixed treatment comparison meta-analysis of randomized controlled trials. Thorax. 2013;68:48–56. | ||
Covelli H, Bhattacharya S, Cassino C, Conoscenti C, Kesten S. Absence of electrocardiographic findings and improved function with once-daily tiotropium in patients with chronic obstructive pulmonary disease. Pharmacotherapy. 2005;25:1708–1718. | ||
Kesten S, Celli B, Decramer M, Leimer I, Tashkin D. Tiotropium HandiHaler® in the treatment of COPD: a safety review. Int J Chron Obstruct Pulmon Dis. 2009;4:397–409. | ||
Verhamme KMC, Afonso ASM, van Noord C, Haag MD, Koudstaal PJ, Brusselle GGO. Tiotropium Handihaler and the risk of cardio- or cerebrovascular events and mortality in patients with COPD. Pulm Pharmacol Ther. 2012;25:19–26. | ||
Wise RA, Anzueto A, Cotton D, et al; TIOSPIR Investigators. Tiotropium respimat inhaler and the risk of death in COPD. N Engl J Med. 2013;369:1491–1501. | ||
Katritsis DE, Zareba W, Camm AJ. Nonsustained ventricular tachycardia. J Am Coll Cardiol. 2012;60:1993–2004. | ||
Buxton AE, Duc J, Berger EE, Torres V. Non-sustained ventricular tachycardia. Cardiol Clin. 2000;18:327–336. | ||
Vlachopoulos C, Aznaoundis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–1327. | ||
Vivodtzev I, Tamisier R, Baguet J-P, Borel JC, Levy P, Pépin J-L. Arterial stiffness in COPD. Chest. 2014;145:861–875. | ||
Pepin J-L, Cockcroft JR, Midwinter D, Sharma S, Rubin DB, Andreas S. Long-acting bronchodilators and arterial stiffness in patients with COPD: a comparison of fluticasone furoate/vilanterol with tiotropium. Chest. 2014;146:1521–1530. | ||
Vestbo J, Anderson J, Brook RD, et al. The study to understand mortality and morbidity in COPD (SUMMIT) study protocol. Eur Respir J. 2013;41:1017–1022. | ||
St Charles M, Blanchette CM, Silver H, Lavallee D, Dalal AA, Mapel D. Adherence to controller therapy for chronic obstructive pulmonary disease: a review. Curr Med Res Opin. 2010;26:2421–2429. | ||
Vestbo J, Anderson JA, Calverley PM, et al. Adherence to inhaled therapy, mortality and hospital admission in COPD. Thorax. 2009;65:939–943. | ||
Toy EL, Beaulieu NU, McHale JM, et al. Treatment of COPD: relationships between daily dosing frequency, adherence, resource use, and costs. Respir Med. 2011;105:434–441. | ||
Gauhar U, Dransfield M, Cooper JA. Sequential comparison of tiotropium to high-dose ipratropium in patients with chronic obstructive pulmonary disease in a practice setting. Int J Chron Obstruct Pulmon Dis. 2009;4:391–395. | ||
Halpern R, Baker CL, Su J, et al. Outcomes associated with initiation of tiotropium or fluticasone/salmeterol in patients with chronic obstructive pulmonary disease. Patient Prefer Adherence. 2011;5:375–388. | ||
Price D, Lee AJ, Sims EJ, et al. Characteristics of patients preferring once-daily controller therapy for asthma and COPD: a retrospective cohort study. Prim Care Respir J. 2013;22:161–168. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.