")
Back to Journals » Journal of Pain Research » Volume 13
Efficacy and Safety of ASP0819 in Patients with Fibromyalgia: Results of a Proof-of-Concept, Randomized, Double-Blind, Placebo-Controlled Trial
Authors Arnold LM , Blauwet MB , Tracy K, Cai N, Walzer M, Blahunka P, Marek GJ
Received 7 August 2020
Accepted for publication 30 October 2020
Published 10 December 2020 Volume 2020:13 Pages 3355—3369
DOI https://doi.org/10.2147/JPR.S274562
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Leslie M Arnold,1 Mary Beth Blauwet,2 Katherine Tracy,2 Na Cai,2 Mark Walzer,2 Paul Blahunka,2 Gerard J Marek2
1University of Cincinnati College of Medicine, Cincinnati, OH, USA; 2Astellas Pharma Global Development, Inc, Northbrook, IL, USA
Correspondence: Gerard J Marek
Astellas Pharma Global Development, Inc, 1 Astellas Way, Northbrook, IL 60062, USA
Tel +1 224-205-5055
Email [email protected]
Purpose: ASP0819 is a novel, non-opioid KCa 3.1 channel opener that reverses abnormal nerve firing of primary sensory afferent nerves. Currently available treatments for fibromyalgia provide only modest relief and are accompanied by a host of adverse side effects.
Patients and Methods: In this phase 2a, double-blind trial (NCT03056690), adults meeting fibromyalgia diagnostic criteria were randomized 1:1 to receive either 15 mg/day of oral ASP0819 (n=91) or placebo (n=95). The primary endpoint was the change from baseline to Week 8 in the mean daily average pain score. Changes in the Fibromyalgia Impact Questionnaire Revised (FIQR) symptoms, function, and overall impact subscales, as well as changes in the patients’ global impression of change, were secondary endpoints; treatment effects on FIQR total score and impact on sleep were exploratory analyses.
Results: There was no statistically significant difference between ASP0819 and placebo for the primary endpoint (P=0.086); however, ASP0819 versus placebo significantly improved daily average pain at Weeks 2, 6, and 7 (all P< 0.05). Numerical improvements were observed on the FIQR total score and several sleep items showed statistically significant improvements with ASP0819 versus placebo. There were no major safety concerns with ASP0819. Headache was the most common treatment-emergent adverse event (TEAE) occurring in both study arms; most TEAEs were mild or moderate in severity and no TEAEs suggestive of potential drug abuse were observed, as assessed by TEAE reporting and/or safety evaluations. Withdrawal effects also were not observed.
Conclusion: ASP0819 demonstrated some signals suggestive of efficacy and had a good tolerability profile in patients with fibromyalgia. Further studies are required to determine if ASP0819 can be a novel non-opioid treatment option in this patient group.
ClinicalTrials.gov Registration: NCT03056690.
Keywords: ASP0819, fibromyalgia, non-opioid treatment, pain, clinical trial
Introduction
Currently available treatments for fibromyalgia provide only modest relief and are accompanied by a host of adverse side effects. Furthermore, the dramatic increase in deaths from opioid-induced overdoses with the opioid crisis has led to the NIH Helping to End Addiction Over the Long-term initiative,1,2 which aims to improve treatments for opioid use disorders and enhance pain management. While only a minority of patients with chronic pain become addicted to opioids, prescription opioid pain management puts patients at increased risk for addiction, overdose, and death.3 Taken together, it is apparent that the development of novel non-opioid analgesic treatments takes on increasing importance.
Fibromyalgia occurs in 2.2% of the United States (US) population4 and presents with chronic widespread pain, fatigue, and sleep disturbances.5–7 The US Food and Drug Administration (FDA) has approved three medications for fibromyalgia treatment: the alpha-2-delta calcium channel ligand pregabalin, and the serotonin/norepinephrine reuptake inhibitors duloxetine and milnacipran.8 Nevertheless, opioid analgesics continue to be used, despite the lack of evidence of their effectiveness for fibromyalgia. Currently, three million US citizens and 16 million citizens worldwide have opioid use disorder.9 Given the ongoing opioid crisis, additional non-opioid analgesics are needed for chronic pain management.
Fibromyalgia pathophysiology is not fully understood, but there is evidence of altered central processing of nociceptive information.6,10,11 However, fibromyalgia likely results from abnormalities in both the peripheral and central nervous systems.12,13 Peripheral, C-fiber abnormalities have been reported in skin biopsies from patients with fibromyalgia.14 Some patients with fibromyalgia have hyperexcitable C nociceptors and structural changes in nociceptive C-fibers, suggesting abnormal ongoing peripheral C nociceptor activity and increased mechanical sensitivity that could contribute to the pain and tenderness reported in fibromyalgia.12 Small-fiber pathology, such as small-fiber polyneuropathy and functional impairment of A-delta- and C-fibers, may also contribute to the widespread pain in patients with fibromyalgia.15–17 Given this peripheral pathophysiology, targeting peripheral mechanisms may be helpful in fibromyalgia treatment strategies.
KCa3.1 is a voltage-insensitive, intermediate conductance calcium-activated potassium channel expressed on numerous cell types and tissues, including dorsal root ganglion cells, microglia, and the gastrointestinal tract.18–21 Opening of the KCa3.1 channel results in a period of reduced excitability after each action potential (ie, potentiates the after-hyperpolarizing current).22 Physiologically, KCa3.1 regulates cellular excitability, thus making KCa3.1 channels a potential therapeutic target in diseases that are associated with abnormal nerve excitation, possibly including fibromyalgia and neuropathic pain.23
ASP0819 is a novel non-opioid KCa3.1 potassium ion channel opener that reverses abnormal nerve firing. The effects of ASP0819 enhance late hyperpolarization and subsequently decrease primary sensory afferent nerve firing rates, thereby potentially reducing the pain experienced by patients with fibromyalgia (Figure 1). ASP0819 demonstrates poor brain penetration and had an acceptable toxicological profile, as well asabsorption, distribution, metabolism, and excretion profiles, in nonclinical studies (data on file). ASP0819 exerted effects in several animal models that may model aspects of fibromyalgia such as the rat vagotomy model24,25 and the reserpine-induced myalgia model.25,26 ASP0819 was also found to be safe and tolerable as indicated by Phase 1 testing in healthy volunteers. Presented here are the results from a phase 2a proof-of-concept study on the analgesic efficacy, safety, and tolerability of ASP0819 in patients with fibromyalgia.
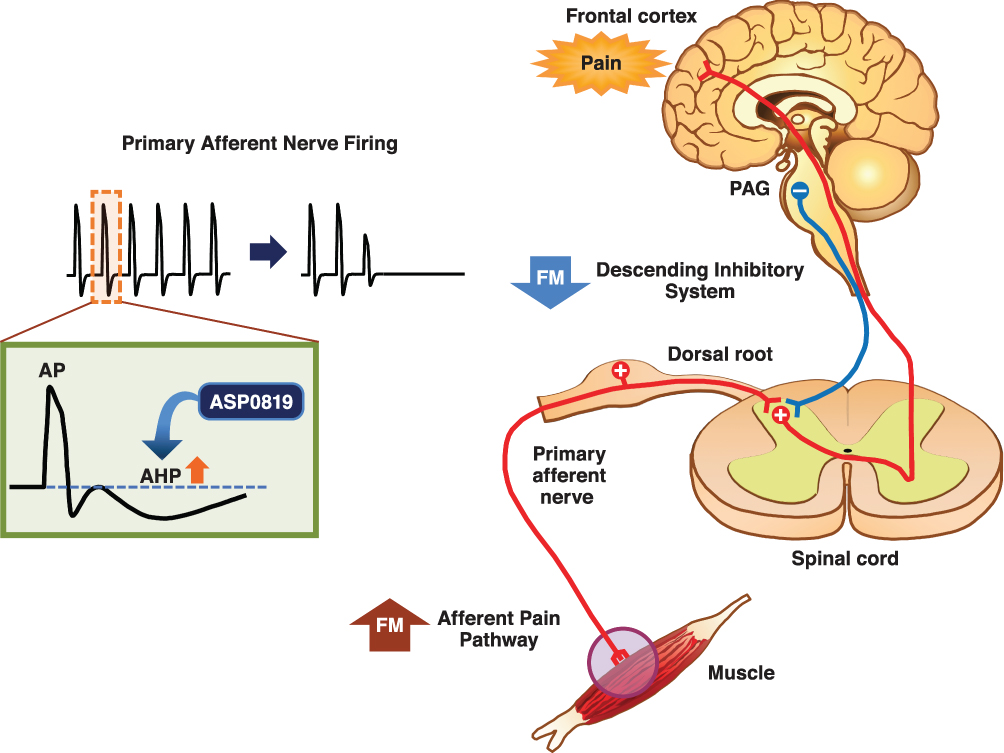 |
Figure 1 Mechanism of action of ASP0819. ASP0819 enhances late hyperpolarization and subsequently decreases firing of primary sensory afferent nerves, which potentially reduces the fibromyalgia pain. Abbreviations: AHP, afterhyperpolarization; AP, action potential; FM, fibromyalgia; PAG, periaqueductal gray. |
Patients and Methods
Study Design and Patients
This phase 2a, randomized, double-blind, placebo-controlled, parallel-group study (NCT03056690) was conducted at 24 sites in the US to assess the analgesic efficacy and safety of ASP0819 in patients with fibromyalgia. The study consisted of the following: a screening period of up to 42 days, which included a washout period and a 7-day baseline diary run-in; a 57-day double-blind, randomized treatment period with site visits at Days 1, 15, 29, and 57; and a 4-week follow-up period consisting of a clinic visit after 2 weeks and a phone call after 4 weeks. The study was carried out in accordance with the Declaration of Helsinki and approved by the relevant Institutional Review Boards. All participants provided written informed consent prior to the initiation of any study procedures.
Male and female patients aged 18 to 80 years with a body mass index of ≤45 kg/m2 were included in the trial if they met the American College of Rheumatology (ACR) 1990 and 2010 fibromyalgia diagnostic criteria at screening.5,7 The core diagnostic criteria for fibromyalgia are defined by the 1990 criteria,7 and while ACR has published newer criteria, the 1990 version is still commonly utilized in clinical trials. To meet ACR 1990 criteria, patients must have had widespread pain for at least 3 months, defined as the presence of pain on the right and left sides of the body, pain above and below the waist, and pain in the axial skeleton (cervical spine, anterior chest, thoracic spine, or low back).7 They must also have had pain in at least 11 of 18 tender point sites on digital palpation performed with an approximate force of 4 kg. Investigators were trained in tender point examination and the assessment of the ACR2010 criteria; these investigators performed this exam and confirmed the diagnosis.
To meet the ACR 2010 criteria, patients needed to have a widespread pain index score of ≥7 and a symptom severity scale score ≥5, or a widespread pain index score 3–6 and a symptom severity scale score ≥9.5 Symptoms must have been present at a similar level for at least 3 months and patients must have been free of any other disorder that could have explained the pain.
In addition, patients must have had a pain score ≥4 on the Fibromyalgia Impact Questionnaire Revised (FIQR) pain item at screening,27 along with a mean daily average pain score of 4–9 (inclusive) on an 11-point (0–10) numerical rating scale (NRS) during the baseline diary run-in period. They were also required to have met prespecified criteria for mean daily average pain scores. Patients had to agree to use only acetaminophen (up to 1000 mg per dose and not to exceed 3000 mg/day) as rescue medication for fibromyalgia pain during the study. Nonsteroidal anti-inflammatory drugs were permitted (with the exception of celecoxib) as needed for non-fibromyalgia pain, such as headache. The concomitant use of medications, therapies, or surgical procedures that could influence the evaluation of the study drug’s efficacy and safety, such as cannabinoids, certain hypnotics, or other medications or procedures that have efficacy in reducing the pain of fibromyalgia, were prohibited throughout the study (Box 1). Patients were not permitted to initiate or change any nonpharmacological interventions (including normal daily exercise routines, chiropractic care, physical therapy, psychotherapy, and massage therapy) over the study period. Stability in nonpharmacological interventions were required for a minimum of 30 days prior to screening. Patients were further required to maintain usual levels of activity for the duration of the study and to wash out from any protocol-excluded drugs they may have been taking prior to baseline. Subjects taking prohibited medications who were willing to discontinue these medications—as clinically indicated and based upon the investigator’s recommendation—underwent a wash-out over a period of five half-lives on a schedule determined by the investigator.
 |
Box 1 Medications, Therapies, or Surgical Procedures Prohibited During the Study |
Key exclusion criteria included receiving an investigational therapy within 28 days or 5 half-lives prior to screening; no meaningful improvement, in the investigator’s opinion, from ≥2 prior treatments for fibromyalgia from at least two pharmacologic classes; pain (including diabetic peripheral neuropathy, post-therapeutic neuralgia, traumatic injury, prior surgery, or complex regional pain syndrome) that would interfere with the assessment of fibromyalgia pain or that required excluded therapies; and/or infectious or inflammatory arthritis, autoimmune disease, or other widespread rheumatic diseases. A comprehensive list of exclusion criteria can be found in the Supplemental Methods.
Treatment
Eligible patients were randomized 1:1 using Interactive Response Technology to receive 15 mg (three capsules of 5 mg) of oral ASP0819 or matching placebo capsules, each given once daily in the morning, with or without food, for 8 weeks. The randomization list and study medication blind were maintained by the Interactive Response Technology system. The treatment code for each randomized subject was provided by the Interactive Response Technology system in the event of a medical emergency requiring knowledge of the treatment assigned to the subject. Subjects were randomized in a blinded fashion such that the investigator, sponsor’s study management team, clinical staff, and the subject did not know which agent was being administered.
The ASP0819 dose was chosen based on nonclinical studies and the phase 1 pharmacokinetic single ascending dose (SAD) and multiple ascending dose (MAD) studies; 15 mg once daily was expected to provide exposure within an efficacious target range and is 3-fold below the no obvious adverse event levels observed in a 13-week dog toxicology study (data on file). The initial dose was administered under the supervision of study center personnel. Compliance was assessed at each clinic visit during the treatment phase by counting unused returned medication.
Endpoints and Assessments
The primary efficacy endpoint was the change from baseline to Week 8 in mean daily average pain score assessed by the NRS (0 to 10 scale). The NRS is a generic pain assessment instrument, which consists of a single question that asks patients to record their daily average pain on an 11-point scale (0=no pain; 10=pain as bad as you can imagine) over the previous 24 hours; weekly mean of these ratings was then calculated. Patients used a handheld e-diary to report daily average pain NRS scores and to capture rescue medication use; these data were automatically transmitted to a central database. Patients completed the NRS each day from the baseline run-in period through Week 10 every evening.
Secondary efficacy endpoints included the number of patients achieving a ≥30% or ≥50% reduction in mean daily average pain score assessed by NRS (0 to 10 scale) from baseline to Week 8 and end of treatment (EOT; Day 57); change from baseline to Weeks 2, 4, 8, and EOT in the FIQR function, symptoms, and overall impact subscales; and overall improvement assessed by patient global impression of change (PGIC)28 at Weeks 2, 4, 8, and EOT. Change from baseline to Weeks 1, 2, 3, 4, 5, 6, 7, and EOT in mean daily average pain scores was an exploratory endpoint. Change from baseline to Weeks 2, 4, 8, and EOT in FIQR total score was also assessed as an exploratory endpoint.
Questionnaires on efficacy, such as FIQR and PGIC, were completed during study visits. The FIQR captures the full spectrum of problems related to fibromyalgia and the responses to therapy, ranging from activities of daily living, overall impact, and symptoms.27 Patients answered each question on an 11-point NRS over a recall period of the last 7 days or the last time the activity was performed (for the physical function domain if the activity was not performed within the 7-day recall period). The PGIC is a self-administered 7-point Likert scale that asks patients to evaluate their fibromyalgia relative to baseline (from “very much improved” to “very much worse”).28
Sleep disturbance was assessed as an exploratory endpoint using change from baseline to each respective week (Weeks 1–8) and from baseline to EOT in daily fibromyalgia sleep diary (FMSD) responses.29 The FMSD is a validated 8-item patient-reported outcome that assesses sleep disturbances specific to patients with fibromyalgia across the domains of falling asleep, staying asleep, and sufficient sleep.29 Each day upon waking, patients rated their sleep quality over the previous night on an 11-point NRS using their handheld e-diary. Each item is rated on an 11-point NRS anchored by “0–not at all” and “10–extremely.”
Safety and tolerability were assessed by monitoring treatment-emergent adverse events (TEAEs), which were coded using MedDRA (v 20), as well as laboratory values, vital signs, and electrocardiograms, and by assessing suicidal ideation, suicidal behavior, and self-injurious behavior without suicidal intent on the Columbia-Suicide Severity Rating Scale (C-SSRS).30 The C-SSRS, a screening tool to identify suicide risk, was performed by trained study center staff via interviews at screening, randomization, and Weeks 2, 4, 8/EOT, and 10. At each site, blinded investigators assessed the adverse events and also decided whether these events were treatment-related.
Statistical Analyses
Considering that many phase 2a studies fail, analyses that bias results toward a false-positive result have profound implications for successfully developing novel medications. Thus, a one-sided 5% significance level (assuming normally distributed data and accounting for the interim analysis for futility) was employed. The effect size of 0.39 was 20% to 30% larger than that determined in a meta-analysis (0.32) of change from baseline in Brief Pain Inventory average pain score31 for duloxetine, or a meta-analysis (0.30) of change from baseline in mean daily average pain NRS for pregabalin versus placebo (data on file).
Efficacy analyses were conducted using the full analysis set (FAS), which consisted of all patients who were randomized and received at least one dose of study drug. Most hypothesis testing was one-sided at the 5% significance level; the two-sided 90% confidence interval (CI) was used when applicable. Unless otherwise stated, there was no imputation of missing data.
Analysis for the primary endpoint of change from baseline to Week 8 in the mean daily average pain NRS used a mixed-model repeated-measures (MMRM) analysis, where the model included the fixed effects for treatment group, center (pooled where necessary), time (study Week 1 to 8), and treatment-by-time interaction, with baseline mean daily average pain NRS and baseline mean daily average pain NRS-by-time interaction as covariates.
A sensitivity analysis for the primary endpoint was conducted using the same MMRM model with multiple imputation based on discontinuation reasons to account for any missing data (using placebo as the reference group for patients who discontinued due to lack of efficacy or TEAEs and standard regression-based multiple imputation for patients with missing data for other reasons). Additionally, an MMRM analysis using data from the per-protocol set (defined as patients from the FAS who met criteria based on adherence to the protocol) was conducted to assess the robustness of the results from the statistical tests based on the FAS.
The additional endpoints, including response reduction of ≥30% and ≥50% from baseline to Week 8 and EOT in mean daily average pain score assessed by NRS, change from baseline to Weeks 2, 4, 8, and EOT in the FIQR, change from baseline to each week from Weeks 1 to 8 in FMSD, and overall patient improvement assessed by PGIC, were analyzed. The Fisher’s Exact Test was used to assess the response rate of mean daily average pain score (≥30% and ≥50% reduction from baseline to Week 8 and to EOT); for the EOT analysis, last observation carried forward (LOCF) was used. For the FIQR subscales, a similar MMRM model was used for the change from baseline to Weeks 2, 4, and 8, and an ANCOVA model, with covariates of baseline FIQR subscale score and center (pooled where necessary), was used for the change from baseline to EOT. An additional analysis using imputation by modified baseline observation carried forward (mBOCF) was conducted for the response rate of the mean daily average pain score (≥30% and ≥50% reduction from baseline to Week 8) and change from baseline in FIQR subscales at Week 8. Specifically, mBOCF was defined as imputation using BOCF for patients who discontinued due to lack of efficacy or TEAE and imputation by LOCF for patients who discontinued for other reasons. A proportional odds model for ordinal data with model term for treatment group was used to assess PGIC at Weeks 2, 4, 8, and EOT. For patients with missing data, an analysis at Weeks 2, 4, and 8 was conducted using mBOCF; an additional analysis at Weeks 2 and 4 used LOCF.
Key exploratory endpoints, including change from baseline to Weeks 2, 4, and 8 in FIQR total score, and change from baseline to each week from Weeks 1 to 8 in FMSD, were analyzed using MMRM, with change from baseline to EOT for both measures analyzed with an ANCOVA model (covariates: baseline score and center [pooled where necessary]). An additional ANCOVA analysis for change from baseline to Week 8 in FIQR total score was performed using mBOCF.
Safety data were summarized using the safety analysis set (SAF) consisting of all patients who were randomized and received at least one dose of study drug.
Results
Study Disposition and Population
Overall, 406 patients provided written informed consent; 182 discontinued during the screening/wash-out period, and 38 discontinued during the 7-day baseline diary run-in period (Figure 2). A total of 186 patients were randomized to receive ASP0819 (n=91) or placebo (n=95); two patients (one in each treatment group) did not take the study drug. Thus, 184 randomized patients (98.9%) were included in the SAF and FAS and 173 patients (93.0%) were included in the per-protocol set. The main reason for exclusion from the per-protocol set was due to current, untreated moderate or severe major depressive disorder/history of any psychotic and/or bipolar disorder (n=8). Overall, 183 patients (n=93, placebo; n=90, ASP0819) were included in the follow-up period. The primary reasons for leaving the study prior to randomization were screening failure, patient withdrawal, and “other.” In total, 156 (83.9%) patients completed the planned double-blind treatment period and 168 patients (90.3%) completed the planned follow-up period; there were no differences between treatment and placebo groups in terms of withdrawals during the double-blind treatment period.
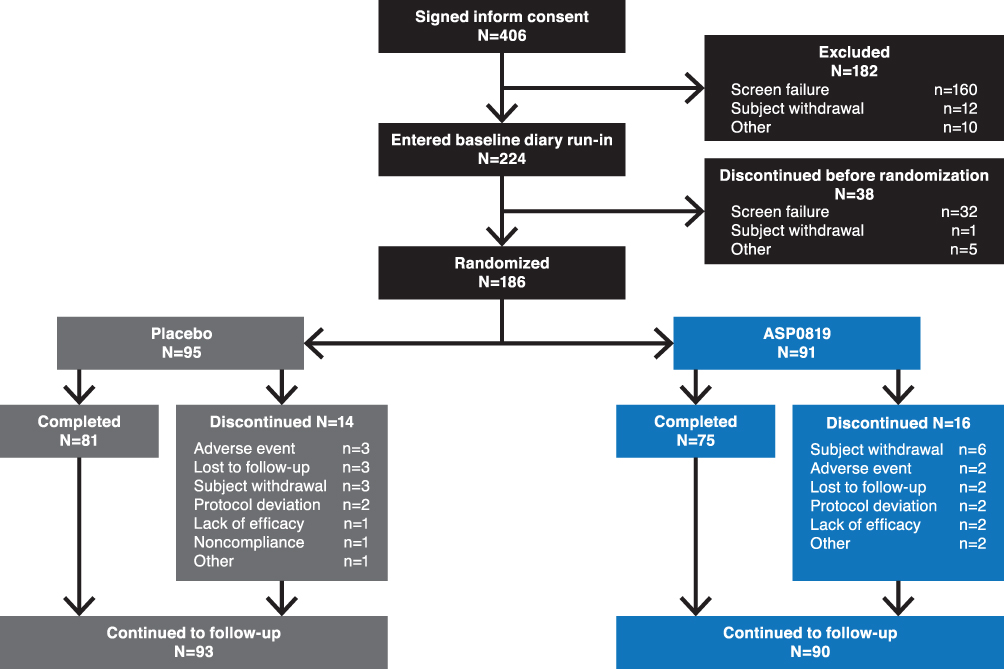 |
Figure 2 Patient disposition. Of the 406 patients who provided informed consent, 186 eligible patients were randomized 1:1 to receive oral ASP0819 15 mg or placebo. Patients who discontinued the study during the treatment period could enter the follow-up period. |
There were no meaningful differences in demographic characteristics between the placebo and ASP0819 treatment groups (Table 1). Most patients were female (96.2%) and white (82.1%). The treatment groups were relatively similar in terms of medical history, although more patients in the ASP0819 group than in the placebo group reported a history of major depressive disorder (21.1% vs 11.7%) and endometriosis (14.8% vs 5.6%), and more patients in the placebo group reported a history of temporomandibular disorder (20.2% vs 5.6%).
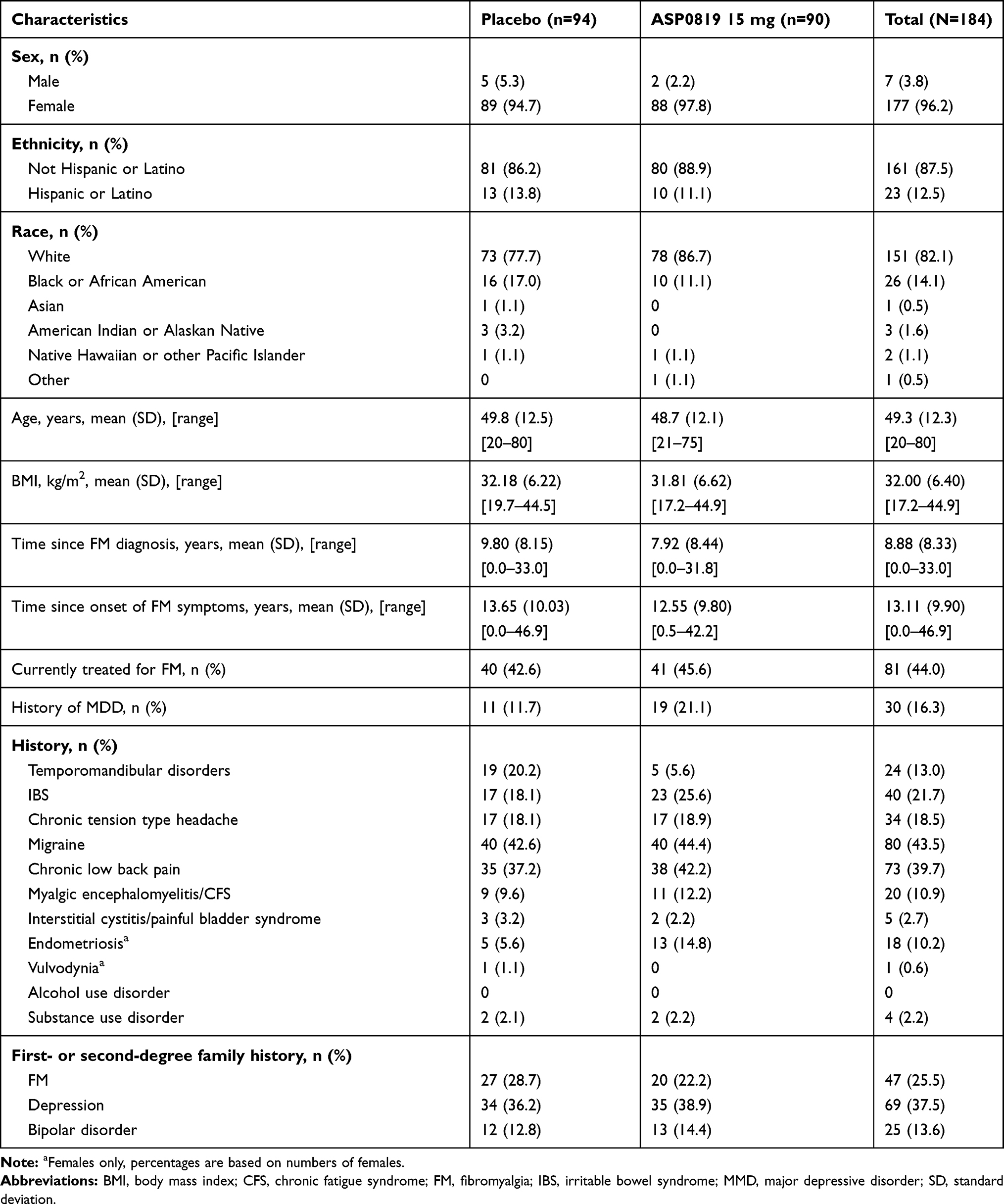 |
Table 1 Demographic and Disease Characteristics (Safety Analysis Set) |
The treatment groups were also similar with respect to fibromyalgia-related baseline characteristics. The mean of the baseline daily average pain score was 6.32 in both treatment groups, but the proportion of patients with severe baseline mean daily average pain score (≥7–10) was higher in the ASP0819 group than in the placebo group (31.1% vs 22.3%). Prior medication used to treat fibromyalgia pain and other types of pain was similar in the ASP0819 and placebo groups.
Efficacy
There was no statistically significant change, only a trend in favor of ASP0819, from baseline to Week 8, in mean daily average pain score (primary endpoint) for ASP0819 versus placebo (−1.60 vs −1.26; treatment difference [SE]: −0.34 [0.25]; two-sided 90% CI: −0.76, 0.07; P=0.086). Similar results were obtained in both sensitivity and per-protocol set analyses (data not shown). However, statistically significant treatment differences in favor of ASP0819 compared with placebo were observed in a change from baseline in mean daily average pain score at Weeks 2, 6, and 7 (Figure 3 and Supplemental Figure 1).
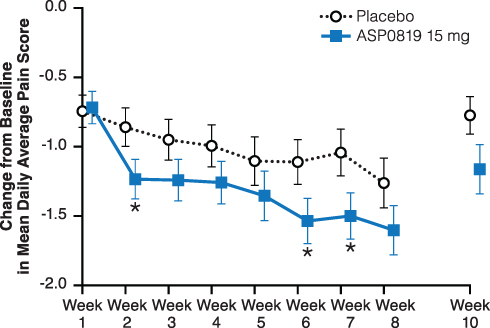 |
Figure 3 Change from baseline in mean daily average pain score during double-blind treatment period and follow-up period (full analysis set). The mean daily average pain score assessed by the Numeric Rating Scale, which consists of a single question that asks patients to record their daily average pain on an 11-point scale (ranging from 0=no pain to 10=pain as bad as you can imagine) over the previous 24 hours. Data from the double-blind period are presented as LS mean ± standard error; data from the follow-up period are presented as mean ± standard error. *Indicates P<0.05. Abbreviation: LS, least squares. |
There were no statistically significant differences between ASP0819 and placebo groups in the responder analysis of mean daily average pain score or for the overall patient improvement assessed by PGIC (Table 2). A statistically significant difference in favor of ASP0819 compared with placebo was seen in the symptoms subscale (Figure 4A) and overall impact subscale (Figure 4B) of the FIQR at Week 4 (least-squares [LS] mean difference: −3.73; P=0.039 and LS mean difference: −1.34; P=0.018, respectively), but not in FIQR function (LS mean difference: −2.97; P=0.103; Figure 4C). While there was a statistically significant difference in the reduction from baseline in the total score at Week 4 (LS mean difference: −4.14; P=0.033), no statistically significant difference was seen in the reduction from baseline in FIQR total score at Week 8 (Table 2). There were numerical improvements for ASP0819 versus placebo, but there were no statistically significant differences in the reduction (improvement) from baseline to Week 8 in FIQR function, symptoms subscale, or overall impact subscale (Table 2).
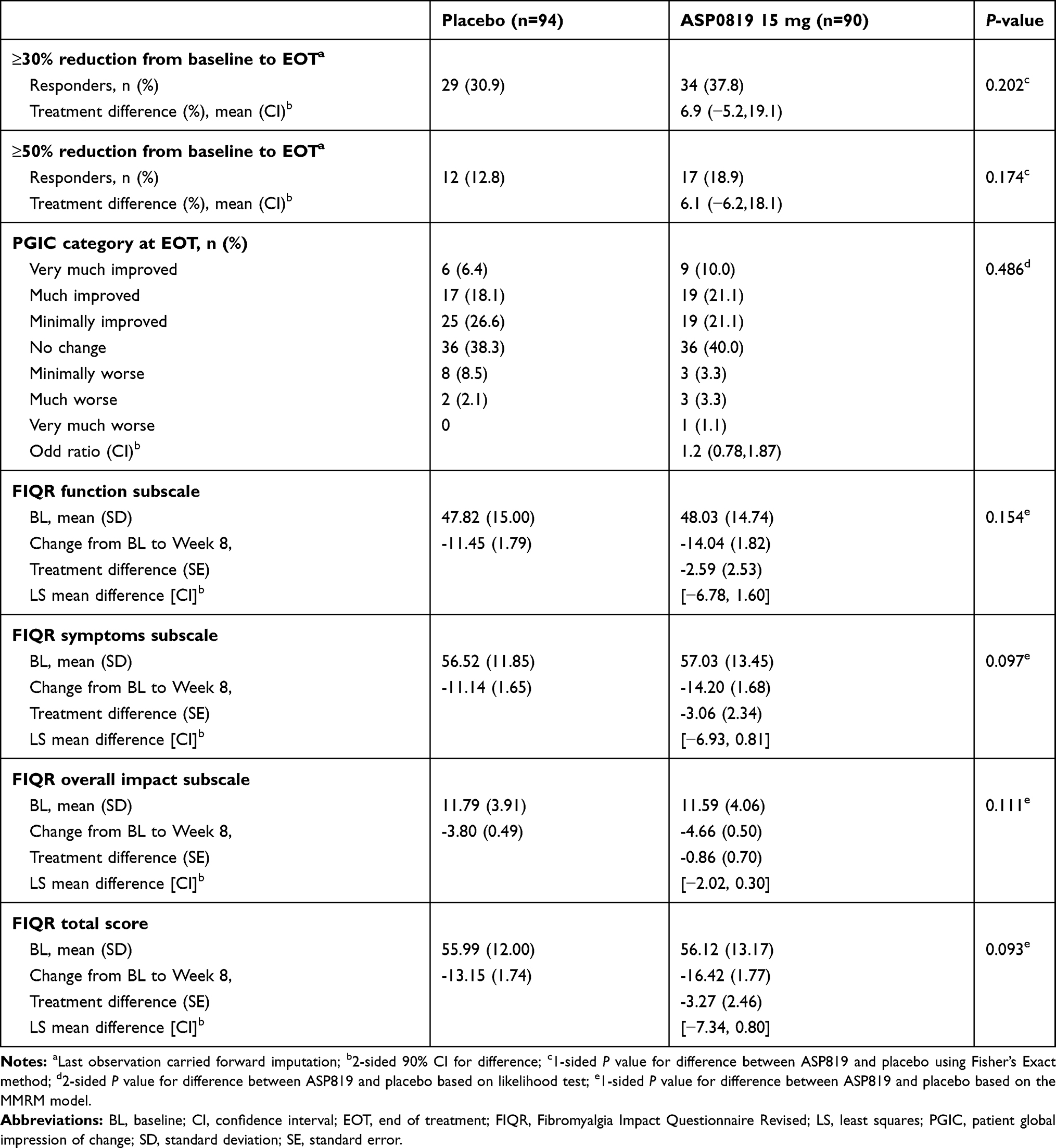 |
Table 2 Additional Efficacy Parameters (Full Analysis Set) |
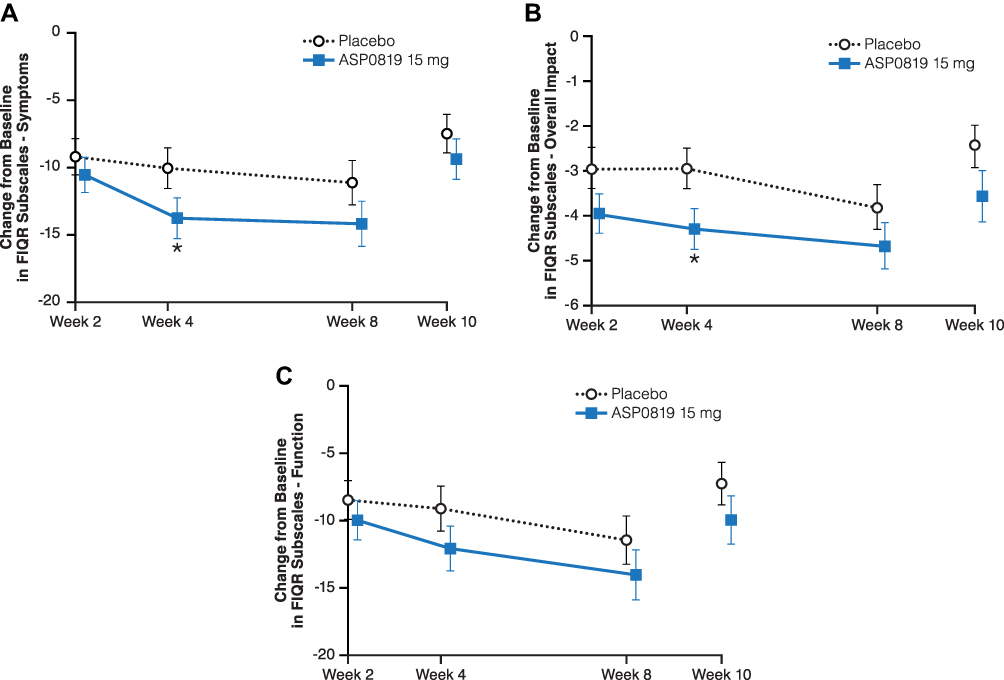 |
Figure 4 Change from baseline during double-blind treatment period and follow-up period in FIQR subscales (full analysis set). Changes from baseline in the mean FIQR subscale scores: (A) Symptom, (B) Overall Impact, and (C) Function are depicted. All questions in each subscale are rated on an 11-point numeric scale, ranging from 0 to 10 with 10 being the worst. Data from the double-blind period are presented as LS mean ± standard error; data from the follow-up period are presented as mean ± standard error. *Indicates P<0.05. Abbreviations: FIQR, Fibromyalgia Impact Questionnaire Revised; LS, least squares. |
Improvement from baseline in FIQR total score was numerically higher in the ASP0819 treatment group compared with the placebo group at all time points (Figure 5). There was no statistically significant difference between ASP0819 and placebo in the reduction (improvement) from baseline to Week 8 in the MMRM analysis (LS mean difference: −3.27; P=0.093); however, in an ANCOVA analysis, the improvement from baseline to Week 8 in FIQR total score was statistically significant for ASP0819 versus placebo using mBOCF imputation (LS mean difference: −4.10; P=0.036) and EOT using LOCF imputation (LS mean difference: −3.75; P=0.050).
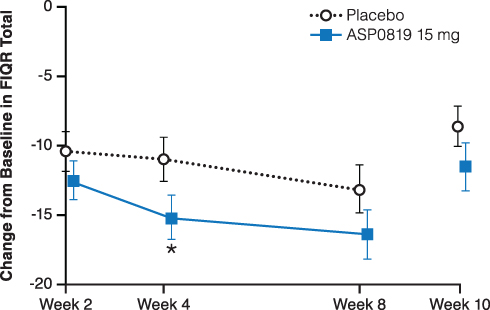 |
Figure 5 Change from baseline during double-blind treatment period and follow-up period in FIQR total (full analysis set). The change from baseline in FIQR total score is depicted. The FIQR total score represents the sum of the three subscale scores: symptom, function, and overall impact. The symptom subscale accounts for 50%, the function subscale accounts for 30%, and the overall impact subscale accounts for 20% of the total score. Data from the double-blind period are presented as LS mean ± standard error; data from the follow-up period are presented as mean ± standard error. *Indicates P<0.05. Abbreviations: FIQR, Fibromyalgia Impact Questionnaire Revised; LS, least squares. |
Statistically significant improvements in favor of ASP0819 compared with placebo were seen in several items of the FMSD beginning at Week 2 (difficulty with falling asleep; P=0.032 [Figure 6A]); at Week 3 (restlessness of sleep, P=0.013 [Figure 6B]; difficulty getting comfortable, P=0.007; and difficulty staying asleep, P=0.013); at Week 6 (degree of deep sleep, P=0.035; and difficulty with beginning the day, P=0.040). Statistically significant improvements for ASP0819 versus placebo were seen for difficulty falling asleep (P=0.018) and restlessness of sleep (P=0.033) at Week 8, and for difficulty falling asleep (P=0.008), restlessness of sleep (P=0.009), and difficulty with beginning the day (P=0.044) at EOT.
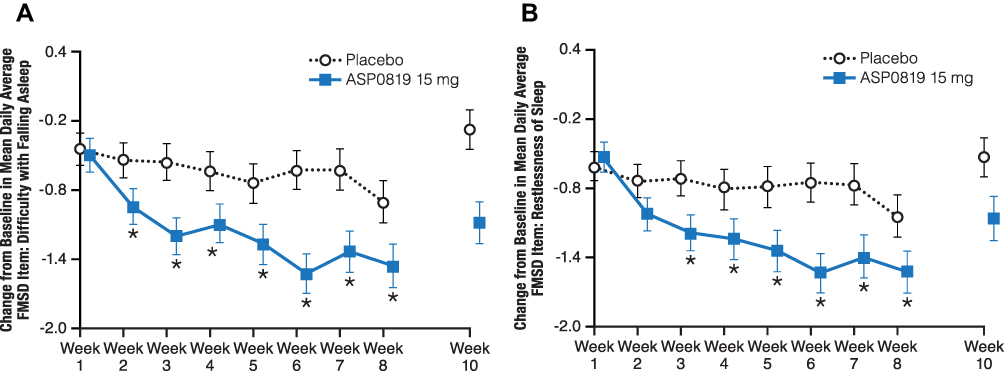 |
Figure 6 Change from baseline during double-blind treatment period and follow-up in mean daily average FMSD Item 1: Difficulty with falling asleep (full analysis set) and Item 2: Restlessness of sleep (full analysis set). The change from baseline in Item 1 of the FMSD, difficulty with falling asleep, is shown in (A). The change from baseline in Item 2 of the FMSD, restlessness of sleep, is displayed in (B). Patients rated their difficulty falling asleep or restlessness of sleep over the previous night on an 11-point numeric rating scale ranging from “0–not at all” to “10–extremely” in an e-diary. Data from double-blind period are presented as LS mean ± standard error; data from the follow-up period are presented as mean ± standard error. *Indicates P<0.05. Abbreviations: FMSD, Fibromyalgia Sleep Diary; LS, least squares. |
Use of Pain Medication
During the double-blind treatment period, concomitant medication for fibromyalgia pain was administered to 5.6% of ASP0819-treated patients and 2.1% of placebo-treated patients. More patients received concomitant medication for fibromyalgia pain during the follow-up period (10.9%) than during the double-blind treatment period (3.8%), and this was similar between the ASP0819 and placebo groups. More patients receiving ASP0819 than placebo reported use of concomitant medication for non-fibromyalgia pain during the double-blind treatment period (31.1% vs 20.2%; Table 3); however, most of these medications were analgesics, which primarily included non-steroidal anti-inflammatory drugs. A total of 17 patients (n=4, placebo; n=13, ASP0819) violated protocol by taking excluded concomitant medication during the study; two of these patients (n=1, placebo; n=1, ASP0819) were excluded from the per-protocol efficacy analyses.
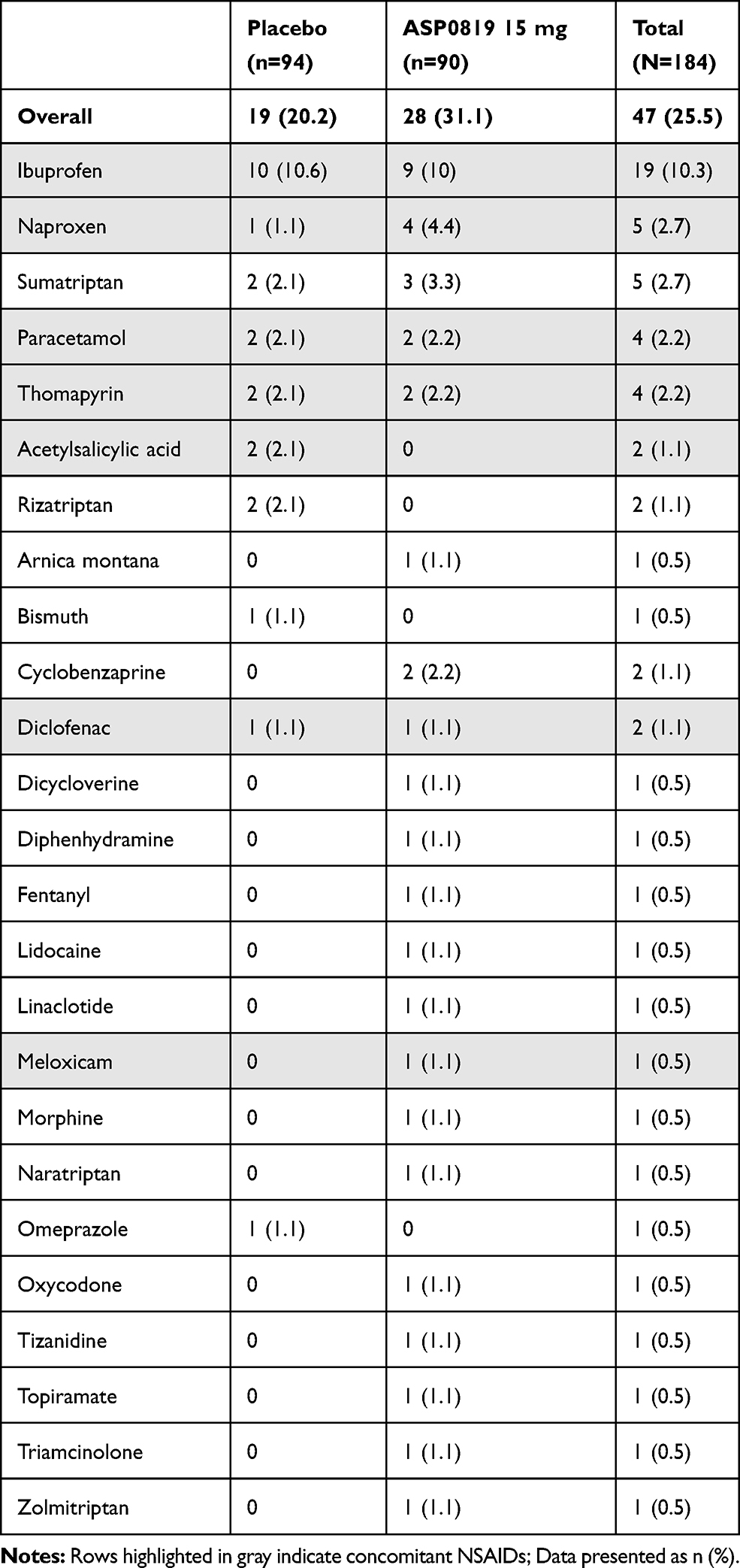 |
Table 3 Concomitant Medications Taken for Non-Fibromyalgia Pain During Double-Blind Treatment Period by WHO Preferred Name |
During the double-blind treatment period, the mean proportion of days with rescue medication (acetaminophen) use was low (24% [placebo] and 22% [ASP0819]). Across the study, there were no statistically significant differences between the treatment groups at any week for the incidence of patients using rescue medication. However, the mean average daily dose of rescue medication was generally lower in the ASP0819 treatment group compared with the placebo group.
Safety
Adverse events were reported by 53 placebo recipients (56.4%) and 62 ASP0819 recipients (68.9%). Drug-related TEAEs, as assessed by the investigator, were reported by more patients in the placebo group (n=29; 30.9%) than in the ASP0819 group (n=22; 24.4%) (Table 4). The most commonly reported TEAE was headache, occurring in a similar proportion of patients in each treatment group. Headache was also the most commonly reported drug-related AE, reported by eight (8.5%) patients in the placebo group and five (5.6%) patients in the ASP0819 treatment group. Most TEAEs were mild or moderate in severity, with severe TEAEs reported by only three placebo recipients (3.2%; gastroenteritis viral, headache, and syncope) and one ASP0819 recipient (1.1%; insomnia). Four patients (three receiving placebo and one receiving ASP0819) reported TEAEs leading to discontinuation of study drug. The events in the placebo group were gastrointestinal hemorrhage, disturbance in attention, anxiety, chills, fatigue, and hot flush. The event leading to discontinuation in the ASP0819 treatment group was insomnia. There were no deaths or serious TEAEs during the study.
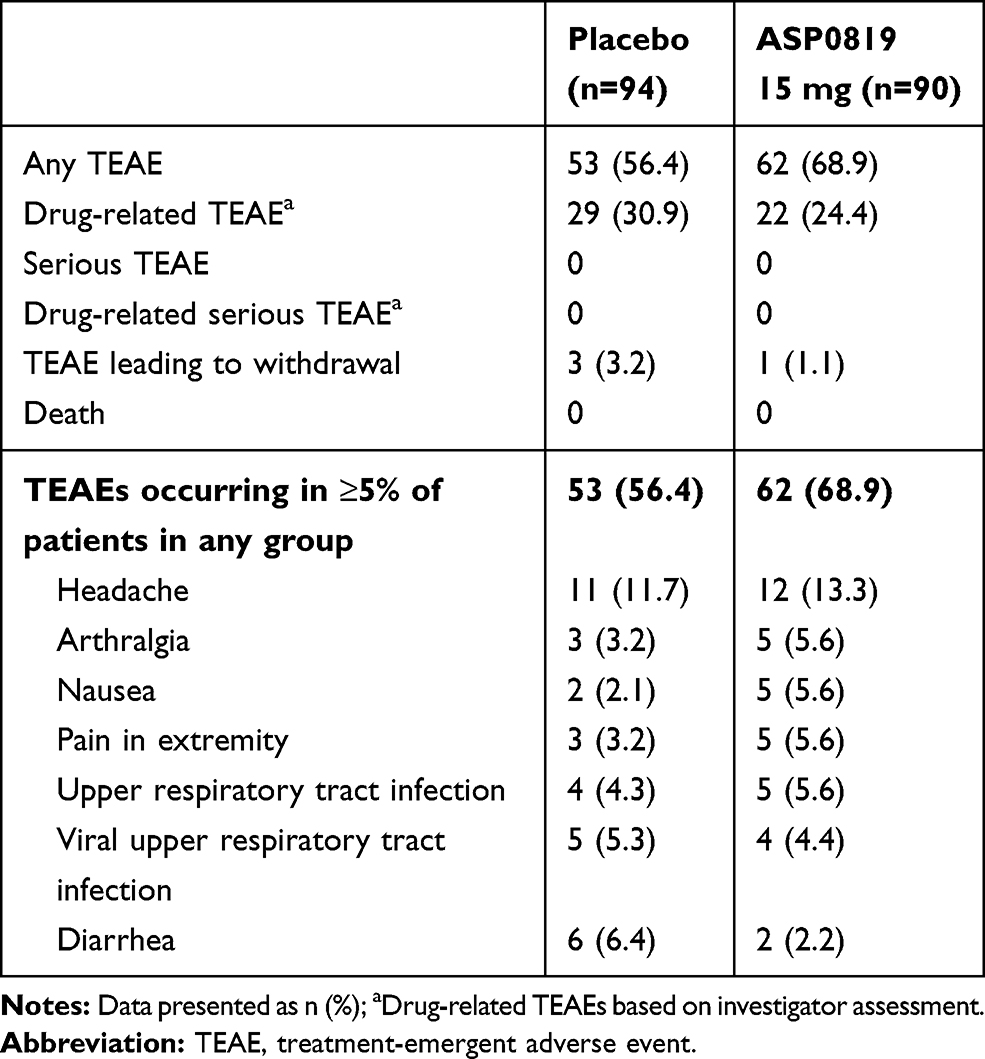 |
Table 4 Overview of TEAEs (Safety Analysis Set) |
There were no clinically relevant changes in laboratory parameters that would indicate an ASP0819-related effect. There were no safety concerns with regard to vital signs or electrocardiogram parameters. No suicidal ideation or suicidal behavior occurred during the double-blind treatment period. One patient in the placebo group reported suicidal ideation in the follow-up period. During the study, there were no patients with TEAEs within the drug abuse and dependence standardized MedDRA Query and no patients had a drug abuse-related TEAE (defined by a predefined Adverse Events of Interest search strategy). During the follow-up period, drug withdrawal-related TEAEs (ie, anxiety, chills, diarrhea, headache, hyperhidrosis, insomnia, nausea, syncope, vomiting) were reported in a total of four patients across the two cohorts (placebo, n=3; ASP0819, n=1).
Discussion
Although this study did not meet its primary efficacy endpoint of change from baseline to Week 8 in mean daily average pain score for ASP0819 versus placebo, there were some signals suggestive of efficacy in patients with fibromyalgia. For example, compared with placebo, ASP0819 demonstrated statistically significant changes in mean daily average pain score at Weeks 2, 6, and 7. Furthermore, there were numerically greater improvements for ASP0819 than for placebo in all secondary endpoints, and statistical significance between groups was achieved on some FIQR subscales at Week 4. However, no statistically significant difference was seen regarding FIQR function at Week 4 and no statistically significant difference was seen in the reduction from baseline in FIQR total score at Week 8. Statistically significant improvements for ASP0819 compared with placebo were also observed for several exploratory endpoints at various time points, including the FIQR total score, and for several FMSD items, including difficulty with falling asleep and restlessness of sleep. Considering ASP0819’s positive impact on sleep, our results support possible further assessment of ASP0189 or similar compounds for such an indication. Indeed, the vast majority of patients with fibromyalgia report disturbed sleep (quality and quantity) as a result of their condition,29,32 the existence of which is associated with reduced quality of life.33,34
Despite not meeting the primary efficacy endpoint in this study, the mean change from baseline compared with placebo in pain NRS score for 15 mg ASP0819 was within the lower part of the range for the mean change in pain NRS scores reported for 300 mg to 450 mg daily pregabalin doses.35,36 However, the evaluation of the efficacy signal should be interpreted with respect to the power of the study sample size. To this end, this PoC trial was powered to test the hypothesis that the change from baseline compared with placebo for ASP8019 would provide at least a 30% improvement for ASP0819 compared with historical pregabalin and duloxetine data (eg, seeking ~39% improvement for ASP0819 vs placebo compared with historical ~30% improvement of pregabalin or duloxetine vs placebo). While progress has been made in identifying new treatments for fibromyalgia, there remains a substantial unmet need for patients who may not respond to, or tolerate, the currently available medications. Therefore, powering studies to demonstrate equivalent efficacy with regard to the standard of care may be a reasonable approach in light of the ongoing opioid crisis.2,9
Notably, ASP0819 was well tolerated in the current study with no major safety concerns with respect to TEAEs, vital signs, electrocardiograms, C-SSRS, and TEAEs sensitive to potential drug abuse or withdrawal effects. Headache was the most commonly reported TEAE in patients treated with either ASP0819 or placebo but was similar in frequency for both study arms. The majority of all the reported TEAEs were considered by the investigator to be mild or moderate in severity.
It should be noted that this is the first study looking for potential efficacy signals of ASP0819 in treating fibromyalgia. However, at this early stage in a drug’s development, it is premature to reject an effective candidate due to marginal significance; a one-sided test was used whenever applicable due to the fact that the current analysis was interested in testing the benefits of ASP0819 over placebo as opposed to the benefits of placebo over ASP0819. It is also worth noting that, in the current study, we tested ASP0819 under the highly stringent hypothesis that ASP0819 would be 30% better than pregabalin or duloxetine. This increased stringency biases the drug development process away from novel medications that may be useful in special populations or medications that may possess similar efficacy to standard of care while possessing improved safety profiles. It is also worth noting that the current study was not designed to test secondary outcomes with the same rigor as the primary outcome. While improvements in sleep were not expected, the current analysis included measures of sleep quality since this is a current unmet medical need for patients with fibromyalgia. More research is needed to determine the clinical significance of the exploratory findings.
There are a number of additional limitations of the present study with respect to interpreting and potential efficacy signal(s) for ASP0819. However, the favorable safety and tolerability profile of ASP0819 compared with placebo in this study raises the question of whether greater efficacy might be observed with higher ASP0819 doses (exposures). Perhaps a longer treatment period at a higher dose might result in an effect size for ASP0819 close to that observed with the FDA-approved medications for fibromyalgia. Interventions to increase subject adherence to the prescribed medication might further decrease the possibility of false-negative results in additional ASP0819 trials. Finally, powering the study according to the hypothesis that ASP0819 possesses similar efficacy to the current standard of care is an additional modification for future studies that will allow for a better understanding of the potential efficacy and safety of ASP0819 with respect to fibromyalgia or other chronic pain syndromes.
Conclusions
In the current study, there was no statistically significant difference between ASP0819 and placebo for the primary endpoint, nor in the change from baseline to Week 8 in the mean daily average pain score. However, ASP0819 demonstrated some signals suggestive of efficacy in this fibromyalgia patient group, along with a good tolerability profile; nevertheless, additional studies are required to determine the efficacy and safety of ASP0819 in fibromyalgia. These additional studies may confirm or refute whether ASP0819 is a novel non-opioid-based option for patients with fibromyalgia.
Abbreviations
ACR, American College of Rheumatology; CI, confidence interval; C-SSRS, Columbia-Suicide Severity Rating Scale; EOT, end of treatment; FAS, full analysis set; FIQR, Fibromyalgia Impact Questionnaire Revised; FMSD, fibromyalgia sleep diary; LOCF, last observation carried forward; LS, least squares; mBOCF, modified baseline observation carried forward; MMRM, mixed model repeated measures; NRS, numerical rating scale; PGIC, patient global impression of change; SAF, safety analysis set; TEAE, treatment-emergent adverse event; ULN, upper limit of normal.
Data Sharing Statement
Access to anonymized individual participant-level data will not be provided for this trial as it meets one or more of the exceptions described on www.clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas.”
Ethics Approval
The Institutional Review Board (IRB) reviewed the ethical, scientific, and medical appropriateness of the study before it was conducted. IRB approval of the protocol, informed consent, and subject information and/or advertising, as relevant, was obtained prior to the authorization of drug shipment to a study center. Any substantial amendments to the protocol required IRB approval prior to the implementation of the changes made to the study design at the site. Please see the Supplemental Materials for more information about the IRBs that approved this study. The study was conducted in accordance with the protocol, Good Clinical Practice (GCP), International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines, applicable regulations and guidelines governing clinical study conduct, and the ethical principles that have their origin in the Declaration of Helsinki.
Acknowledgments
Astellas Pharma Global Development, Inc. provided funding for this trial and was involved in the development of the study protocol, and in data collection, analysis, and interpretation. Editorial and writing assistance, under the guidance of the authors, was provided by Patrick Tucker, PhD and Elizabeth Hermans, PhD of OPEN Health Medical Communications (Chicago, IL) and funded by the study sponsor. The corresponding author had full access to all of the data in the study and final responsibility for the decision to submit the paper for publication.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. LMA, MBB, KT, MW, PB, and GM were involved in the conception and design of the study and provided guidance in the analysis and interpretation of the data, and contributions to the writing and revision of the manuscript. NC was integral in data collection and statistical analysis and contributed to the development and review of the manuscript.
Funding
Astellas Pharma Global Development, Inc. provided funding for this trial and was involved in the development of the study protocol, and in data collection, analysis, and interpretation.
Disclosure
L. Arnold has consulted for Pfizer, Eli Lilly, Astellas, Teva, Allergan, Daiichi Sankyo, Jazz Pharmaceuticals, Zynerba, Prismic, Cinrx, Innovative Med Concepts, and Lundbeck and has received research support from Pfizer, Eli Lilly, Daiichi Sankyo, Axsome, Tonix, Allergan, Aptinyx, Teva, Astellas, and Cefaly; and reports grants and personal fees from Astellas, during the conduct of the study; personal fees from Pfizer, Eli Lilly, Teva, Allergan, Daiichi Sankyo, Jazz Pharmaceuticals, Zynerba, Prismic, Cinrx, Innovative Med Concepts, and Lundbeck, and grants from Pfizer, Eli Lilly, Daiichi Sankyo, Axsome, Tonix, Allergan, Aptinyx, Teva, and Cefaly, outside the submitted work.
Mark Walzer and Paul Blahunka have a patent pending: Methods of treating sleep disorders associated with pain (PCT2020/043047).
K. Tracy and P. Blahunka are former employees of Astellas. All other authors are current employees of Astellas Pharma Global Development. The authors report no other potential conflicts of interest for this work.
References
1. Collins FS, Koroshetz WJ, Volkow ND. Helping to end addiction over the long-term: the research plan for the NIH HEAL initiative. JAMA. 2018;320(2):129–130. doi:10.1001/jama.2018.8826
2. Volkow ND, Collins FS. The role of science in addressing the opioid crisis. N Engl J Med. 2017;377(4):391–394. doi:10.1056/NEJMsr1706626
3. NIH. Improving opioid prescribing: opioid prescribers can play a key role in stopping the opioid overdose epidemic. Published 2017. Available from: https://www.drugabuse.gov/publications/improving-opioid-prescribing/improving-opioid-prescribing.
4. Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17(8):356. doi:10.1007/s11916-013-0356-5
5. Wolfe F, Clauw DJ, Fitzcharles MA, et al. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res (Hoboken). 2010;62(5):600–610. doi:10.1002/acr.20140
6. Wolfe F, Clauw DJ, Fitzcharles M-A, et al. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR preliminary diagnostic criteria for fibromyalgia. J Rheumatol. 2011;38(6):1113–1122. doi:10.3899/jrheum.100594
7. Wolfe F, Smythe HA, Yunus MB, et al. The American college of rheumatology 1990 criteria for the classification of fibromyalgia. Report of the multicenter criteria committee. Arthritis Rheum. 1990;33(2):160–172. doi:10.1002/art.1780330203
8. Arnold LM, Clauw DJ, Dunegan LJ, Turk DC. A framework for fibromyalgia management for primary care providers. Mayo Clin Proc. 2012;87(5):488–496. doi:10.1016/j.mayocp.2012.02.010
9. Azadfard M, Huecker MR, Leaming JM. Opioid Addiction. Treasure Island, FL: StatPearls; 2020.
10. Arnold LM, Bennett RM, Crofford LJ, et al. AAPT diagnostic criteria for fibromyalgia. J Pain. 2019;20:611–628.
11. Staud R. Brain imaging in fibromyalgia syndrome. Clin Exp Rheumatol. 2011;29(6 Suppl 69):S109–117.
12. Serra J, Collado A, Sola R, et al. Hyperexcitable C nociceptors in fibromyalgia. Ann Neurol. 2014;75(2):196–208. doi:10.1002/ana.24065
13. Staud R, Smitherman ML. Peripheral and central sensitization in fibromyalgia: pathogenetic role. Curr Pain Headache Rep. 2002;6(4):259–266. doi:10.1007/s11916-002-0046-1
14. Kim SH, Kim DH, Oh DH, Clauw DJ. Characteristic electron microscopic findings in the skin of patients with fibromyalgia: preliminary study. Clin Rheumatol. 2008;27(2):219–223. doi:10.1007/s10067-007-0739-2
15. Swiecka M, Maslinska M, Kwiatkowska B. Small fiber neuropathy as a part of fibromyalgia or a separate diagnosis? Int J Clin Rheumatol. 2018;13:353.
16. Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain. 2013;154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
17. Üçeyler N, Sommer C, Walitt B, Häuser W. Anticonvulsants for fibromyalgia. Cochrane Database Syst Rev. 2013;10.
18. Boettger MK, Till S, Chen MX, et al. Calcium-activated potassium channel SK1- and IK1-like immunoreactivity in injured human sensory neurones and its regulation by neurotrophic factors. Brain. 2002;125(Pt 2):252–263. doi:10.1093/brain/awf026
19. Hogan Q, Lirk P, Poroli M, et al. Restoration of calcium influx corrects membrane hyperexcitability in injured rat dorsal root ganglion neurons. Anesth Analg. 2008;107(3):1045–1051. doi:10.1213/ane.0b013e31817bd1f0
20. Lu R, Flauaus C, Kennel L, et al. KCa3.1 channels modulate the processing of noxious chemical stimuli in mice. Neuropharmacology. 2017;125:386–395. doi:10.1016/j.neuropharm.2017.08.021
21. Mongan LC, Hill MJ, Chen MX, et al. The distribution of small and intermediate conductance calcium-activated potassium channels in the rat sensory nervous system. Neuroscience. 2005;131(1):161–175. doi:10.1016/j.neuroscience.2004.09.062
22. Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14(13):1437–1457. doi:10.2174/092986707780831186
23. Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 2014;37(3):146–158. doi:10.1016/j.tins.2013.12.002
24. Khasar SG, Miao JP, Janig W, Levine JD. Modulation of bradykinin-induced mechanical hyperalgesia in the rat by activity in abdominal vagal afferents. Eur J Neurosci. 1998;10(2):435–444. doi:10.1046/j.1460-9568.1998.00030.x
25. Takeshita N, Oe T, Kiso T, Kakimoto S. A KCa3.1 channel opener, ASP0819, modulates nociceptive signal processing from peripheral nerves in fibromyalgia. J Pain Res. 2020.
26. Nagakura Y, Oe T, Aoki T, Matsuoka N. Biogenic amine depletion causes chronic muscular pain and tactile allodynia accompanied by depression: a putative animal model of fibromyalgia. Pain. 2009;146(1–2):26–33. doi:10.1016/j.pain.2009.05.024
27. Bennett RM, Friend R, Jones KD, Ward R, Han BK, Ross RL. The revised fibromyalgia impact questionnaire (FIQR): validation and psychometric properties. Arthritis Res Ther. 2009;11(4):R120. doi:10.1186/ar2783
28. Rampakakis E, Ste-Marie PA, Sampalis JS, Karellis A, Shir Y, Fitzcharles MA. Real-life assessment of the validity of patient global impression of change in fibromyalgia. RMD Open. 2015;1(1):e000146. doi:10.1136/rmdopen-2015-000146
29. Kleinman L, Mannix S, Arnold LM, et al. Assessment of sleep in patients with fibromyalgia: qualitative development of the fibromyalgia sleep diary. Health Qual Life Outcomes. 2014;12:111. doi:10.1186/s12955-014-0111-6
30. Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). New York: The Research Foundation for Mental Hygiene; 2009.
31. Poquet N, Lin C. The brief pain inventory (BPI). J Physiother. 2016;62(1):52. doi:10.1016/j.jphys.2015.07.001
32. Cetin B, Sunbul EA, Toktas H, Karaca M, Ulutas O, Gulec H. Comparison of sleep structure in patients with fibromyalgia and healthy controls. Sleep Breath. 2020:1–8.
33. Türkoğlu G, Selvi Y. The relationship between chronotype, sleep disturbance, severity of fibromyalgia, and quality of life in patients with fibromyalgia. Chronobiol Int. 2020;37(1):68–81. doi:10.1080/07420528.2019.1684314
34. Theadom A, Cropley M, Humphrey KL. Exploring the role of sleep and coping in quality of life in fibromyalgia. J Psychosom Res. 2007;62(2):145–151. doi:10.1016/j.jpsychores.2006.09.013
35. Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled monotherapy trial of pregabalin in patients with fibromyalgia. J Pain. 2008;9(9):792–805. doi:10.1016/j.jpain.2008.03.013
36. Mease PJ, Russell IJ, Arnold LM, et al. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol. 2008;35(3):502–514.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.