")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Effects of Voriconazole on the Pharmacokinetics of Vonoprazan in Rats
Authors Shen J, Wang B, Wang S , Chen F, Meng D, Jiang H, Zhou Y, Geng P, Zhou Q, Liu B
Received 25 March 2020
Accepted for publication 19 May 2020
Published 4 June 2020 Volume 2020:14 Pages 2199—2206
DOI https://doi.org/10.2147/DDDT.S255427
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jiquan Shen,1,* Bo Wang,1,* Shuanghu Wang,2,3 Feifei Chen,2 Deru Meng,2 Hui Jiang,2 Yunfang Zhou,2 Peiwu Geng,2 Quan Zhou,2 Bin Liu1
1Department of Orthopaedics, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, Zhejiang 323000, People’s Republic of China; 2The Laboratory of Clinical Pharmacy, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, Zhejiang 323000, People’s Republic of China; 3School of Pharmaceutical Science, Guangdong Provincial Key Laboratory of New Drug Screening, Southern Medical University, Guangzhou 510515, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Quan Zhou
The Laboratory of Clinical Pharmacy, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui 323000, Zhejiang, People’s Republic of China
Tel/Fax +86 578 278 0081
Email [email protected]
Bin Liu
Department of Orthopaedics, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui 323000, Zhejiang, People’s Republic of China
Email [email protected]
Purpose: The purpose of this study was to examine the effects of voriconazole on the pharmacokinetics of vonoprazan.
Methods: Fifteen Sprague-Dawley rats were randomly divided into three groups: five rats in each group, including control group, single-dose group (a single dose of 30 mg/kg of voriconazole), and multiple-dose group (multiple doses of 30 mg/(kg•day) per dose of voriconazole). Each group of rats was given an oral dose of 10 mg/kg vonoprazan 30 min after the administration of voriconazole or vehicle. After the oral administration of vonoprazan, 50 μL of blood was collected into 1.5-mL heparinized tubes via the caudal vein. The concentration of vonoprazan in plasma was quantified by ultra-performance liquid chromatography/tandem mass spectrometry. Both in vitro effects of voriconazole on vonoprazan and the mechanism of the observed inhibition were studied in rat liver microsomes.
Results: When orally administered, voriconazole increased the area under the plasma concentration–time curve (AUC), prolonged the elimination half-life (t1/2), and decreased the clearance (CL) of vonoprazan; there was no significant difference between the single-dose and multiple-dose groups. Voriconazole inhibited the metabolism of vonoprazan at an IC50 of 2.93 μM and showed mixed inhibition. The results of the in vivo experiments were consistent with those of the in vitro experiments.
Conclusion: Our findings provide the evidence of drug–drug interactions between voriconazole and vonoprazan that could occur with pre-administration of voriconazole. Thus, clinicians should pay attention to the resulting changes in pharmacokinetic parameters and accordingly, adjust the dose of vonoprazan in clinical settings.
Keywords: cytochrome P450, drug–drug interaction, pharmacokinetic, proton pump inhibitor
Introduction
Proton pump inhibitors (PPIs) suppress gastric acid secretion and are used for the treatment of gastric acid-related diseases such as gastroesophageal reflux disease, peptic ulcers, erosive esophagitis, and reflux esophagitis.1–3 Traditional PPIs, such as omeprazole, lansoprazole, and esomeprazole, have a slow onset of action and short plasma half-life, and most of them are metabolized by the liver microsomal enzyme, CYP2C19, which shows both genetic and phenotypic polymorphisms.4,5 All these factors may lead to insufficient inhibition of acid secretion at night and may cause a large variation in efficacy among patients. Vonoprazan is an orally available, reversible potassium-competitive acid blocker that was approved for the treatment of acid-related diseases in December 2014 in Japan.6 It inhibits K+ binding sites on H+-K +-ATPase and suppresses gastric acid secretion.7 Compared to traditional PPIs, vonoprazan has a more rapid onset of action, higher affinity, slower dissociation, longer plasma half-life, more long-lasting acid suppression resulting in prolonged inhibition of acid secretion, and is unaffected by CYP2C19 polymorphism, which results in less variation in efficacy among patients.8–10 Vonoprazan is metabolized by different metabolic pathways to form four major metabolites, M-I, M-II, M-III, and M-IV-Sul.11,12 In a correlation study, vonoprazan was found to be mainly metabolized by the CYP isoforms, CYP2B6, CYP3A4, CYP2C19, and CYP2D6 to M-I, M-III, and N-demethylated vonoprazan (a presumed metabolite), while the other pathway involved sulfation by SULT2A1 to M-IV-Sul.11,13 We hypothesized that the metabolism of vonoprazan might be altered when it is co-administered with drugs that can alter the metabolic activity of CYP3A4, CYP2B6, or CYP2C19.
Voriconazole is a second-generation, synthetic, broad-spectrum antifungal drug used for treating a variety of invasive fungal diseases because of its pharmacokinetic characteristics, such as rapid onset of action and high bioavailability.14–16 Voriconazole is mainly metabolized by CYP2C19 and, to a lesser extent, by CYP2C9, as well as CYP3A4.17 Additionally, voriconazole is a potent inhibitor of CYP3A4, CYP2B6, CYP2C9, and CYP2C19.18,19 A clinical study showed that voriconazole could significantly increase the exposure to oral buprenorphine by inhibiting hepatic CYP3A4.20 PPIs and voriconazole are often used together for the treatment of inpatients with gastrointestinal problems and invasive fungal infections.21–23 Treatment of human liver microsomes with five PPIs (rabeprazole, omeprazole, pantoprazole, lansoprazole, and esomeprazole) resulted in a significant increase in the systemic exposure of voriconazole.24 Different types of PPIs have different effects on the pharmacokinetic parameters of voriconazole. The findings of a retrospective clinical study revealed that the voriconazole trough level was significantly increased with the co-administration of PPIs such as omeprazole, lansoprazole, and esomeprazole, but not pantoprazole and ilaprazole.23 However, in another clinical study, neither lansoprazole nor ilaprazole appeared to affect the pharmacokinetic parameters of voriconazole in renal transplant recipients.15
Although studies have demonstrated the potential for CYP450-mediated interactions with the co-administration of voriconazole and PPIs, the effects of voriconazole on the pharmacokinetics of PPIs remain unclear.25–29 In addition, there are no reports of clinical studies on the combination of vonoprazan with voriconazole. As voriconazole and vonoprazan are metabolized by the same enzymes, and both drugs are co-administered in clinical settings, we aimed to examine the effects of voriconazole on the pharmacokinetics of vonoprazan. The pharmacokinetic parameters of vonoprazan, with or without voriconazole pretreatment in rats, were analyzed using a sensitive and reliable ultra-performance liquid chromatography/tandem mass spectrometry (UPLC/MS-MS) system. The effect of voriconazole on the metabolic stability of vonoprazan was determined using rat liver microsomes, and the mode of inhibition of vonoprazan by voriconazole was studied in vitro.
Materials and Methods
Chemicals and Biologicals
Vonoprazan (purity > 98%) and voriconazole (purity > 98%) were purchased from the Beijing Sunflower Technology Development Co. Ltd. (Beijing, China). M-I was purchased from the WuXi PharmaTech Co. Ltd. (Jiangsu, China). Diazepam (internal standard (IS); purity > 98%) was purchased from the Tianjin KingYork Pharmaceutical Co. Ltd. (Tianjin, China). Acetonitrile and methanol were purchased from Merck Co. Ltd. (Darmstadt, Germany). Carboxy methylcellulose sodium salt (CMC-Na) was purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai China). Formic acid was obtained from Sigma-Aldrich (St. Louis, MO, USA). Ultrapure water was obtained by using a Milli-Q water purification system (Millipore, Billerica, MA, USA). Rat liver microsomes (RLM) were prepared in our laboratory. All other chemicals and biologicals were of analytical grade or higher.
Animals and Treatment
Male Sprague-Dawley rats were obtained from the Experimental Animal Center of Wenzhou Medical University (Wenzhou, China). The animals were housed in a breeding room at 25°C with 60 ± 5% humidity and a 12 h/12 h dark-light cycle. Tap water and diet were provided ad libitum. The rats were acclimated to the above conditions for two weeks before initiating the animal experiments. All of the experimental procedures followed the guidelines for the care and use of laboratory animals and were approved by the Animal Experimental Ethical Inspection of Laboratory Animal Center, Wenzhou Medical University.
Instruments and Operation Conditions
The measurement of vonoprazan parameters was performed by using a UPLC-MS/MS system, which possessed an ACQUITY I Class UPLC and a XEVO TQD triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA). Chromatographic analysis of vonoprazan was performed with a CORTECS C18 column (2.1 × 50 mm, 1.6 µm) maintained at 40°C. The mobile phase consisted of 0.1% formic acid, 5 mM ammonium formate and acetonitrile. The elution process had a linear gradient: it started with acetonitrile increasing from 10% to 30% (0 to 0.5 min); rapidly increasing from 30% to 95% (0.5 to 1.0 min), which was maintained at 95% (1.0 to 2.0 min); and then decreasing to 10% (2.0 to 2.6 min). The flow rate was 0.4 mL/min, and the total run time was 3 min.
The mass scan mode was the positive multiple reaction monitoring mode. The precursor ion and product ion were m/z 347.08→205.06 for vonoprazan M-I, m/z 346.04→314.97 for vonoprazan, and m/z 285.10→193.10 for IS. The optimal MS parameters were defined as follows: the cone voltages were set at 40 V, 20 V, and 35 V for M-I, vonoprazan, and IS, respectively; the collision energies were set at 15 V, 10 V, and 30 V for M-I, vonoprazan, and IS, respectively.
In vivo Pharmacokinetics
Fifteen Sprague-Dawley rats, each weighing 230–250 g, were selected and divided into three groups, five rats in each group, including control group (orally administered 30 mg/kg/day CMC-Na solution for 14 consecutive days), single-dose group (orally administered a single dose of 30 mg/kg of voriconazole, dissolved in CMC-Na solution, on the 14th day), and multiple-dose group (orally administered multiple doses of 30 mg/kg/day of voriconazole for 14 consecutive days). After oral administration for 14 consecutive days, each group of rats was administered an oral gavage at a dose of 10 mg/kg vonoprazan (dissolved in CMC-Na solution) 30 min after the administration of voriconazole or vehicle. Blood (50 μL) was collected from the tail veins into heparinized glass capillary tubes for analysis at 5, 15, and 30 min and 1, 2, 3, 4, 6, 8, 12, and 24 h after vonoprazan administration. To the collected blood sample, 20 μL of IS and 100 μL of acetonitrile were added in a 1.5 mL microcentrifuge tube. The mixture was vortexed for 30 s and then centrifuged at 12,000 rpm for 10 min. Subsequently, the supernatant was transferred into a separate sample bottle. Five microliters of this supernatant were immediately analyzed using a sensitive and reliable LC-MS/MS method.
In vitro Pharmacokinetics
The procedure for preparing RLM was based on the methods described by Wang et al.30,31 The 200-µL incubation system contained vonoprazan, voriconazole, 0.44 mg/mL RLM, 1 mM of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), and 100 mM potassium phosphate buffer (pH 7.4). To determine the half-maximal inhibitory concentration (IC50) of voriconazole for inhibiting vonoprazan metabolism, 0.01, 0.1, 1, 10, 25, 50, and 100 µM of voriconazole and 10 µM of vonoprazan (close to its Km value) were selected. To determine the mechanisms underlying the inhibitory effect of voriconazole on vonoprazan based on the IC50 value, 0, 1.5, 3, 6, and 12 μM of voriconazole, and 5, 10, 20, and 40 μM of vonoprazan, based on the Km value in the RLM system, were selected.
The incubation was performed at 37°C for 50 min, followed by cooling to −80°C to terminate all of the reactions at the same time, after which 200 µL of acetonitrile and 20 µL of IS (500 ng/mL) were added to the mixture. After vortex mixing for 30 s and centrifugation at 13,000 rpm for 5 min, the supernatant was obtained for LC-MS/MS analysis.
Statistical Analysis
The pharmacokinetic parameters, including the maximal plasma concentration (Cmax), the maximum plasma time (Tmax), the apparent volume of distribution (Vz/F), the area under the plasma concentration–time curve (AUC), the elimination half-life (t1/2), the plasma clearance (CL) and the mean residence time (MRT), were analyzed using DAS (Drug and Statistics) software (Version 3.2.8, The People’s Hospital of Lishui, China). The IC50 and Lineweaver–Burk plot were obtained using GraphPad Prism (Version 8; Graphpad Software Inc., San Diego, CA, USA).
All pharmacokinetic parameters are expressed as the means ± standard deviation (SD). Statistical analyses of the main pharmacokinetic parameters were performed using the one-way ANOVA using SPSS (version 16.0; SPSS Inc., Chicago, IL, USA). Values of P < 0.05 were considered to be statistically significant.
Results
Method Validation
The concentrations of the calibration curves ranged from 0.1 to 100 ng/mL for vonoprazan, with both correlation coefficients higher than 0.9977. The LLOQ was set at 0.1 ng/mL for vonoprazan with acceptable accuracy and precision.
Effects of Voriconazole on the in vivo Pharmacokinetics of Vonoprazan
The mean plasma concentration–time curves of vonoprazan in the control, single-dose, and multiple-dose groups are shown in Figure 1. The corresponding pharmacokinetic parameters are summarized in Table 1. Pretreatment with a single dose and multiple doses of voriconazole led to a significant increase in the values of AUC(0-t), AUC(0-∞), MRT(0-t), and MRT(0-∞) of vonoprazan, whereas the value of CLz/F had significantly decreased compared to that of the control group. However, there was no significant change in the Cmax and Tmax values of vonoprazan after pretreatment with voriconazole (P > 0.05). These results demonstrate that pretreatment with a single dose or multiple doses of voriconazole inhibited vonoprazan metabolism in rats.
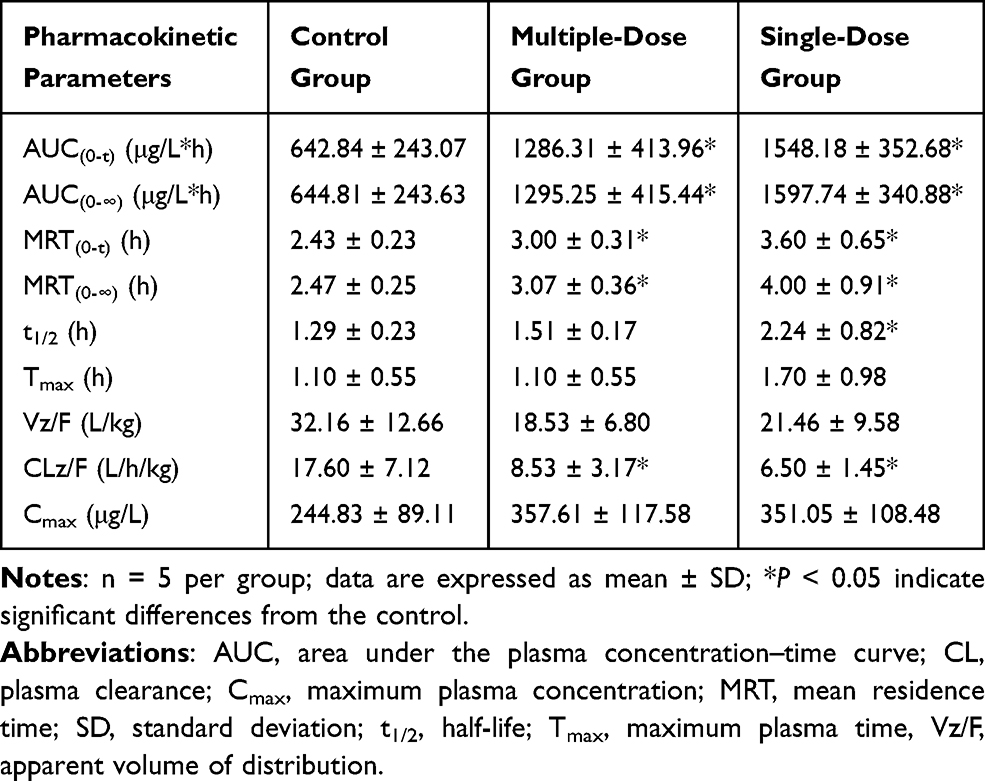 |
Table 1 Main Pharmacokinetic Parameters of Vonoprazan in Rats |
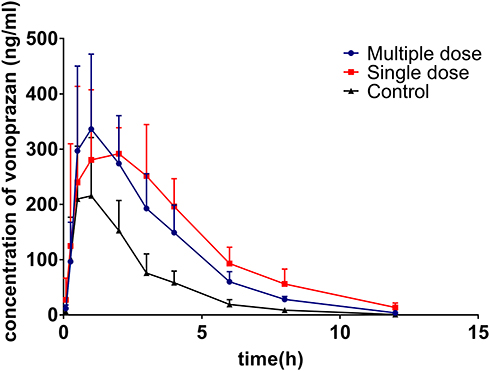 |
Figure 1 Mean plasma concentration–time curves of vonoprazan in control, single-dose, and multiple-dose groups. |
Effects of Voriconazole on the in vitro Pharmacokinetics of Vonoprazan
The inhibitory effect of voriconazole on vonoprazan in RLM was examined in terms of the peak area ratio of the metabolites of vonoprazan. The Michaelis–Menten kinetics and the IC50 value of vonoprazan in RLM are shown in Figure 2. The mechanism of inhibition by voriconazole in RLM is illustrated by the Lineweaver–Burk plot shown in Figure 3. The IC50, Ki, and αKi values of vonoprazan in RLM are 2.93 μM, 2.69 μM, and 5.32 μM, respectively. These results indicate that there was a significant in vitro inhibitory effect of voriconazole on vonoprazan.
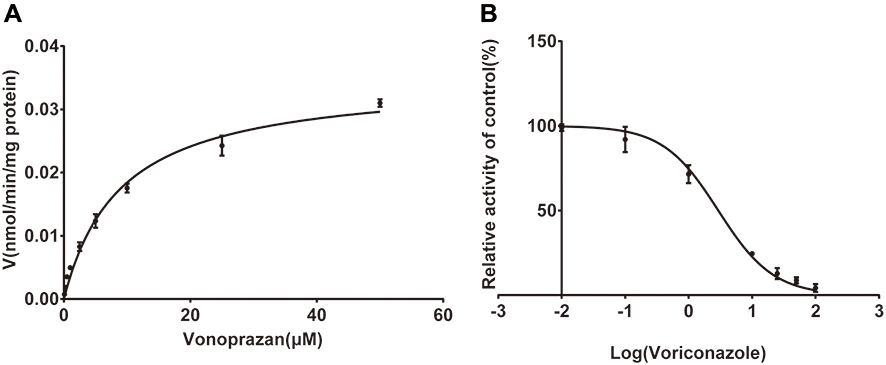 |
Figure 2 Michaelis–Menten kinetics (A) and the IC50 value (B) of vonoprazan in rat liver microsomes. |
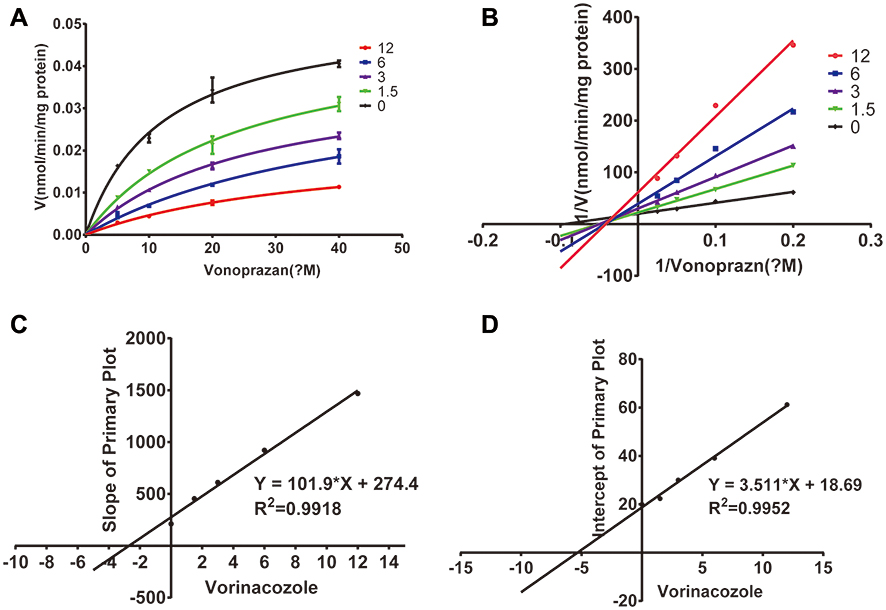 |
Figure 3 Michaelis-Menten model (A), Lineweaver–Burk plot (B) and the secondary plot for Ki (C) and αKi (D) in the inhibition of vonoprazan metabolism by various concentrations of voriconazole in rat liver microsomes. |
Discussion
The objective of this study was to demonstrate changes in the pharmacokinetics of vonoprazan after the administration of voriconazole in rats. After oral administration, vonoprazan was absorbed quickly and reached the highest plasma concentration at approximately 1.1 h in rats. In addition, vonoprazan was quickly eliminated from plasma with an elimination t1/2 of 1.29 ± 0.23 h in this study. Drug–drug interactions are generally determined according to pharmacokinetic properties, and their effects can be observed during absorption, distribution, metabolism, or excretion.32 Such interactions are increasingly recognized as important clinical events as they can produce irrelevant, synergistic, additive, or antagonistic results.33,34 However, as drug combinations are commonly used when treating patients in clinical practice, it is important to identify any potential drug–drug interactions so that drug dosages can be adjusted.
When rats were pretreated with a single dose or multiple doses of voriconazole at 30 mg/kg each dose, the AUC (0-∞) value of vonoprazan ranged from 644.81 ± 243.63 to 1548.18 ± 352.68 μg/(L•h) (single-dose group) and 644.81 ± 243.63 to 1286.31 ± 413.96 μg/(L•h) (multiple-dose group), which implied that the extent of absorption of vonoprazan may increase. Compared with the control group, the CLz/F value of vonoprazan had decreased by 2.7- and 2.1-fold in the single-dose and multiple-dose groups, respectively. These data indicate that the pharmacokinetic parameters of vonoprazan were significantly affected by voriconazole in rats. A Phase I clinical study on the effects of multiple oral doses of clarithromycin (a potent CYP3A4 inhibitor) on the pharmacokinetics of a single oral dose of vonoprazan revealed that the mean ratio of the geometric means of AUC(0-∞) and Cmax of vonoprazan increased by approximately 1.58- and 1.35-fold, respectively.35 These results indicate that the metabolism of vonoprazan may be altered when it is co-administered with potent CYP3A4 inducers. In addition, although the changes in the pharmacokinetic parameters of vonoprazan were greater in the single-dose group than in the multiple-dose group, the differences between the two groups were not significant, which shows that the inhibition was not dependent on the frequency or the duration of administration of voriconazole. We performed in vitro experiments to further evaluate the effect of voriconazole on the pharmacokinetics of vonoprazan metabolites. The results show that voriconazole inhibited vonoprazan metabolism at IC50 < 10 μM in vitro, and the in vivo results also inferred this inhibition effect of voriconazole. Furthermore, the Lineweaver–Burk plot suggested that the inhibition was of a mixed type, which included competitive and non-competitive inhibition (Ki ≠ αKi).36
The metabolism of vonoprazan is complex: it is known that CYP3A4, CYP2B6, CYP2C19, and CYP2D6 participate in the metabolism of vonoprazan. Vonoprazan has been shown to inhibit the anti-platelet function of clopidogrel and prasugrel more potently than esomeprazole, and it inhibits not only CYP3A4, but also CYP2C19.37 However, the findings of subsequent studies suggest that the pharmacodynamic drug interaction of vonoprazan and clopidogrel or prasugrel is not likely to be caused by the inhibition of CYP2B6, CYP2C19, or CYP3A4 by vonoprazan.38,39 Another study had previously demonstrated that pre-administration of vonoprazan increased the plasma levels and altered the pharmacokinetic profiles of gefitinib, erlotinib, and osimertinib.40 Vonoprazan has been found to be more effective than lansoprazole in the treatment of duodenal and gastric ulcers, erosive esophagitis, and Helicobacter pylori infections.41–43 Therefore, in view of the efficacy of vonoprazan, the co-administration of vonoprazan and voriconazole in inpatients with invasive fungal infections, and the inhibition of enzymes metabolizing vonoprazan by voriconazole, it is important to understand the effects of voriconazole on the pharmacokinetics of vonoprazan.
Conclusion
In summary, the data from this study clearly illustrate that voriconazole alters the pharmacokinetic parameters of vonoprazan. When orally administered, voriconazole could increase the AUC, prolong the t1/2, and decrease the CL of vonoprazan; there was no significant difference between the single-dose and multiple-dose groups that received voriconazole. Our results also indicate that the inhibition of vonoprazan metabolism by voriconazole may be of mixed type. Therefore, when administering voriconazole before vonoprazan, clinicians should pay attention to these interactions and adjust the dose of vonoprazan to avoid toxicity.
Data Sharing Statement
The data used to support the findings of this study are included within the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mermelstein J, Mermelstein AC, Chait MM. Proton pump inhibitors for the treatment of patients with erosive esophagitis and gastroesophageal reflux disease: current evidence and safety of dexlansoprazole. Clin Exp Gastroenterol. 2016;9:163–172. doi:10.2147/CEG.S91602
2. Savarino V, Marabotto E, Zentilin P, et al. The appropriate use of proton-pump inhibitors. Minerva Med. 2018;109(5):386–399. doi:10.23736/S0026-4806.18.05705-1
3. Savarino V, Marabotto E, Zentilin P, et al. Proton pump inhibitors: use and misuse in the clinical setting. Expert Rev Clin Pharmacol. 2018;11(11):1123–1134. doi:10.1080/17512433.2018.1531703
4. El Rouby N, Lima JJ, Johnson JA. Proton pump inhibitors: from CYP2C19 pharmacogenetics to precision medicine. Expert Opin Drug Metab Toxicol. 2018;14(4):447–460. doi:10.1080/17425255.2018.1461835
5. Kang H, Kim BJ, Choi G, Kim JG. Vonoprazan versus proton pump inhibitors for the management of gastroesophageal reflux disease: a protocol for a systematic review with meta-analysis. Medicine (Baltimore). 2018;97(39):e12574. doi:10.1097/MD.0000000000012574
6. Garnock-Jones KP. Vonoprazan: first global approval. Drugs. 2015;75(4):439–443. doi:10.1007/s40265-015-0368-z
7. Shin JM, Inatomi N, Munson K, et al. Characterization of a novel potassium-competitive acid blocker of the gastric H,K-ATPase, 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamin e monofumarate (TAK-438). J Pharmacol Exp Ther. 2011;339(2):412–420. doi:10.1124/jpet.111.185314
8. Echizen H. The first-in-class potassium-competitive acid blocker, vonoprazan fumarate: pharmacokinetic and pharmacodynamic considerations. Clin Pharmacokinet. 2016;55(4):409–418. doi:10.1007/s40262-015-0326-7
9. Ashida K, Sakurai Y, Hori T, et al. Randomised clinical trial: vonoprazan, a novel potassium-competitive acid blocker, vs. lansoprazole for the healing of erosive oesophagitis. Aliment Pharmacol Ther. 2016;43(2):240–251. doi:10.1111/apt.13461
10. Ashida K, Sakurai Y, Nishimura A, et al. Randomised clinical trial: a dose-ranging study of vonoprazan, a novel potassium-competitive acid blocker, vs. lansoprazole for the treatment of erosive oesophagitis. Aliment Pharmacol Ther. 2015;42(6):685–695. doi:10.1111/apt.13331
11. Yamasaki H, Kawaguchi N, Nonaka M, et al. In vitro metabolism of TAK-438, vonoprazan fumarate, a novel potassium-competitive acid blocker. Xenobiotica. 2017;47(12):1027–1034. doi:10.1080/00498254.2016.1203505
12. Yoneyama T, Teshima K, Jinno F, Kondo T, Asahi S. A validated simultaneous quantification method for vonoprazan (TAK-438F) and its 4 metabolites in human plasma by the liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1015(2016):42–49. doi:10.1016/j.jchromb.2016.01.051
13. Kogame A, Takeuchi T, Nonaka M, et al. Disposition and metabolism of TAK-438 (vonoprazan fumarate), a novel potassium-competitive acid blocker, in rats and dogs. Xenobiotica. 2017;47(3):255–266. doi:10.1080/00498254.2016.1182667
14. Mangal N, Hamadeh IS, Arwood MJ, et al. Optimization of voriconazole therapy for the treatment of invasive fungal infections in adults. Clin Pharmacol Ther. 2018;104(5):957–965. doi:10.1002/cpt.1012
15. Lin XB, Li ZW, Yan M, et al. Population pharmacokinetics of voriconazole and CYP2C19 polymorphisms for optimizing dosing regimens in renal transplant recipients. Br J Clin Pharmacol. 2018;84(7):1587–1597. doi:10.1111/bcp.13595
16. Driscoll TA, Frangoul H, Nemecek ER, et al. Comparison of pharmacokinetics and safety of voriconazole intravenous-to-oral switch in immunocompromised adolescents and healthy adults. Antimicrob Agents Chemother. 2011;55(12):5780–5789. doi:10.1128/AAC.05010-11
17. Grün B, Krautter S, Riedel KD, Mikus G. Inhibition of the active principle of the weak opioid tilidine by the triazole antifungal voriconazole. Br J Clin Pharmacol. 2009;68(5):712–720. doi:10.1111/j.1365-2125.2009.03498.x
18. Scholz I, Oberwittler H, Riedel KD, et al. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br J Clin Pharmacol. 2009;68(6):906–915. doi:10.1111/j.1365-2125.2009.03534.x
19. Ohbuchi M, Yoshinari K, Kaneko H, et al. Coordinated roles of pregnane X receptor and constitutive androstane receptor in autoinduction of voriconazole metabolism in mice. Antimicrob Agents Chemother. 2013;57(3):1332–1338. doi:10.1128/AAC.01900-12
20. Fihlman M, Hemmila T, Hagelberg NM, et al. Voriconazole greatly increases the exposure to oral buprenorphine. Eur J Clin Pharmacol. 2018;74(12):1615–1622. doi:10.1007/s00228-018-2548-8
21. Yasu T, Konuma T, Kato S, Kurokawa Y, Takahashi S, Tojo A. Different effects of lansoprazole and rabeprazole on the plasma voriconazole trough levels in allogeneic hematopoietic cell transplant recipients. Ann Hematol. 2016;95(11):1845–1851. doi:10.1007/s00277-016-2782-z
22. Qi F, Zhu L, Li N, Ge T, Xu G, Liao S. Influence of different proton pump inhibitors on the pharmacokinetics of voriconazole. Int J Antimicrob Agents. 2017;49(4):403–409. doi:10.1016/j.ijantimicag.2016.11.025
23. Yan M, Wu ZF, Tang D, et al. The impact of proton pump inhibitors on the pharmacokinetics of voriconazole in vitro and in vivo. Biomed Pharmacother. 2018;108:60–64. doi:10.1016/j.biopha.2018.08.121
24. Niece KL, Boyd NK, Akers KS. In vitro study of the variable effects of proton pump inhibitors on voriconazole. Antimicrob Agents Chemother. 2015;59(9):5548–5554. doi:10.1128/AAC.00884-15
25. Wood N, Tan K, Purkins L, et al. Effect of omeprazole on the steady-state pharmacokinetics of voriconazole. Br J Clin Pharmacol. 2003;56:56–61. doi:10.1046/j.1365-2125.2003.02000.x
26. Dai DP, Wang SH, Geng PW, Hu GX, Cai JP. In vitro assessment of 36 CYP2C9 allelic isoforms found in the Chinese population on the metabolism of glimepiride. Basic Clin Pharmacol Toxicol. 2014;114(4):305–310. doi:10.1111/bcpt.12159
27. Dai DP, Wang SH, Li CB, et al. Identification and functional assessment of a new CYP2C9 allelic variant CYP2C9*59. Drug Metab Dispos. 2015;43(8):1246–1249. doi:10.1124/dmd.115.063412
28. Dai DP, Wang YH, Wang SH, et al. In vitro functional characterization of 37 CYP2C9 allelic isoforms found in Chinese Han population. Acta Pharmacol Sin. 2013;34(11):1449–1456. doi:10.1038/aps.2013.123
29. Al-Ghobashy MA, Kamal SM, El-Sayed GM, et al. Determination of voriconazole and co-administered drugs in plasma of pediatric cancer patients using UPLC-MS/MS: A key step towards personalized therapeutics. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1092:489–498. doi:10.1016/j.jchromb.2018.06.043
30. Wang SH, Dong YW, Su K, et al. Effect of codeine on CYP450 isoform activity of rats. Pharm Biol. 2017;55(1):1223–1227. doi:10.1080/13880209.2017.1297466
31. Wang SH, Wang ZY, Chen DX, et al. Effect of acute paraquat poisoning on CYP450 isoforms activity in rats by cocktail method. Int J Clin Exp Med. 2015;8(10):19100–19106.
32. Wang S, Zhang Z, Yu Z, Han C, Wang X. Pharmacokinetic study of delavinone in mice after intravenous and oral administration by UPLC-MS/MS. Biomed Res Int. 2019;2019:3163218.
33. Lin G, Wang C, Qiu X, et al. Differential effects of ketoconazole, itraconazole and voriconazole on the pharmacokinetics of imatinib and its main metabolite GCP74588 in rat. Drug Dev Ind Pharm. 2014;40(12):1616–1622. doi:10.3109/03639045.2013.838582
34. Egger SS, Meier S, Leu C, et al. Drug interactions and adverse events associated with antimycotic drugs used for invasive aspergillosis in hematopoietic SCT. Bone Marrow Transplant. 2010;45(7):1197–1203. doi:10.1038/bmt.2009.325
35. Jenkins H, Jenkins R, Patat A. Effect of multiple oral doses of the potent CYP3A4 inhibitor clarithromycin on the pharmacokinetics of a single oral dose of vonoprazan: a Phase I, open-label, sequential design study. Clin Drug Investig. 2017;37(3):311–316. doi:10.1007/s40261-016-0488-6
36. Sun M, Tang Y, Ding T, Liu M, Wang X. Inhibitory effects of celastrol on rat liver cytochrome P450 1A2, 2C11, 2D6, 2E1 and 3A2 activity. Fitoterapia. 2014;92:1–8. doi:10.1016/j.fitote.2013.10.004
37. Kagami T, Yamade M, Suzuki T, et al. Comparative study of effects of vonoprazan and esomeprazole on antiplatelet function of clopidogrel or prasugrel in relation to CYP2C19 genotype. Clin Pharmacol Ther. 2018;103(5):906–913. doi:10.1002/cpt.863
38. Nishihara M. Inhibitory effect of vonoprazan on the metabolism of [(14)C]prasugrel in human liver microsomes. Eur J Drug Metab Pharmacokinet. 2019;44(5):713–717. doi:10.1007/s13318-019-00554-y
39. Nishihara M, Yamasaki H, Czerniak R, Jenkins H. In vitro assessment of potential for CYP-inhibition-based drug-drug interaction between vonoprazan and clopidogrel. Eur J Drug Metab Pharmacokinet. 2019;44(2):217–227. doi:10.1007/s13318-018-0521-7
40. Yasumuro O, Uchida S, Kashiwagura Y, et al. Changes in gefitinib, erlotinib and osimertinib pharmacokinetics under various gastric pH levels following oral administration of omeprazole and vonoprazan in rats. Xenobiotica. 2018;48(11):1106–1112. doi:10.1080/00498254.2017.1396379
41. Hirai A, Takeuchi T, Takahashi Y, et al. Comparison of the effects of vonoprazan and lansoprazole for treating endoscopic submucosal dissection-induced artificial ulcers. Dig Dis Sci. 2018;63(4):974–981. doi:10.1007/s10620-018-4948-0
42. Miwa H, Uedo N, Watari J, et al. Randomised clinical trial: efficacy and safety of vonoprazan vs. lansoprazole in patients with gastric or duodenal ulcers–results from two Phase 3, non-inferiority randomised controlled trials. Aliment Pharmacol Ther. 2017;45(2):240–252. doi:10.1111/apt.13876
43. Murakami K, Sakurai Y, Shiino M, Funao N, Nishimura A, Asaka M. Vonoprazan, a novel potassium-competitive acid blocker, as a component of first-line and second-line triple therapy for Helicobacter pylori eradication: a Phase III, randomised, double-blind study. Gut. 2016;65(9):1439–1446. doi:10.1136/gutjnl-2015-311304
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.