")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Effects of the POMC System on Glucose Homeostasis and Potential Therapeutic Targets for Obesity and Diabetes
Authors Yang D , Hou X , Yang G, Li M , Zhang J, Han M , Zhang Y, Liu Y
Received 30 June 2022
Accepted for publication 13 September 2022
Published 25 September 2022 Volume 2022:15 Pages 2939—2950
DOI https://doi.org/10.2147/DMSO.S380577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Dan Yang,1,2 Xintong Hou,1,2 Guimei Yang,1,2 Mengnan Li,1,2 Jian Zhang,1,2 Minmin Han,1,2 Yi Zhang,3 Yunfeng Liu1
1Department of Endocrinology, First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2First Clinical Medical College, Shanxi Medical University, Taiyuan, People’s Republic of China; 3Department of Pharmacology, Shanxi Medical University, Taiyuan, People’s Republic of China
Correspondence: Yi Zhang, Department of Pharmacology, Shanxi Medical University, Taiyuan, People’s Republic of China, Email [email protected] Yunfeng Liu, Department of Endocrinology, First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China, Tel +86 18703416196, Email [email protected]
Abstract: The hypothalamus is indispensable in energy regulation and glucose homeostasis. Previous studies have shown that pro-opiomelanocortin neurons receive both central neuronal signals, such as α-melanocyte-stimulating hormone, β-endorphin, and adrenocorticotropic hormone, as well as sense peripheral signals such as leptin, insulin, adiponectin, glucagon-like peptide-1, and glucagon-like peptide-2, affecting glucose metabolism through their corresponding receptors and related signaling pathways. Abnormalities in these processes can lead to obesity, type 2 diabetes, and other metabolic diseases. However, the mechanisms by which these signal molecules fulfill their role remain unclear. Consequently, in this review, we explored the mechanisms of these hormones and signals on obesity and diabetes to suggest potential therapeutic targets for obesity-related metabolic diseases. Multi-drug combination therapy for obesity and diabetes is becoming a trend and requires further research to help patients to better control their blood glucose and improve their prognosis.
Keywords: POMC neurons, central signals, peripheral signals, glucose homeostasis, obesity, diabetes
Introduction
Type 2 diabetes1 is a pressing global health concern. Notably, the incidence is higher among patients with obesity, and the number of affected individuals is predicted to reach 700 million in 2045.2 Currently, islet β cell dysfunction and insulin resistance are considered to be the fundamental pathologic mechanisms of type 2 diabetes.3 In addition to the disease, poor lifestyle and compliance render it difficult for individuals to maintain normal glucose levels. Therefore, the risk of microcirculation and macrovascular complications is extremely high, substantially increasing the incidence of cardiovascular and cerebrovascular disorders.4 Most research has focused on the peripheral mechanisms of pancreatic β-cell dysfunction; however, the central nervous system (CNS) also plays an indispensable role in regulating insulin sensitivity and glucose balance.5
The hypothalamus is involved in energy metabolism by responding to signals such as hormones, neurotransmitters and metabolites.6 This effect is more prominent in pro-opiomelanocortin (POMC) neurons.7 Specifically, POMC is the common precursor of many neuropeptides, including α-melanocyte-stimulating hormone (α-MSH), beta-endorphin (β-endorphin), and adrenocorticotropic hormone. It can respond to energy metabolism by sensing these central signals.8 Moreover, POMC neurons can also receive peripheral signals (Figure 1), such as leptin, insulin, adiponectin, glucagon-like peptide-1 (GLP-1), and glucagon-like peptide-2 (GLP-2). Also, POMC affects glucose metabolism through its corresponding receptors and related signaling pathways.6 In addition, POMC can sense glucose levels and regulate the excitability of POMC neurons in response to glucose.9 Abnormalities in any of these processes may lead to metabolic diseases such as obesity and type 2 diabetes.10,11
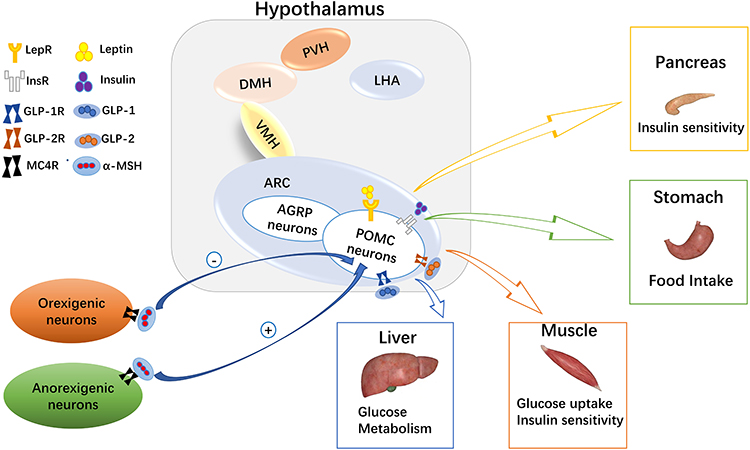 |
Figure 1 Melanocortin system in glucose homeostasis. In the arcuate nucleus of the hypothalamus, there are many hormone receptors on pro-opiomelanocortin (POMC) neurons. Leptin, insulin, glucagon-like peptide (GLP)-1, and GLP-2 act on liver, skeletal muscle, and the pancreas to regulate blood glucose levels by binding with their corresponding receptors. At the same time, α-melanocyte stimulating hormone (α-MSH) on orexigenic or anorexigenic neurons binds with the melanocortin 4 receptor (MC4R) to regulate appetite and glucose homeostasis. |
Currently, the treatment of diabetes remains a challenge, and multi-drug combinations for obesity and diabetes are increasingly becoming a trend. For example, metformin in combination with GLP-1 receptor agonists can lead to weight loss in patients with type 2 diabetes and obesity while controlling blood glucose. In addition to traditional peripheral hypoglycemic drugs, some drugs have been observed to maintain blood glucose stability and improve diabetes symptoms through central regulation. However, the mechanism of POMC regulation on glucose homeostasis is still unclear. Based on the influence of the POMC system on appetite, body weight, and energy expenditure, studying the mechanism could help us identify novel therapeutic strategies for managing obesity and type 2 diabetes. Therefore, in the study, we primarily explored the effects of these hormones and signaling molecules on the onset and progression of obesity and diabetes to identify novel therapeutic targets for obesity-related metabolic diseases.
POMC Neurons Sense Peripheral Signals
Leptin and Its Receptor
Leptin is a protein encoded by obesity genes and secreted by white fat cells, which regulates energy balance throughout the body and exerts potent anti-obesity effects. Notably, it can affect blood pressure, sympathetic excitability, and blood sugar levels.12,13 Previous animal and clinical studies have demonstrated that leptin also has an anti-diabetic effect, revealing that this effect is primarily mediated by stimulating leptin receptors in the CNS, subsequently activating POMC neurons and melanocortin 4 receptor (MC4R).5 Leptin plays a pleiotropic role by specifically binding to leptin receptors and subsequently activating the related signal pathways, ultimately reducing food intake and hepatic glucose production.11,14 Additionally, both leptin deficiency and leptin resistance can exacerbate the onset of type 1 and type 2 diabetes.15
The Leptin-POMC system is critical for glucose metabolism. Leptin can increase the expression of the POMC genes and regulate the excitation of the POMC neurons.16 Furthermore, leptin receptors are abundant in the POMC neurons. They can sense changes in peripheral energy metabolism at an early stage.17 Mobbs et al reported that POMC gene overexpression could effectively improve hyperglycemia and insulin resistance in leptin-deficient mice so that the blood glucose level tended to be normal and the symptoms of diabetes were improved.16,18 Recent studies have suggested that Zucker (FA/FA) rats with leptin receptor deficiency were obese, hyperinsulinemic, and leptin-resistant.19 In addition, the expression of the POMC gene was reduced.20 Leptin receptor deficiency could partly result in the re-expression of these receptors in the POMC neurons and reverse hyperinsulinemia and hyperglycemia in mice.21 Furthermore, leptin resistance occurs when they cannot fully sense the leptin levels or when the leptin signal transduction pathway is damaged. Thus, the body is insensitive to leptin, which manifests as increased appetite, obesity, type 2 diabetes, and other metabolic-related diseases.22 Notably, leptin also shows sex differences in regulating glucose homeostasis. In the POMC neurons, leptin has a considerable effect on energy balance and fat distribution in women and on glucose homeostasis in men. Thus, leptin receptor deficiency is associated with reduced energy expenditure in women, whereas men present glucose intolerance and insulin resistance.14,23
The central leptin-POMC system has potent anti-obesity and anti-diabetic effects, and the POMC neurons can improve blood glucose levels by affecting the leptin-related signaling pathways (Figure 2). The first is the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signal pathway,24,25 leptin binds to a leptin receptor to activate JAK2 and subsequently phosphorylate STAT3. Consequently, STAT3 binds to the POMC gene promoters to upregulate POMC expression. The src homology region 2-containing protein tyrosine phosphatase 2-mitogen-activated protein kinase (SHP2-MAPK) pathway26 may be crucial in mediating the chronic effects of leptin on glucose regulation. Specifically, SHP2 binds to phosphorylated Tyr985 in the leptin receptor through its Src homology region 2 domain and stimulates the activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2). In the POMC neurons, the PI3K signaling pathway27,28 is particularly vital for regulating glucose metabolism. Forkhead box protein O1 (FoxO1) is a negative regulator of leptin signaling, and leptin inhibits FoxO1 activity through the PI3K pathway in the hypothalamus and promotes STAT3 binding to the POMC promoter, thereby upregulating POMC expression, reducing food intake, and increasing insulin sensitivity.
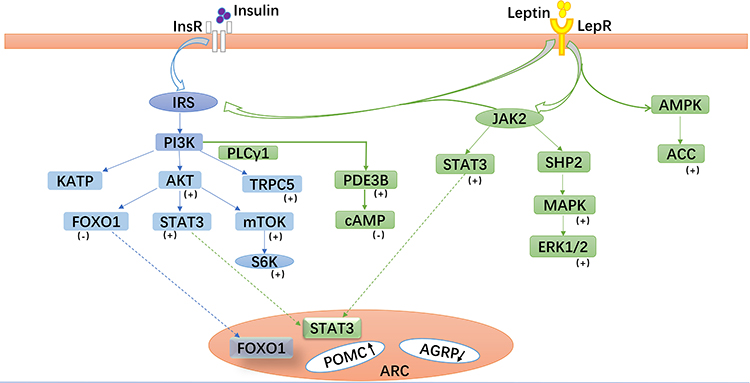 |
Figure 2 Leptin- and insulin-related signaling pathways in pro-opiomelanocortin (POMC) neurons. Leptin and insulin bind to their corresponding receptors and activate related signaling pathways, especially the phosphoinositide 3-kinase (PI3K) signaling pathway, which play important roles in glucose metabolism mediated by POMC neurons in the arcuate nucleus. Abbreviations: (+), Positive regulatory factors; (-), Negative regulatory factors; IRS, insulin receptor substrate; JAK2, Janus tyrosine kinase 2; STAT3, signal transducer and activator of transcription 3; FoxO1, forkhead box protein O1; TRPC5, transient receptor potential channels-5; mTOR, mammalian target of rapamycin; S6K, S6 kinase; PLCγ1, phospholipase C-γ1; PDE3B, phosphodiesterase 3B; SHP2, Src homologous region protein tyrosine phosphatase 2; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1/2; AMPK, AMP-dependent kinase; ACC, acetyl-CoA carboxylase. |
Moreover, leptin also regulates the mechanistic target of rapamycin/S6 kinase (mTOR/S6K) pathway,29,30 which may be a downstream target of the PI3K pathway. Chronic activation of the mTOR/S6K pathway leads to leptin resistance and obesity. Abnormal glucose metabolism may occur when the body cannot sense these signaling pathways or any of these signals is damaged. Therefore, these signals may become new targets for diabetes treatment. Recent studies have found that tea saponin, a substance extracted from tea leaves, can reduce the expression of POMC mRNA in the hypothalamus of obese mice. Additionally, tea saponin can reduce hypothalamic inflammation and improve central leptin sensitivity and signaling. Furthermore, treatment with tea saponin can increase the adiponectin level and improve insulin sensitivity in obese mice.31 These results suggest that tea saponin could have a role in anti-obesity and anti-diabetes based on the central POMC system. Therefore, further studies should be conducted to investigate the mechanisms underlying the leptin-POMC system to identify therapeutic strategies to treat obesity and diabetes.
Insulin and Its Receptor
Insulin is the only hormone in the body that can reduce blood glucose. It can regulate glucose balance through peripheral and central pathways and has been widely used in clinical practice. Over the years, the central regulatory mechanism of insulin has been gradually revealed. In 1855, Claude Bernard first observed that the brain was involved in glucose homeostasis in animals.32 In 1973, Roth et al first identified insulin receptors (IR) in the brain and suggested that injecting insulin into the hypothalamus could reduce blood glucose.33 Furthermore, peripheral insulin is transported to the brain via the median eminence and cerebrospinal fluid, which is essential for regulating glucose homeostasis across the blood-brain barrier.34,35 Neurogliaform cells in the cortex may be one of the sources of insulin in the brain.36 When the blood-brain barrier is damaged and insulin in the brain is reduced, intranasal insulin administration can increase insulin concentration in the brain and affect peripheral insulin sensitivity.37 In addition, insulin receptors in the brain are mainly expressed in the POMC neurons. Deficiency in insulin receptors results in their re-expression in the POMC neurons, resulting in increased hepatic glucose production in mice.38 Also, abnormalities in insulin and the insulin receptors in the brain may lead to glucose metabolic diseases.
As shown in Figure 2, the PI3K signaling pathway39 is particularly important in glucose metabolism. When the hypothalamus senses glucose level changes, peripheral insulin crosses the blood-brain barrier and binds to its receptors on the POMC neurons. This leads to the phosphorylation of insulin receptor substrate (IRS) protein and the activation of PI3K, consequently modulating the excitability of the POMC neurons.40,41 After activating the POMC neurons, insulin promotes the expression and secretion of α-MSH, which specifically binds to the melanocortin-3 receptor (MC3R) or MC4R. Thus, it can reduce food intake, improve the basal metabolic rate, and lower blood glucose.
Insulin resistance can lead to reduced insulin sensitivity and glucose utilization in the body. Moreover, compensatory secretion of insulin leads to hyperinsulinemia, which can lead to type 2 diabetes and obesity. In recent years, studies have found that some central factors are associated with insulin resistance, such as obesity,42 hypothalamic inflammation,43 endoplasmic reticulum stress,44 and the cilia and autophagy mechanism,45 which can prevent insulin from transmitting peripheral metabolic information to the center, resulting in impaired glucose metabolism. A long-term high-fat diet (HFD) or hypothalamic inflammation can activate some inflammatory factors such as toll-like receptor-4 (TLR4), tumor necrosis factor (TNF), myeloid differentiation factor 88 (MyD88), and nuclear factor-kappa B (NF-κB). These inflammatory factors can affect insulin and leptin activity, leading to glucose metabolism disorders.43 In addition, the loss of cilia and autophagy in the POMC neurons can disrupt insulin signaling, prevent insulin-dependent glucose uptake, and eventually lead to obesity and insulin resistance in mice.45
Recently, novel molecules and mechanisms related to insulin resistance have gradually been revealed. For instance, inositol-requiring enzyme 1α (IRE1α),46 spliced X-box binding protein 1 (XBP1s),47 and other positive regulatory factors can activate the POMC neurons, enhance peripheral insulin sensitivity, improve glucose tolerance, inhibit liver glucose production, and thus, prevent obesity and type 2 diabetes. In contrast, negative regulatory factors, such as transforming growth factor-β,42 ethanol,48 and resistin,49 can lead to type 2 diabetes. Resistin, a kind of adipokine, activates the TLR4-NF-κB pathway of the POMC neurons (Figure 3), up-regulates suppressor of cytokine signaling 3 and protein tyrosine phosphatase 1B expression, and inhibits the activation of the insulin receptor, AKT, and ERK1/2 in the POMC neurons. This inhibits IRS-1 phosphorylation, leading to insulin resistance and symptoms associated with peripheral type 2 diabetes.50 Consequently, the central regulatory mechanism of insulin is highly complex and must be further explored. We suggest that there might be several potential targets in the insulin receptors and related signal pathway mechanisms to maintain glucose homeostasis.
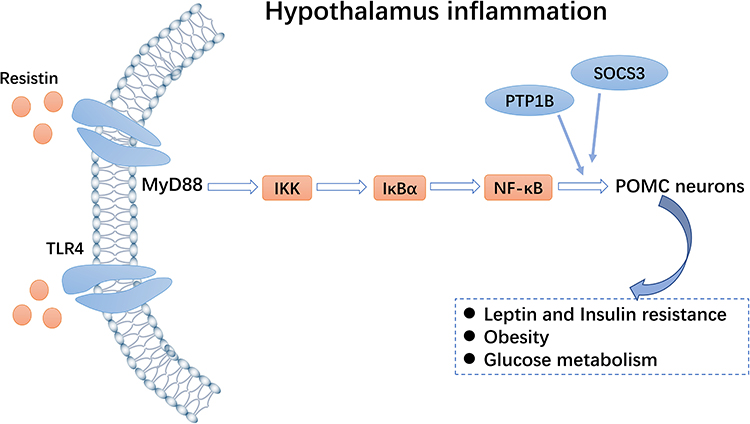 |
Figure 3 Toll-like receptor 4 (TLR4)-NF-κB pathway. Resistin can activate the TLR4-NF-κB pathway of pro-opiomelanocortin (POMC) neurons and inhibit expression of the insulin receptor, thereby regulating glucose homeostasis. |
Currently, based on the effects of leptin and insulin, some factors such as Smad7, hypoxia-inducible factor (HIF), liver kinase B1 (LKB1), sirtuin 6 (Sirt6), and steroid receptor coactivator-2 (SRC-2) can mediate hypothalamic inflammation through the POMC system, thus, affecting insulin resistance. A TGF-β inhibitor, Smad7,51 is widely expressed in tissues throughout the body. Yuan et al52 demonstrated that an HFD increased the expression of Smad7 and that Smad7 overexpression in the POMC neurons markedly reduced insulin sensitivity, leading to impaired glucose tolerance. This change may be due to the activation of the Smad7-AKT pathway, resulting in decreased insulin signaling in the hypothalamus. In contrast, glucose tolerance and insulin resistance improved when Smad7 was knocked out. The HIF is a transcriptional activator and is involved in the regulation of body weight and glucose homeostasis.53 HIF-1-alpha (HIF1α) is associated with hypothalamic inflammation, apoptosis, and autophagy. Thus, HIF1α deletion leads to decreased POMC neuron activity and impaired glucose uptake,54 whereas HIF-2-alpha deletion leads to weight gain in an age-dependent manner, accompanied by abnormal glucose and lipid metabolism.55 LKB1 is a metabolic regulator with potential anti-inflammatory activity.56 Hypothalamic LKB1 overexpression can reduce hypothalamic inflammation and increase insulin sensitivity. After HFD induction, LKB1 deficient mice showed increased appetite and obesity, increased leptin resistance, decreased POMC neuron expression, and worsening hypothalamic inflammation.57 Additionally, Sirt6, a member of the Sirtuin family, has a vital role in maintaining glucose metabolism.58 The overexpression of Sirt6 can reverse HFD-induced obesity in mice. Conversely, the loss of Sirt6 can affect leptin activity in the POMC neurons, which impairs glucose homeostasis.59 Furthermore, SRC-260 is a molecule that regulates nuclear receptors and transcription factors. Also, SRC-2 can reduce energy consumption during fasting in the POMC neurons. Moreover, SRC-2 can co-activate FoxO1 to inhibit POMC gene expression and affect glucose balance, which is a defensive mechanism to prevent severe hypoglycemia. Overall, these factors, as hypothalamic inflammatory regulators, could become a therapeutic target for HFD-induced obesity and metabolic diseases.
Adiponectin and Adiponectin Receptor 1
Adiponectin is an adipocyte-derived hormone with anti-inflammatory and insulin-sensitizing effects.61 The peripheral mechanism of adiponectin has been thoroughly studied, however, the regulation of central energy homeostasis and glucose metabolism remains unclear. Peripheral adiponectin can be transported to the brain through the blood-brain barrier. Rapid central injection of adiponectin significantly reduces food intake and increases energy expenditure, whereas chronic infusion improves glucose homeostasis and does not appear to affect food intake.62–64 Subsequently, adiponectin receptors 1 and 2 are also expressed in the POMC and neuropeptide Y (NPY)-expressing neurons of the hypothalamus and peripheral organs.65 In the POMC neurons, adiponectin and leptin can exert synergistic regulatory effects on glucose homeostasis by activating the AMP-activated protein kinase (AMPK) pathway and PI3K pathway.66,67 Previous studies have shown that the action of adiponectin on the POMC neurons correlates with glucose levels. Elevated blood glucose levels result in an AMPK pathway-dependent inhibition of POMC neurons and increased food intake. Conversely, low blood glucose levels exert the opposite effect through phosphorylation of the PI3K pathway, thereby suppressing appetite.68 In addition, adiponectin, as an anti-inflammatory factor, can also play a hypoglycemic role by affecting the hypothalamic inflammatory response and reversing proinflammatory signals.69 Based on the insulin-sensitizing and anti-inflammatory properties of adiponectin, this hormone may be a crucial target for treating metabolic-related diseases.
Glucagon-Like Peptide-1 and -2 and Their Receptors
The gut-derived endocrine hormones, GLP-170 and GLP-2,71 control energy balance and glucose homeostasis in the brain and pancreas. The receptors, GLP-1 receptor (GLP-1R)72 and GLP-2 receptor (GLP-2R),73 are expressed in the endocrine cells, as well as widely expressed in the POMC and NPY neurons in the arcuate nucleus (ARC). Specifically, GLP-1 and GLP-2 play a vital role in the regulation of glucose homeostasis through the gut-brain axis. Many studies have been conducted on GLP-1, whereas GLP-2 is rarely studied.
Injecting GLP-1 directly into the ARC can increase glucose-stimulated insulin secretion, reduce glucose production in the liver, and inhibit glucose uptake in ATP-sensitive potassium (KATP) channel-dependent manner, thereby limiting postprandial glucose fluctuations.74,75 In contrast, a relatively high glycemic status was observed after the application of central GLP-1R antagonists, suggesting that the activation of central GLP-1R is essential for glucose homeostasis.76 Liraglutide, a GLP-1R agonist, can directly bind to GLP-1R on the cell membrane to activate the hypothalamic POMC neurons.77 Liraglutide has also been shown to improve the excitability of the POMC neurons, especially when combined with leptin,78 and better suppress appetite and reduce blood glucose.
In the POMC neurons, GLP-2 binds to the specific G-protein-coupled receptor GLP-2R to regulate appetite and glucose metabolism.71 Previous studies have shown that intraventricular injection of GLP-2 can reduce food intake and inhibit hepatic glucose production. A POMC-GLP2R KO mice model has been established to illustrate the regulation of GLP-2R on glucose in the POMC neurons. In the POMC neurons, GLP-2R deletion led to postprandial glucose intolerance in mice, whereas the fasting glucose levels remained unchanged.79,80 Meanwhile, the insulin level of the mice increased, and insulin resistance occurred.81 Moreover, GLP-2R deficiency increases glucagon secretion, exacerbating hyperglycemia and impaired glucose tolerance in leptin-deficient mice71 The PI3K-AKT-FOXO1 axis (Figure 4) is a critical signaling pathway that regulates energy metabolism in the brain.82 GLP-2 activates PI3K signals and directly regulates the excitability of the POMC neurons.83 This suggests that the activation of GLP-2R in the POMC neurons is required for GLP-2 to promote glucose homeostasis and increase insulin sensitivity.80,81
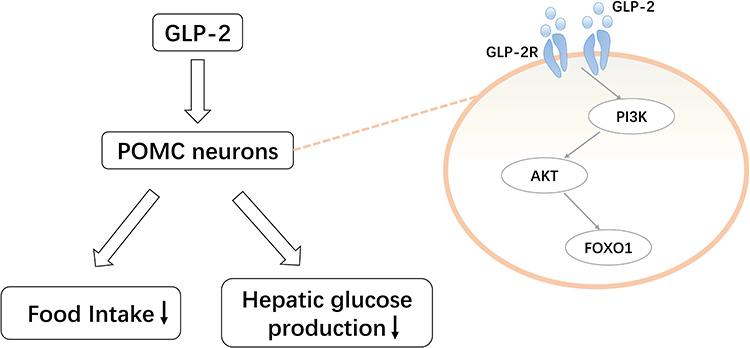 |
Figure 4 Pro-opiomelanocortin (POMC)-mediated regulation of glucagon-like peptide (GLP)-2 on glucose homeostasis. GLP-2 activates the phosphoinositide 3-kinase (PI3K)-AKT-forkhead box protein O1 (FOXO1) pathway by binding to the GLP-2 receptor expressed in POMC neurons. This reduces food intake and hepatic glucose production. |
Currently, GLP-1R agonists and dipeptidyl peptidase IV inhibitors have been widely used in the treatment of type 2 diabetes.84,85 Thus, based on the central role of GLP-1 and GLP-2, we should explore more possibilities and targets for diabetes.
POMC Neurons Integrate Central Signals
Melanocortin and Melanocortin 4 Receptor
Melanocortin is a POMC neuron-derived peptide that affects food intake, glucose homeostasis, and the release of certain inflammatory factors. MC3R and MC4R receptors are mainly involved in energy metabolism, and MC4R mainly affects food intake and energy balance.9 α-MSH can bind to MC4R to reduce insulin release and increase insulin sensitivity.86 Injection of the MC4R agonist into the lateral ventricle reduced plasma insulin levels and improved insulin sensitivity in leptin-deficient mice.87 Conversely, MC4R antagonists injection might result in high appetite and hyperinsulinemia.88 In addition, the absence or mutations in MC4R leads to increased food intake and body weight, prompting inflammatory cytokine secretion and eventually leading to insulin resistance and type 2 diabetes.89 Based on these studies, we suggest that α-MSH could contribute to the management of patients with obesity and type 2 diabetes.
Based on the POMC neurons and MC4R, many studies have been conducted on their anti-obesity effects.90 For obese patients with type 2 diabetes, α-MSH-based therapy may be promising to managing obesity and type 2 diabetes. The MC4R agonists have been gradually used as weight-loss drugs but have certain side effects on the cardiovascular system, leading to an increased heart rate and blood pressure.91–93 Therefore, α-MSH and its analogs and the MC4R agonists still need to be continuously explored to minimize the occurrence of adverse reactions.
Orexin-A and OX-A Receptor Type 1
Orexin-A is a hypothalamic neuropeptide comprising 33 amino acids, which can be involved in the regulation of energy homeostasis, sleep state, drug addiction, tumor treatment, and other aspects.94,95 Here, we mainly discuss its regulatory effects on appetite and glucose homeostasis. Like leptin and insulin, orexin-A can enter the brain through the blood-brain barrier, and OX-A receptor type 1(OX-1R) and cannabinoid receptor 1(CB1R) are widely expressed in the POMC neurons.96 Therefore, orexin-A can bind to OX-1R in the POMC neurons and lead to weight gain by stimulating the endocannabinoid signaling pathway.97,98 Morello et al demonstrated that OX-A expression was increased in the ARC of obese mice. OX-A-induced overeating in mice depended on the activation of CB1R and ERK1/2, which inhibited POMC and α-MSH production. The use of CB1R antagonist AM251 and the ERK1/2 inhibitors PD98059 and SB334867 exhibited the opposite result, reducing food intake and weight loss in obese mice.99,100
Currently, there are relatively few studies on the regulation of blood glucose by OX-A through the POMC system. Therefore, we recommend that more studies focus on its effect on blood glucose. Further research should be conducted on the potential synergies between the OX-1R and CB1R antagonists, and MC4R agonists to provide novel targets for the treatment of obesity and type 2 diabetes.
Melanin Concentrating Hormone and its receptors
Melanin concentrating hormone (MCH) is a 19-amino acid neuropeptide that can affect food intake, glucose metabolism, obesity and other processes.101 There are two corresponding receptors in the brain, namely melanin concentrating hormone receptor 1(MCHR1) and melanin concentrating hormone receptor 2, and these effects are mainly reflected in MCHR1.102,103 Obesity can occur after chronic intravenous MCH. Oral or intravenous administration of the MCH-R1 antagonist reduced appetite and body weight, mainly in HFD-induced obese mice, but not in mice fed a normal diet.101 As for glucose homeostasis, the POMC system interacts with sirtuin 1 (SIRT1) to regulate energy homeostasis and insulin sensitivity. Also, MCH regulates the SIRT1/FoxO1 pathway and reduces the activity of the POMC neurons, inducing binge eating, obesity, insulin resistance, and glucose intolerance.104 In addition, the MCH neurons can sense the glucose signal, mediated by the KATP channels, and are negatively regulated by uncoupling protein 2, a mitochondrial protein that reduces ATP production, which regulates peripheral glucose homeostasis.105 Therefore, the MCH system is also a crucial target for the treatment of obesity and diabetes and warrants further attention.
Opioid and Opioid Receptors
Opioids affect blood glucose in addition to their traditional analgesic effects.106 The endogenous opioids include β-endorphin, leu-enkephalin, met-enkephalin, and dynorphin.107 These substances can be produced in various systems throughout the body and act as neurotransmitters or hormones involved in the body’s energy regulation. Similarly, opioid receptors can also be expressed in tissues and cells throughout the body.108 The regulation of blood glucose by opioids is affected by glucose concentration.106,109 In the case of hyperglycemia, the injection of β-endorphin and µ-opioid receptor agonists can reduce hypoglycemia without affecting food intake.110,111 In contrast, the β-endorphin levels increase during hypoglycemia, consequently inducing an increase in the blood glucose levels.108
Dynorphin-A, an endogenous κ-opioid receptor agonist, can increase food intake and body weight when acting on the POMC neurons, while directly inhibiting the POMC neurons by activating the G-protein-coupled inwardly-rectifying K+ channels.112 Additionally, obesity affects opioid sensitivity and the expression of its receptors and increases β-endorphin levels.108 However, the mechanisms by which these processes occur must be explored to identify their therapeutic viability against obesity and type 2 diabetes.
Serotonin and Serotonin 2C Receptors
In the POMC neurons, the serotonin 2C receptor (5-HT2CR) is involved in the regulation of central glucose homeostasis.113 The selective loss of 5-HT2CR directly impairs the glucose balance, which is manifested by elevated glucagon, insulin, and blood glucose. In addition, lorcaserin,114 as a 5-HT2CR agonist, can improve insulin sensitivity, inhibit hepatic glucose production and improve blood glucose control in T2D mice. Burke et al reported that 5-HT2CR and MC4R are required for lorcaserin to regulate blood glucose. Collectively, the 5-HT2CR agonists improve the symptoms and blood glucose levels of patients with type 2 diabetes through the MC4R signaling pathway,115 serving as an effective and novel strategy in the treatment of type 2 diabetes.
Ephrins B1 and B2
The POMC neurons can receive specific central signals by receiving a glutamatergic input.116 Ephrins B1 and B2117 are abundantly expressed in the POMC neurons, bind to their corresponding receptors, and act on the glutamate synapses, affecting feeding and glucose homeostasis. Gervais et al118 observed that ephrinB1 deficiency in the POMC-expressing progenitor exhibited impaired glucose tolerance, whereas ephrinB2 had no apparent effect on glucose homeostasis in deficient mice. Additionally, ephrinB2 deficiency in mice affected the energy balance in a sex-dependent manner, with gluconeogenesis and feeding behavior impaired in male and female mice, respectively. Thus, the EphrinB1- and EphrinB2-encoding genes may be potential targets for the treatment of diabetes, although the regulatory mechanism of hypothalamic neuronal circuits on glucose homeostasis is not well understood.
Conclusions and Prospects
The hypothalamus neuronal network is intricate, among which the POMC neurons are particularly critical to glucose homeostasis. Several recent studies have demonstrated that POMC neurons can affect appetite, energy metabolism, and insulin sensitivity through various central and peripheral signals. The lack of these hormones or their corresponding receptors or the disruption of their signaling pathways can lead to metabolic abnormalities, resulting in obesity, type 2 diabetes, and other metabolic syndromes. Therefore, the specific mechanisms underlying these processes require further investigation. Currently, in addition to the traditional peripheral blood hypoglycemic drugs, some hypoglycemic drugs with a central action are gradually emerging. Multi-drug combination therapy for obesity and diabetes is gaining attention and must be continuously explored to help patients to better control their blood glucose and improve their prognosis.
Abbreviations
ARC, the arcuate nucleus; AMPK, AMP-activated protein kinase; CB1R, cannabinoid receptor 1; CNS, central nervous system; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; FoxO1, forkhead box protein O1; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; GLP-2, glucagon-like peptide-2; GLP-2R, GLP-2 receptor; HFD, high-fat diet; HIF, hypoxia inducible factor; HIF1α, HIF-1-alpha; IRS, insulin receptor substrate; JAK/STAT, Janus kinase/signal transducer and activator of transcription; KATP, ATP sensitive potassium; LKB1, liver kinase B1; Sirt6, Sirtuin 6; MC3R, melanocortin-3 receptor; MC4R, melanocortin 4 receptor; MCH, melanin-enhanced hormone; MCHR1, melanin-enhanced hormone receptor 1; mTOR/S6K, the mechanistic target of rapamycin/S6 kinase; NF-κB, nuclear factor-kappa B; NPY, neuropeptide Y; OX-1R, OX-A receptor type 1; POMC, pro-opiomelanocortin; SHP2-MAPK, src homology region 2-containing protein tyrosine phosphatase 2-mitogen-activated protein kinase; SIRT1, Sirtuin 1; SRC-2, steroid receptor coactivator-2; TGF-β, transforming growth factor-β; TLR4, toll-like receptor-4; α-MSH, α-melanocyte-stimulating hormone; β-endorphin, beta-endorphin; 5-HT2CR, serotonin 2C receptor.
Acknowledgments
The coauthors thank the National Natural Science Foundation of China (81973378, 82073909), Research Project Supported by Shanxi Scholarship Council of China (2020-0172) for their support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas. Diabetes Res Clin Pract. 2019;157:107843. doi:10.1016/j.diabres.2019.107843
2. Sun H, Saeedi P, Karuranga S, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119
3. DeFronzo RA, Ferrannini E, Groop L, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019. doi:10.1038/nrdp.2015.19
4. Labazi H, Trask AJ. Coronary microvascular disease as an early culprit in the pathophysiology of diabetes and metabolic syndrome. Pharmacol Res. 2017;123:114–121. doi:10.1016/j.phrs.2017.07.004
5. da Silva AA, Do Carmo JM, Hall JE. CNS regulation of glucose homeostasis: role of the leptin-melanocortin system. Curr Diab Rep. 2020;20(7):29. doi:10.1007/s11892-020-01311-1
6. Myers MG
7. Candler T, Kuhnen P, Prentice AM, Silver M. Epigenetic regulation of POMC; implications for nutritional programming, obesity and metabolic disease. Front Neuroendocrinol. 2019;54:100773. doi:10.1016/j.yfrne.2019.100773
8. Zhan C. POMC neurons: feeding, energy metabolism, and beyond. Adv Exp Med Biol. 2018;1090:17–29.
9. Goit RK, Taylor AW, Lo ACY. The central melanocortin system as a treatment target for obesity and diabetes: a brief overview. Eur J Pharmacol. 2022;924:174956. doi:10.1016/j.ejphar.2022.174956
10. Hill JW, Elias CF, Fukuda M, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11(4):286–297. doi:10.1016/j.cmet.2010.03.002
11. Myers MG
12. Park HK, Ahima RS. Physiology of leptin: energy homeostasis, neuroendocrine function and metabolism. Metabolism. 2015;64(1):24–34. doi:10.1016/j.metabol.2014.08.004
13. Nakano M, Asakawa A, Inui A. Long-term correction of type 1 and 2 diabetes by central leptin gene therapy independent of effects on appetite and energy expenditure. Indian J Endocrinol Metab. 2012;16(Suppl 3):S556–561. doi:10.4103/2230-8210.105572
14. Shi H, Strader AD, Sorrell JE, Chambers JB, Woods SC, Seeley RJ. Sexually different actions of leptin in proopiomelanocortin neurons to regulate glucose homeostasis. Am J Physiol Endocrinol Metab. 2008;294(3):E630–639. doi:10.1152/ajpendo.00704.2007
15. Munzberg H, Morrison CD. Structure, production and signaling of leptin. Metabolism. 2015;64(1):13–23. doi:10.1016/j.metabol.2014.09.010
16. Zhang Y, Scarpace PJ. Circumventing central leptin resistance: lessons from central leptin and POMC gene delivery. Peptides. 2006;27(2):350–364. doi:10.1016/j.peptides.2005.01.024
17. Wardlaw SL. Hypothalamic proopiomelanocortin processing and the regulation of energy balance. Eur J Pharmacol. 2011;660(1):213–219. doi:10.1016/j.ejphar.2010.10.107
18. Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47(2):294–297. doi:10.2337/diab.47.2.294
19. Bray GA. The Zucker-fatty rat: a review. Fed Proc. 1977;36(2):148–153.
20. Kim EM, O’Hare E, Grace MK, Welch CC, Billington CJ, Levine AS. ARC POMC mRNA and PVN alpha-MSH are lower in obese relative to lean Zucker rats. Brain Res. 2000;862(1–2):11–16. doi:10.1016/S0006-8993(00)02060-6
21. Huo L, Gamber K, Greeley S, et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 2009;9(6):537–547. doi:10.1016/j.cmet.2009.05.003
22. Kahn BB, Minokoshi Y. Leptin, GABA, and glucose control. Cell Metab. 2013;18(3):304–306. doi:10.1016/j.cmet.2013.08.015
23. Faulkner LD, Dowling AR, Stuart RC, Nillni EA, Hill JW. Reduced melanocortin production causes sexual dysfunction in male mice with POMC neuronal insulin and leptin insensitivity. Endocrinology. 2015;156(4):1372–1385. doi:10.1210/en.2014-1788
24. Ernst MB, Wunderlich CM, Hess S, et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci. 2009;29(37):11582–11593. doi:10.1523/JNEUROSCI.5712-08.2009
25. Darnell JE
26. Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes. 2009;58(3):536–542. doi:10.2337/db08-0822
27. Hill JW, Xu Y, Preitner F, et al. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology. 2009;150(11):4874–4882. doi:10.1210/en.2009-0454
28. Belgardt BF, Husch A, Rother E, et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008;7(4):291–301. doi:10.1016/j.cmet.2008.01.006
29. Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron. 2012;75(3):425–436. doi:10.1016/j.neuron.2012.03.043
30. Smith MA, Katsouri L, Irvine EE, et al. Ribosomal S6K1 in POMC and AgRP neurons regulates glucose homeostasis but not feeding behavior in mice. Cell Rep. 2015;11(3):335–343. doi:10.1016/j.celrep.2015.03.029
31. Yu Y, Wu Y, Szabo A, et al. Teasaponin reduces inflammation and central leptin resistance in diet-induced obese male mice. Endocrinology. 2013;154(9):3130–3140. doi:10.1210/en.2013-1218
32. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology. 2007;22:241–251. doi:10.1152/physiol.00010.2007
33. Dodd GT, Tiganis T. Insulin action in the brain: roles in energy and glucose homeostasis. J Neuroendocrinol. 2017;29:10. doi:10.1111/jne.12513
34. Schwartz MW, Sipols A, Kahn SE, et al. Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am J Physiol. 1990;259(3 Pt 1):E378–383. doi:10.1152/ajpendo.1990.259.3.E378
35. Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. doi:10.1126/scitranslmed.3003748
36. Molnar G, Farago N, Kocsis AK, et al. GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci. 2014;34(4):1133–1137. doi:10.1523/JNEUROSCI.4082-13.2014
37. Choi JH, Kim MS. Homeostatic regulation of glucose metabolism by the central nervous system. Endocrinol Metab. 2022;37(1):9–25. doi:10.3803/EnM.2021.1364
38. Lin HV, Plum L, Ono H, et al. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes. 2010;59(2):337–346. doi:10.2337/db09-1303
39. Klockener T, Hess S, Belgardt BF, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci. 2011;14(7):911–918. doi:10.1038/nn.2847
40. Qiu J, Zhang C, Borgquist A, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014;19(4):682–693. doi:10.1016/j.cmet.2014.03.004
41. Shin AC, Filatova N, Lindtner C, et al. Insulin receptor signaling in POMC, but Not AgRP, neurons controls adipose tissue insulin action. Diabetes. 2017;66(6):1560–1571. doi:10.2337/db16-1238
42. Yan J, Zhang H, Yin Y, et al. Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat Med. 2014;20(9):1001–1008. doi:10.1038/nm.3616
43. Jais A, Bruning JC. Hypothalamic inflammation in obesity and metabolic disease. J Clin Invest. 2017;127(1):24–32. doi:10.1172/JCI88878
44. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18(1):59–68. doi:10.1016/j.molmed.2011.07.010
45. Avalos Y, Hernandez-Caceres MP, Lagos P, et al. Palmitic acid control of ciliogenesis modulates insulin signaling in hypothalamic neurons through an autophagy-dependent mechanism. Cell Death Dis. 2022;13(7):659. doi:10.1038/s41419-022-05109-9
46. Yao T, Deng Z, Gao Y, et al. Ire1alpha in pomc neurons is required for thermogenesis and glycemia. Diabetes. 2017;66(3):663–673. doi:10.2337/db16-0533
47. Williams KW, Liu T, Kong X, et al. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 2014;20(3):471–482. doi:10.1016/j.cmet.2014.06.002
48. Lindtner C, Scherer T, Zielinski E, et al. Binge drinking induces whole-body insulin resistance by impairing hypothalamic insulin action. Sci Transl Med. 2013;5(170):170ra114. doi:10.1126/scitranslmed.3005123
49. Weissmann L, Quaresma PG, Santos AC, et al. IKKepsilon is key to induction of insulin resistance in the hypothalamus, and its inhibition reverses obesity. Diabetes. 2014;63(10):3334–3345. doi:10.2337/db13-1817
50. Wang Z, Do Carmo JM, da Silva AA, et al. Role of SOCS3 in POMC neurons in metabolic and cardiovascular regulation. Am J Physiol Regul Integr Comp Physiol. 2019;316(4):R338–R351. doi:10.1152/ajpregu.00163.2018
51. Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–635. doi:10.1038/39369
52. Yuan F, Yin H, Deng Y, et al. Overexpression of Smad7 in hypothalamic POMC neurons disrupts glucose balance by attenuating central insulin signaling. Mol Metab. 2020;42:101084. doi:10.1016/j.molmet.2020.101084
53. Gaspar JM, Velloso LA. Hypoxia inducible factor as a central regulator of metabolism - implications for the development of obesity. Front Neurosci. 2018;12:813. doi:10.3389/fnins.2018.00813
54. Gaspar JM, Mendes NF, Correa-da-Silva F, et al. Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain Behav Immun. 2018;73:550–561. doi:10.1016/j.bbi.2018.06.020
55. Wang Z, Khor S, Cai D. Age-dependent decline of hypothalamic HIF2alpha in response to insulin and its contribution to advanced age-associated metabolic disorders in mice. J Biol Chem. 2019;294(13):4946–4955. doi:10.1074/jbc.RA118.005429
56. Wu Z, Xi P, Zhang Y, et al. LKB1 up-regulation inhibits hypothalamic inflammation and attenuates diet-induced obesity in mice. Metabolism. 2021;116:154694. doi:10.1016/j.metabol.2020.154694
57. Wu Z, Han J, Xue J, et al. Deletion of liver kinase B1 in POMC neurons predisposes to diet-induced obesity. Life Sci. 2020;258:118204. doi:10.1016/j.lfs.2020.118204
58. Tang Q, Liu Q, Yang X, et al. Sirtuin 6 supra-physiological overexpression in hypothalamic pro-opiomelanocortin neurons promotes obesity via the hypothalamus-adipose axis. FASEB J. 2021;35(3):e21408. doi:10.1096/fj.202002607
59. Tang Q, Gao Y, Liu Q, et al. Sirt6 in pro-opiomelanocortin neurons controls energy metabolism by modulating leptin signaling. Mol Metab. 2020;37:100994. doi:10.1016/j.molmet.2020.100994
60. Yang Y, He Y, Liu H, et al. Hypothalamic steroid receptor coactivator-2 regulates adaptations to fasting and overnutrition. Cell Rep. 2021;37(10):110075. doi:10.1016/j.celrep.2021.110075
61. Koch CE, Lowe C, Legler K, et al. Central adiponectin acutely improves glucose tolerance in male mice. Endocrinology. 2014;155(5):1806–1816. doi:10.1210/en.2013-1734
62. Coope A, Milanski M, Araujo EP, et al. AdipoR1 mediates the anorexigenic and insulin/leptin-like actions of adiponectin in the hypothalamus. FEBS Lett. 2008;582(10):1471–1476. doi:10.1016/j.febslet.2008.03.037
63. Qi Y, Takahashi N, Hileman SM, et al. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10(5):524–529. doi:10.1038/nm1029
64. Park S, Kim DS, Kwon DY, Yang HJ. Long-term central infusion of adiponectin improves energy and glucose homeostasis by decreasing fat storage and suppressing hepatic gluconeogenesis without changing food intake. J Neuroendocrinol. 2011;23(8):687–698. doi:10.1111/j.1365-2826.2011.02165.x
65. Guillod-Maximin E, Roy AF, Vacher CM, et al. Adiponectin receptors are expressed in hypothalamus and colocalized with proopiomelanocortin and neuropeptide Y in rodent arcuate neurons. J Endocrinol. 2009;200(1):93–105. doi:10.1677/JOE-08-0348
66. Thundyil J, Pavlovski D, Sobey CG, Arumugam TV. Adiponectin receptor signalling in the brain. Br J Pharmacol. 2012;165(2):313–327. doi:10.1111/j.1476-5381.2011.01560.x
67. Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569–574. doi:10.1038/nature02440
68. Suyama S, Maekawa F, Maejima Y, Kubota N, Kadowaki T, Yada T. Glucose level determines excitatory or inhibitory effects of adiponectin on arcuate POMC neuron activity and feeding. Sci Rep. 2016;6:30796. doi:10.1038/srep30796
69. Posey KA, Clegg DJ, Printz RL, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296(5):E1003–1012. doi:10.1152/ajpendo.90377.2008
70. Drucker DJ, Habener JF, Holst JJ. Discovery, characterization, and clinical development of the glucagon-like peptides. J Clin Invest. 2017;127(12):4217–4227. doi:10.1172/JCI97233
71. Amato A, Baldassano S, Mule F. GLP2: an underestimated signal for improving glycaemic control and insulin sensitivity. J Endocrinol. 2016;229(2):R57–66. doi:10.1530/JOE-16-0035
72. Ten Kulve JS, van Bloemendaal L, Balesar R, et al. Decreased hypothalamic glucagon-like peptide-1 receptor expression in type 2 diabetes patients. J Clin Endocrinol Metab. 2016;101(5):2122–2129. doi:10.1210/jc.2015-3291
73. Guan X, Shi X, Li X, et al. GLP-2 receptor in POMC neurons suppresses feeding behavior and gastric motility. Am J Physiol Endocrinol Metab. 2012;303(7):E853–864. doi:10.1152/ajpendo.00245.2012
74. Halawi H, Khemani D, Eckert D, et al. Effects of liraglutide on weight, satiation, and gastric functions in obesity: a randomised, placebo-controlled pilot trial. Lancet Gastroenterol Hepatol. 2017;2(12):890–899. doi:10.1016/S2468-1253(17)30285-6
75. Roh E, Song DK, Kim MS. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med. 2016;48:e216. doi:10.1038/emm.2016.4
76. Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. 2014;124(10):4473–4488. doi:10.1172/JCI75276
77. Jones GL, Wittmann G, Yokosawa EB, et al. Selective restoration of pomc expression in glutamatergic POMC neurons: evidence for a dynamic hypothalamic neurotransmitter network. eNeuro. 2019;6:2. doi:10.1523/ENEURO.0400-18.2019
78. Bjorbaek C, Hollenberg AN. Leptin and melanocortin signaling in the hypothalamus. Vitam Horm. 2002;65:281–311.
79. Arora S. Role of neuropeptides in appetite regulation and obesity--a review. Neuropeptides. 2006;40(6):375–401. doi:10.1016/j.npep.2006.07.001
80. Baldassano S, Amato A, Mule F. Influence of glucagon-like peptide 2 on energy homeostasis. Peptides. 2016;86:1–5. doi:10.1016/j.peptides.2016.09.010
81. The GX. CNS glucagon-like peptide-2 receptor in the control of energy balance and glucose homeostasis. Am J Physiol Regul Integr Comp Physiol. 2014;307(6):R585–596. doi:10.1152/ajpregu.00096.2014
82. Shi X, Zhou F, Li X, et al. Central GLP-2 enhances hepatic insulin sensitivity via activating PI3K signaling in POMC neurons. Cell Metab. 2013;18(1):86–98. doi:10.1016/j.cmet.2013.06.014
83. Taher J, Baker C, Alvares D, Ijaz L, Hussain M, Adeli K. GLP-2 dysregulates hepatic lipoprotein metabolism, inducing fatty liver and VLDL overproduction in male hamsters and mice. Endocrinology. 2018;159(9):3340–3350. doi:10.1210/en.2018-00416
84. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27(4):740–756. doi:10.1016/j.cmet.2018.03.001
85. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi:10.1016/S0140-6736(06)69705-5
86. Girardet C, Butler AA. Neural melanocortin receptors in obesity and related metabolic disorders. Biochim Biophys Acta. 2014;1842(3):482–494. doi:10.1016/j.bbadis.2013.05.004
87. Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141(9):3072–3079. doi:10.1210/endo.141.9.7665
88. Hill JW, Faulkner LD. The role of the melanocortin system in metabolic disease: new developments and advances. Neuroendocrinology. 2017;104(4):330–346. doi:10.1159/000450649
89. Huszar D, Lynch CA, Fairchild-Huntress V, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi:10.1016/S0092-8674(00)81865-6
90. Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–543. doi:10.1210/er.2009-0037
91. Kievit P, Halem H, Marks DL, et al. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013;62(2):490–497. doi:10.2337/db12-0598
92. Mark AL, Correia M, Morgan DA, Shaffer RA, Haynes WG. State-of-The-art-lecture: obesity-induced hypertension: new concepts from the emerging biology of obesity. Hypertension. 1999;33(1 Pt 2):537–541. doi:10.1161/01.HYP.33.1.537
93. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. doi:10.1056/NEJMoa0803085
94. Koch M, Varela L, Kim JG, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519(7541):45–50. doi:10.1038/nature14260
95. Tsunematsu T, Yamanaka A. The role of orexin/hypocretin in the central nervous system and peripheral tissues. Vitam Horm. 2012;89:19–33.
96. Laburthe M, Voisin T, El Firar A. Orexins/hypocretins and orexin receptors in apoptosis: a mini-review. Acta Physiol. 2010;198(3):393–402. doi:10.1111/j.1748-1716.2009.02035.x
97. Yang D, Xu L, Guo F, Sun X, Zhang D, Wang M. Orexin-A and endocannabinoid signaling regulate glucose-responsive arcuate nucleus neurons and feeding behavior in obese rats. Neuropeptides. 2018;69:26–38. doi:10.1016/j.npep.2018.04.001
98. Morello G, Imperatore R, Palomba L, et al. Orexin-A represses satiety-inducing POMC neurons and contributes to obesity via stimulation of endocannabinoid signaling. Proc Natl Acad Sci U S A. 2016;113(17):4759–4764. doi:10.1073/pnas.1521304113
99. Nishimura Y, Mabuchi K, Taguchi S, et al. Involvement of orexin-A neurons but not melanin-concentrating hormone neurons in the short-term regulation of food intake in rats. J Physiol Sci. 2014;64(3):203–211. doi:10.1007/s12576-014-0312-0
100. Blais A, Drouin G, Chaumontet C, et al. Impact of Orexin-A treatment on food intake, energy metabolism and body weight in mice. PLoS One. 2017;12(1):e0169908. doi:10.1371/journal.pone.0169908
101. Griffond B, Risold PY. MCH and feeding behavior-interaction with peptidic network. Peptides. 2009;30(11):2045–2051. doi:10.1016/j.peptides.2009.07.008
102. Cheon HG. Antiobesity effects of melanin-concentrating hormone receptor 1 (MCH-R1) antagonists. Handb Exp Pharmacol. 2012;209:383–403.
103. Shearman LP, Camacho RE, Sloan Stribling D, et al. Chronic MCH-1 receptor modulation alters appetite, body weight and adiposity in rats. Eur J Pharmacol. 2003;475(1–3):37–47. doi:10.1016/S0014-2999(03)02146-0
104. Al-Massadi O, Quinones M, Clasadonte J, et al. MCH Regulates SIRT1/FoxO1 and Reduces POMC neuronal activity to induce hyperphagia, adiposity, and glucose intolerance. Diabetes. 2019;68(12):2210–2222. doi:10.2337/db19-0029
105. Kong D, Vong L, Parton LE, et al. Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab. 2010;12(5):545–552. doi:10.1016/j.cmet.2010.09.013
106. Surwit RS, McCubbin JA, Kuhn CM, Cochrane C, Feinglos MN. Differential glycemic effects of morphine in diabetic and normal mice. Metabolism. 1989;38(3):282–285. doi:10.1016/0026-0495(89)90089-9
107. Benarroch EE. Endogenous opioid systems: current concepts and clinical correlations. Neurology. 2012;79(8):807–814. doi:10.1212/WNL.0b013e3182662098
108. Koekkoek LL, van der Gun LL, Serlie MJ, la Fleur SE. The clash of two epidemics: the relationship between opioids and glucose metabolism. Curr Diab Rep. 2022;22(7):301–310. doi:10.1007/s11892-022-01473-0
109. Lux F, Brase DA, Dewey WL. Differential effects of subcutaneous and intrathecal morphine administration on blood glucose in mice: comparison with intracerebroventricular administration. J Pharmacol Exp Ther. 1988;245(1):187–194.
110. Tzeng TF, Lo CY, Cheng JT, Liu IM. Activation of mu-opioid receptors improves insulin sensitivity in obese Zucker rats. Life Sci. 2007;80(16):1508–1516. doi:10.1016/j.lfs.2007.01.016
111. Giugliano D, Ceriello A, Salvatore T, Paolisso G, D’Onofrio F, Lefebvre P. Beta-endorphin infusion restores acute insulin responses to glucose in type-2 diabetes mellitus. J Clin Endocrinol Metab. 1987;64(5):944–948. doi:10.1210/jcem-64-5-944
112. Pennock RL, Hentges ST. Direct inhibition of hypothalamic proopiomelanocortin neurons by dynorphin A is mediated by the mu-opioid receptor. J Physiol. 2014;592(19):4247–4256. doi:10.1113/jphysiol.2014.275339
113. Berglund ED, Liu C, Sohn JW, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061–5070. doi:10.1172/JCI70338
114. Burke LK, Ogunnowo-Bada E, Georgescu T, et al. Lorcaserin improves glycemic control via a melanocortin neurocircuit. Mol Metab. 2017;6(10):1092–1102. doi:10.1016/j.molmet.2017.07.004
115. Zhou L, Sutton GM, Rochford JJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6(5):398–405. doi:10.1016/j.cmet.2007.10.008
116. Wang D, He X, Zhao Z, et al. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat. 2015;9:40. doi:10.3389/fnana.2015.00040
117. Blits-Huizinga CT, Nelersa CM, Malhotra A, Liebl DJ. Ephrins and their receptors: binding versus biology. IUBMB Life. 2004;56(5):257–265. doi:10.1080/15216540412331270076
118. Gervais M, Labouebe G, Picard A, Thorens B, Croizier S. EphrinB1 modulates glutamatergic inputs into POMC-expressing progenitors and controls glucose homeostasis. PLoS Biol. 2020;18(11):e3000680. doi:10.1371/journal.pbio.3000680
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.