")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Effects of Simvastatin on the Metabolism of Vonoprazan in Rats Both in vitro and in vivo
Authors Hong Y , Dai DP, Cai JP , Wang SH, Wang YR, Zhao FL , Zhou S, Zhou Q, Geng PW, Zhou YF, Xu X, Shi JH, Luo QF
Received 9 March 2022
Accepted for publication 2 June 2022
Published 9 June 2022 Volume 2022:16 Pages 1779—1789
DOI https://doi.org/10.2147/DDDT.S365610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Yun Hong,1,* Da-Peng Dai,2,* Jian-Ping Cai,2 Shuang-Hu Wang,3 Yi-Ran Wang,1,4 Fang-Ling Zhao,2,4 Shan Zhou,2 Quan Zhou,3 Pei-Wu Geng,3 Yun-Fang Zhou,3 Xue Xu,1 Ji-Hua Shi,1 Qing-Feng Luo1
1Department of Gastroenterology, Beijing Hospital, National Center of Gerontology; Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, 100730, People’s Republic of China; 2The Key Laboratory of Geriatrics, Beijing Institute of Geriatrics, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing Hospital/National Center of Gerontology of National Health Commission, Beijing, 100730, People’s Republic of China; 3Laboratory of Clinical Pharmacy, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, 323020, People’s Republic of China; 4Peking University Fifth School of Clinical Medicine, Beijing, 100730, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qing-Feng Luo, Department of Gastroenterology, Beijing Hospital, National Center of Gerontology; Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, 100730, People’s Republic of China, Tel + 86 138 1151 9095, Email [email protected]
Purpose: To study the potential drug–drug interactions between simvastatin and vonoprazan and to provide the scientific basis for rational use of them in clinical practice.
Methods: An incubation system was established with rat liver microsomes, and the main metabolite of vonoprazan M-I was detected by UPLC-MS/MS. The IC50 value of simvastatin was then calculated and its inhibitory mechanism against vonoprazan was also analyzed. Twelve SD rats were randomly divided into 2 groups, then they were given simvastatin or saline for 2 weeks continuously. On the day of the experiment, both groups were intragastrically administered with vonoprazan once, followed by the collection of blood at different time points. Then the plasma concentration of vonoprazan and M-I in rats were detected by UPLC-MS/MS.
Results: In vitro experiments revealed that simvastatin could inhibit the metabolism of vonoprazan, and its inhibition type belonged to the mixed non-competitive and competitive inhibition model. In vivo experiments in rats demonstrated that the area under concentration time curve (AUC) of vonoprazan was decreased but the clearance (CLz/F) of it was increased in the simvastatin administrated group, as compared to those of the control group. However, M-I in simvastatin treated group exhibited the higher AUC and lower CLz/F values compared to those in the control group. These data indicated that multiple doses of simvastatin administration could reduce the plasma concentration of vonoprazan and accelerate its metabolic rate in rats.
Conclusion: Simvastatin could inhibit the metabolism of vonoprazan in vitro but multiple doses of simvastatin exhibited the opposite effect In vivo. Altogether, our data indicated that an interaction existed between simvastatin and vonoprazan and additional cares might be taken when they were co-administrated in clinic.
Keywords: vonoprazan, simvastatin, drug–drug interactions, liquid chromatography-tandem mass spectrometry, rat liver microsomes
Introduction
As one of the drugs with wide clinical application, simvastatin is mainly used to treat dyslipidemia and it can reduce the concentrations of total cholesterol, triglyceride and low-density lipoprotein (LDL) in plasma, accomplished by increasing the concentration of high-density lipoprotein (HDL).1 It has been reported that simvastatin is the substrates for cytochrome P450 (CYP) enzymes, P-glycoprotein (P-gp) and organic anion-transporting polypeptide transporter (OATP1B1), and it is mainly metabolized via CYP3A4/5, with CYP2C8/9/19 and CYP2D6 also involved in partial metabolism.2 Recently, drug–drug interactions between simvastatin and other drugs were reported, such as simvastatin could inhibit the metabolism of verapamil and apatinib and increase their bioavailability by inhibiting CYP3A and P-glycoprotein (P-gp) efflux pumps.3,4
Vonoprazan, a potassium-competitive acid blocker (P-CAB), is a novel reversible proton pump inhibitor (PPI) that is used for the treatment or the prevention of gastric acid-related clinical conditions, such as erosive esophagitis, gastroesophageal reflux, gastroduodenal ulcer, aspirin or NSAID-induced peptic ulcer, and it can also be used for the eradication of Helicobacter pylori.5 It is reported that vonoprazan has a promising prospect for clinical application as it can be absorbed faster and has a longer half-life with stronger and longer-lasting antigastric acid secretion effect, compared to the traditional PPIs.6
Previous reports revealed that vonoprazan was mainly metabolized in two different pathways, the oxidization pathway by CYP enzyme isoforms (CYP3A4, CYP2B6, CYP2C19, CYP2D6) and the non-oxidization pathway by sulfotransferase (SULT2A1). With CYP enzyme, especially for CYP3A4, vonoprazan can be converted into 5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrole-3-carboxylic acid (M-I) by the oxidative deamidation. We provided 3D models of chemical structure of vonoprazan and M-I for reference (Figure 1). As the main primary metabolite of vonoprazan, M-I can be further metabolized to the secondary metabolites, such as 5-(2-fluorophenyl)-1H-pyrrole-3-carboxylic acid (M-II), M-III and M-IV-Sul. However, none of these metabolites is pharmacologically active.7–9
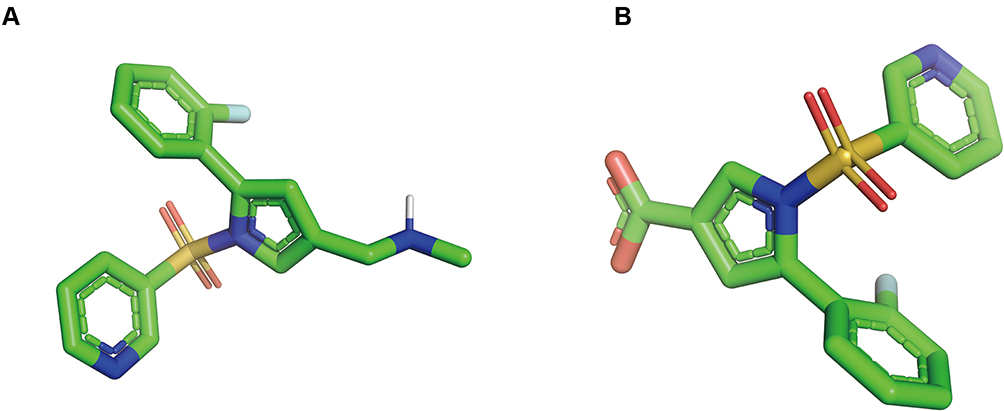 |
Figure 1 Structure displays of vonoprazan and M-I. (A) 3D model of chemical structure of vonoprazan; (B) 3D model of chemical structure of M-I. |
Like other CYP3A4-mediated drugs, the metabolism of vonoprazan could be inhibited by some strong CYP3A4 inhibitor drugs. For example, voriconazole was reported to inhibit vonoprazan metabolism by inhibiting the enzymatic activity of cytochrome P450.10 To date, no reports were found regarding the potential interactions between CYP3A4-mediated drugs simvastatin and vonoprazan. Considering for the wide usage of simvastatin and the possibility of co-administration of simvastatin and acid inhibitory drugs in clinic, we performed the drug-drug interaction study in rat model both in vitro and In vivo. Our data provide the first-hand evidence of the interactions between these two drugs and indicated that more cares might be taken when using them simultaneously in clinic.
Materials and Methods
Drugs and Reagents
Simvastatin and vonoprazan were purchased from Sunflower Technology Development Co. (Beijing, China). M-I was got from Wuxi Medical Technology Co. (Wuxi, China). Diazepam was purchased from Tianjin Golden York Pharmaceutical Co. (Tianjin China). Methanol and acetonitrile (chromatographic grade) were obtained from Merck GmbH (Darmstadt, Germany). Formic acid (chromatographic grade) was gotten from Sigma-Aldrich (St. Louis, Missouri, USA). Other chemical reagents were all analytically pure or guaranteed reagent.
Experimental Animals
The experimental animals were healthy SPF-grade SD male rats with 250±20 g, provided by the Laboratory Animal Center of Wenzhou Medical University. Animals were fed rat chow once in the morning and once in the evening, and water was consumed freely at daily temperature of 20–25°C, 12 hours of light and relative humidity of 40–60% for 1 week before the drug administrations. All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act, and the Office of Laboratory Animal Welfare.
All experimental procedures were approved by the Animal Experimental Ethics Inspection Department of the Laboratory Animal Center in Wenzhou Medical University (approval No. wydw2019-650).
Chromatography and Conditions
ACQUITY I-Class UPLC and Waters XEVO TQD MS (Milford, MA, USA) were used for the detection of vonoprazan and M-I with ACQUITY UPLC BEH C18 column (50 mm×2.1 mm, 1.7 μm) (Milford, MA, USA) at temperature of 40°C. The mobile phase consisted of acetonitrile and 0.1% formic acid in gradient proportion with running time of 3 minutes at flow rate of 0.4 mL/min. The injection volume of sample was 2 μL. The scanning method was multiple reaction monitoring (MRM) with detection in positive ion mode and an ESI+ ion source. Other mass spectrometry parameters were listed as following: capillary voltage 2.0 kV; ion source temperature 150 °C; argon flow rate: 0.15 mL/min. The other conditions of mass spectrometry were referred to Shen.10
In vitro Incubation Experiments
Rat liver microsomes (RLM) were prepared by differential centrifugation as described previously9 and its protein concentration were determined as 28 mg/mL. The volume of incubation system was 190 μL, containing 10 μM vonoprazan (close to its Km, Figure 2), 2 μL RLM, 20 μL potassium phosphate buffer (1 M, pH = 7.4), 100, 50, 10, 5, 1, 0.1, 0.01, and 0 μM simvastatin. Three parallels were set up for each group and the above operations were performed on ice. Then the mixture was vortexed and pre-incubated in a water bath at 37°C for 5 min, and the reaction was initiated by adding 10 μL of NADPH (1 mM) and incubated at 37°C for 30 min with shaking in water bath. At the end, 200 μL acetonitrile and 20 μL internal standard diazepam (500 ng/mL) were added to terminate the reaction, following by vortex and centrifugation at 13,000 rpm for 5 minutes. Subsequently, 150 μL of supernatant was taken out and 2μL of sample was injected into LC-MS/MS machine for the detection and quantification of M1 which were used for calculation of IC50 value by GraphPad Prism 9.0.
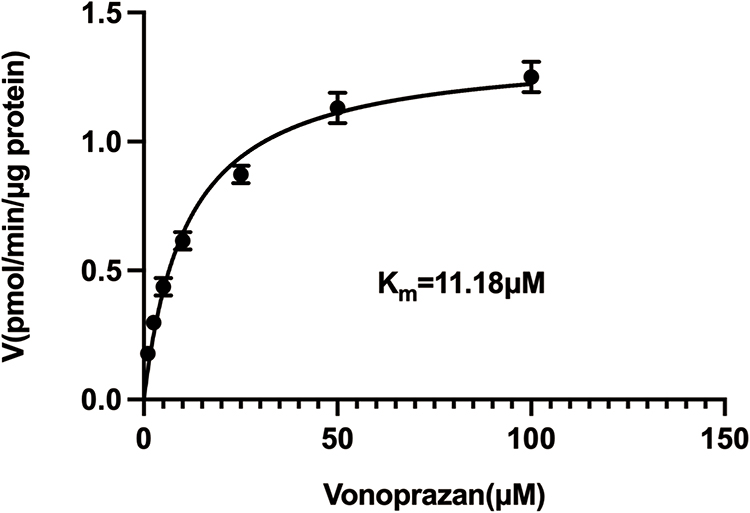 |
Figure 2 Michaelis-Menten kinetics of vonoprazan on cytochrome P450 enzymes in rat liver microsomes. |
In order to investigate the inhibition mechanism of simvastatin on vonoprazan, these two drugs were incubated together at different concentration gradients, with 2.5, 5, 10 and 20 μM of vonoprazan and 0, 1.5, 3, 6 and 12 μM of simvastatin. The incubation procedure was the same as that mentioned above.
In vivo Pharmacokinetic Experiments
Twelve healthy SPF-grade SD male rats were randomly and equally divided into 2 groups: animals in the simvastatin group were injected intraperitoneally with 50 mg/kg of simvastatin solution once a day for 2 weeks, followed by 12 hours of fasting in advance then administrated by intragastrical gavage of vonoprazan (10 mg/kg); animals in the control group were injected with saline for 2 weeks, followed by gavage administration of the same dose of vonoprazan in the same period. Next, at 0.083h, 0.25h, 0.5h, 1h, 2h, 3h, 4h, 6h, 12h, and 24h after administration, 300 μL of blood was collected from the tail vein and placed into EP tubes containing 20 μL sodium heparin. After the centrifugation at 4°C, 4000 rpm for 10 minutes, 100 μL of plasma samples were taken out and mixed with 100 μL of acetonitrile and 20 μL of diazepam (500 ng/mL) to precipitate proteins. Then the mixtures were vortexed and centrifuged at 4°C, 4000 rpm for 5 minutes. 150 μL supernatant was transferred into the sample bottle and 2 μL of sample was analyzed by LC-MS/MS machine for metabolites quantification.
Molecular Docking Method
We retrieved the crystal structure of CYP3A4 from the RCSB PDB database (https://www.rcsb.org/) and the molecular structures of simvastatin and vornolazem from Pubchem (https://pubchem.ncbi.nlm.nih.gov/), and then we performed structure optimization using Pymol (Version 2.5. 2) and molecular docking by AutoDock Vina (Version 1.2.0), choosing the result with the highest affinity among the docked structures we simulated.11,12 Finally, we visualized docking results on Pymol and found the sites where the drug interactions with CYP through hydrogen bonding.
Statistical Analysis
The IC50 value, Lineweaver-Burk plot and plasma concentration-time curves were obtained by GraphPad prism software (version 9.0, San Diego, CA, USA). DAS (Drug and Statistics) software (version 3.2.8; Lishui People’s Hospital, Lishui, China) was used to plot the drug-time curves and to calculate the changes of vonoprazan and M-I in rat plasma over time with the non-atrial model of statistical moment parameters. Statistical analysis of the main pharmacokinetic parameters, such as area under the drug-time curve (AUC), clearance (CLz/F), and peak concentration (Cmax), was performed using SPSS software (version 25.0; SPSS Inc., Chicago, IL, USA). All data were assessed distribution by using QQ plots. When comparing two independent samples, the Independent Samples t-test was used for normally distributed data (parametric test) and Mann–Whitney test was used for the non-normal distributed dataset (non-parametric test) and *P<0.05 indicates the statistical difference.
Results
Chromatograms of Vonoprazan Metabolite
Under conditions listed in the methods section, the peak emergence times of vonoprazan metabolite M-I and internal standard diazepam were 1.45 minutes and 1.70 minutes, respectively. As shown in Figure 3 that no interference could be detected among input drugs vonoprazan, M-I and diazepam, or in the blank rat liver microsomes.
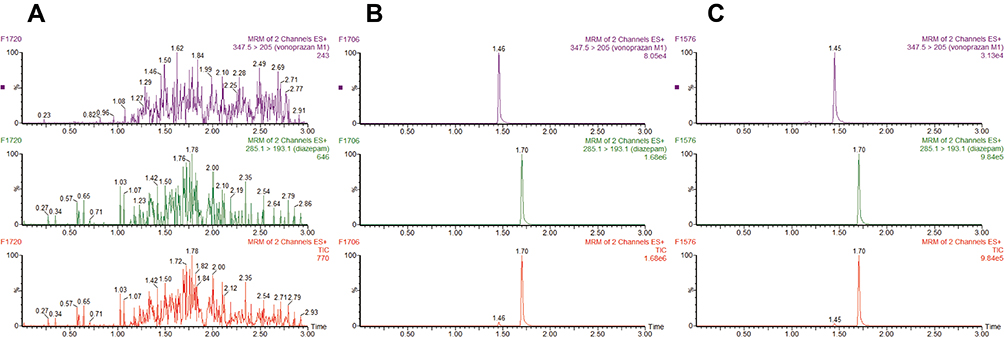 |
Figure 3 UPLC-MS/MS chromatograms of vonoprazan metabolite M-I and internal standard (IS) diazepam. (A) Blank rat microsomal system; (B) Standard M-I was mixed with denatured microsomes; (C) M-I and diazepam were detected after the incubation of rat liver microsomes with vonoprazan. |
Inhibition of Vonoprazan by Simvastatin in vitro
In vitro pharmacokinetic data revealed that simvastatin could inhibit the metabolism of vonoprazan with IC50 value of 6.444 μM (Figure 4). Lineweaver-Burk plots for simvastatin inhibition of vonoprazan metabolism in rat liver microsomes indicated that the mechanism behind this inhibition was a mixture of non-competitive and competitive inhibition with Ki=13.46 μM and αKi=5.54 μM (Figure 5).
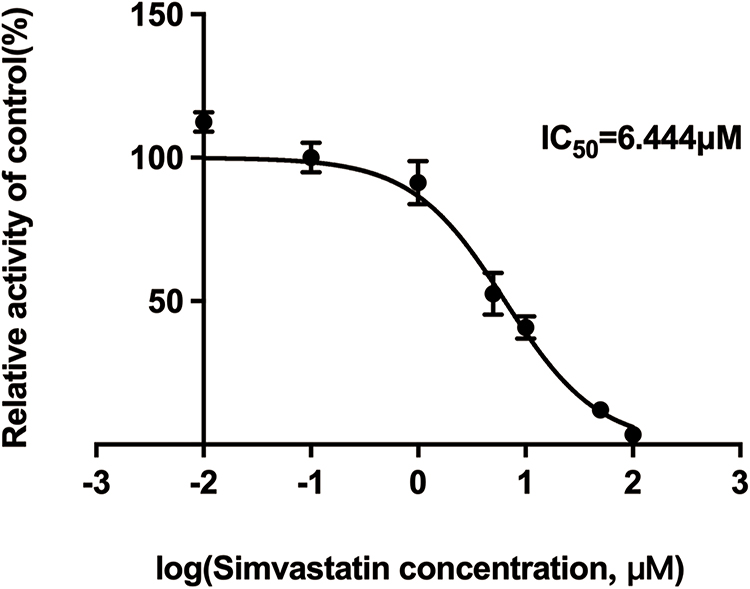 |
Figure 4 IC50 profile of simvastatin on cytochrome P450 enzymes in rat liver microsomes. |
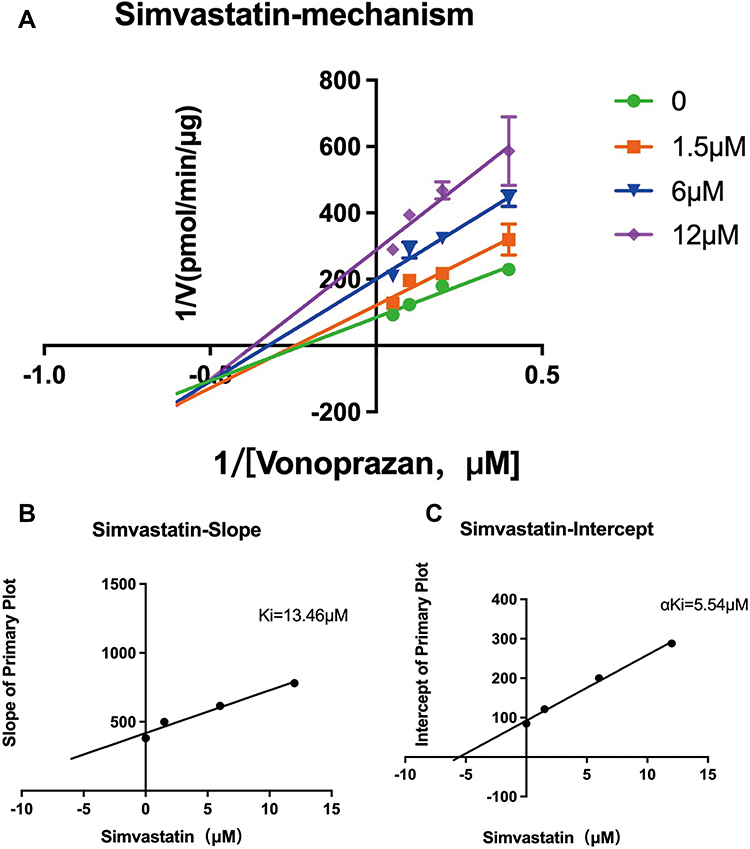 |
Figure 5 Lineweaver-Burk plots for simvastatin inhibition of vonoprazan metabolism in rat liver microsomes. (A) Lineweaver–Burk plots for simvastatin (0, 1.5, 6, 12 μM) inhibition of vonoprazan (2.5, 5, 10, 20 μM) in rat liver microsomes. Data are shown with the mean standard deviation of three parallel experiments; (B) Slope of primary plot; (C) Intercept of primary plot. |
Effect of Multiple Doses of Simvastatin on Vonoprazan in vivo
The mean plasma concentration-versus-time curves of vonoprazan and its main metabolite M-1 in rat after administration by gavage of simvastatin are shown in Figure 6. Detailed pharmacokinetic parameters of drugs are illustrated in Table 1. As compared to those of control group, the AUC(0-t) and AUC(0-∞) of vonoprazan in the simvastatin pretreated group decreased to 33.4% and 34.5%, respectively; the peak concentration Cmax of vonoprazan decreased to 25.4%, while the clearance CLz/F of vonoprazan increased by 1.65-fold. On the contrary, the AUC(0-t) and AUC(0-∞) of M-I in the simvastatin pretreated group were 1.28-fold and 1.35-fold higher than those in the control group, respectively, but the clearance CLz/F of M-I decreased to 77.9% of control group. These data indicated that multiple doses of simvastatin could significantly induce the metabolism of vonoprazan in rats by decreasing the plasma concentration of vonoprazan and increasing that of its metabolite.
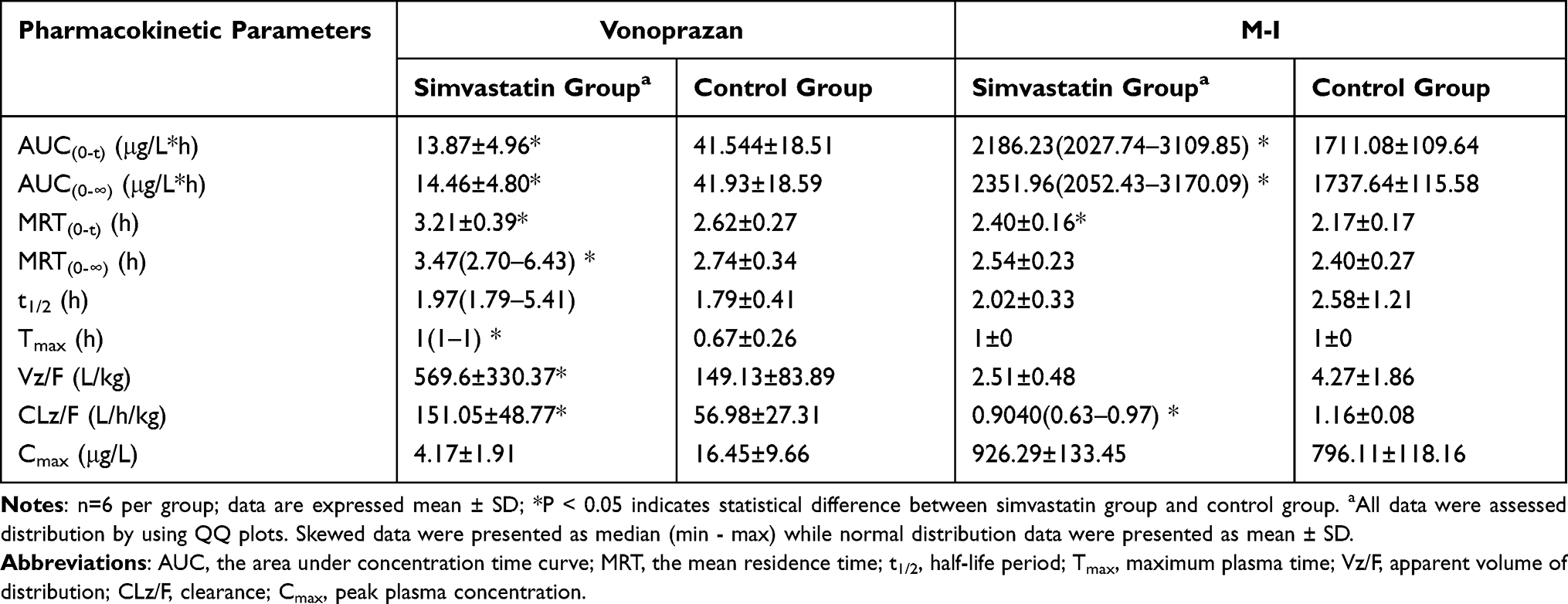 |
Table 1 Main Pharmacokinetic Parameters of Vonoprazan and M-I in vivo |
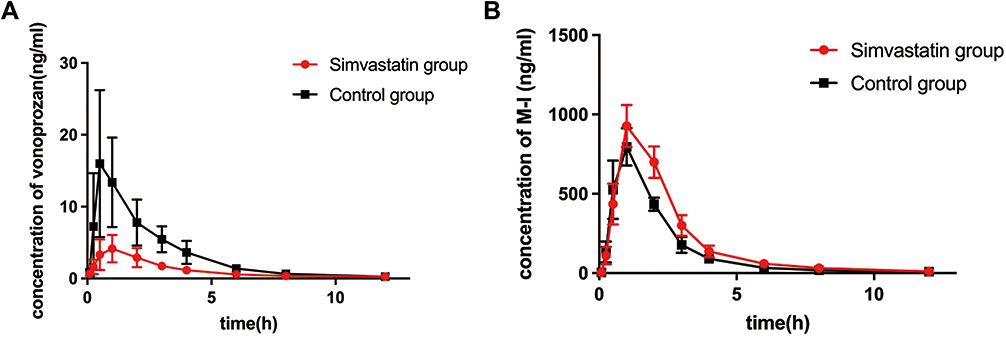 |
Figure 6 The concentration-time curves of vonoprazan (A) and M-I (B) in rat (n = 6). The simvastatin group was injected with simvastatin intraperitoneally for 2 weeks and then administered with vonoprazan intragastrically; The control group was injected with saline only for 2 weeks and followed by administrated with same dose of vonoprazan to that of simvastatin pretreated group. |
Molecular Docking Prediction of Simvastatin and Vonoprozan
To better understand the mechanism behind the interaction of simvastatin and vonoprazan, molecular docking analysis were also conducted in this study according to the methods previously reported.13–15 Our simulation results on Pymol showed that simvastatin interacted with arginine (ARG) at position 106 of CYP3A4 via hydrogen bonding, while vonoprazan interacted with heme (HEM) at position 601 of CYP3A4, with the sites of action 9.2 Å apart (Figure 7). Probably, this spatial positional proximity may be one of the reasons for the ease of interaction between the two drugs.
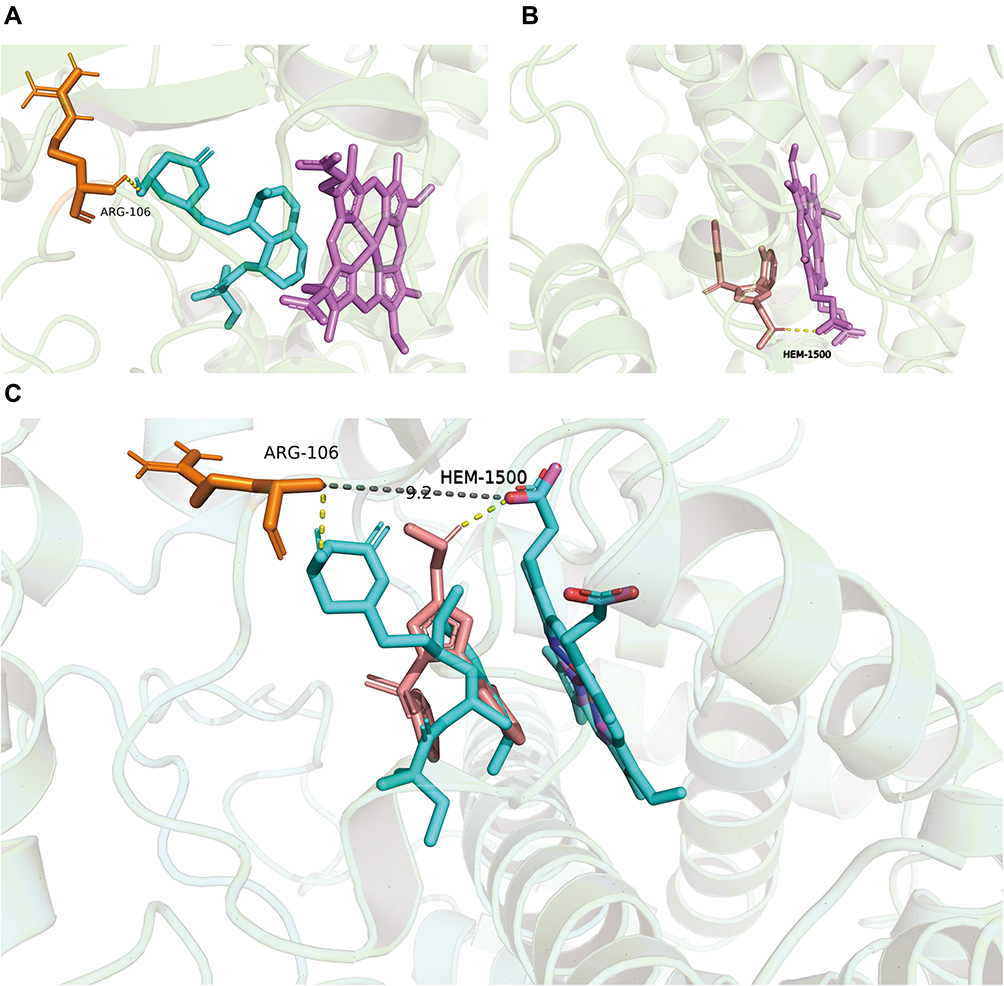 |
Figure 7 Molecular docking scheme of simvastatin and vonoprozan. (A) Action site between simvastatin and CYP3A4 via hydrogen bonding; (B) Action site between vonoprazan and CYP3A4 via hydrogen bonding; (C) Aligned docking results of the two groups. |
Discussion
Traditional PPIs are widely used for the treatment of gastric acid-related diseases, however, a series of deficiencies have been reported recently, such as instability under acidic conditions, relatively slow onset of action, and unstable and short duration of acid-suppressive effects. Specially, traditional PPIs are mainly metabolized by the hepatic CYP2C19, and about 63.75% of the Chinese population are poor metabolizers who carry the typical defective alleles CYP2C19*2 or CYP2C19*3. These two variants can significantly reduce the drug metabolic activity of CYP2C19, and that is the main reason for the individual differences in pharmacokinetics of PPIs in the oriental Asian population.16,17 In contrast, vonoprazan is mainly metabolized by CYP3A4 and is more stable than traditional PPIs that exhibits a promising prospect in clinic, especially for Asian populations.18
The main advantages of vonoprazan over conventional PPIs include: (1) Vonoprazan has stronger and more lasting anti-gastric acid secretion effect than common PPIs. As a lipophilic weak base with high pKa value (9.3), vonoprazan can immediately be protonated when exposing it to the acidic environment. Due to the highly selective and slow dissociation rate for binding to H+-K+-ATPase in the range of weak acid to neutral pH, vonoprazan exhibited 358–2357 times greater H+-K+-ATPase activity than that of lansoprazole. It is not only less affected by pH value, but also can better control of nocturnal acid secretion; (2) Vonoprazan absorbs faster than traditional PPIs, and it can reach maximum plasma concentration within 1.5–2.0 hours of oral administration; (3) Vonoprazan has shorter onset time than conventional PPIs, with acid-suppressive effects occurring within 24 hours of administration of 20 mg; (4) Vonoprazan exhibits longer half-life of approximately 7.7 hours, compared with that of 1–1.5 hours for conventional PPIs; (5) Vonoprazan has high stability, whereas the activation of conventional PPIs is acid-dependent; (6) Vonoprazan has higher population tolerance.19–22
In spite of the advantages listed above, vonoprazan still has some potential to interact with other drugs, especially for CYP3A4 mediated drugs. As the most highly expressed metabolic enzyme in the liver, CYP3A4 is involved in the in vivo metabolism of most of the clinically used drugs. It has been reported that many drugs can also inhibit or induce changes in CYP3A4 activity or expression, thus cause drug toxicity reactions or make the treatment ineffective.23,24 However, little is known about the drug-drug interactions between vonoprazan and other drugs metabolized by CYP3A4.
In this study, the influence of simvastatin on the metabolism of vonoprazan was investigated for the first time. Our data indicated that simvastatin could inhibit the metabolism of vonoprazan in a mixed model of non-competitive and competitive inhibition in vitro (Figure 5). However, in vivo experiments showed that plasma concentration of vonoprazan was decreased and its clearance was increased significantly in the simvastatin pretreated group. These data inferred that simvastatin could significantly induce the metabolism of vonoprazan in rats. Wang et al also reported similar results when they investigated the interactions between simvastatin and sinomenine. They found that simvastatin could inhibit the metabolism of sinomenine in vitro, while multiple doses of simvastatin could induce the metabolism of sinomenine In vivo.25 They proved that CYP3A1/2 was responsible for the metabolism of sinomenine in rats and the mRNA and protein expression levels of CYP3A1/2 in rat livers were significantly increased in the simvastatin pretreated group.25 Because CYP3A1/2 in rats is highly homologous to CYP3A4 in humans, we believe that the induction effect of simvastatin to vonoprazan metabolism in rats might also be due to the higher expression level of CYP3A1/2 in multidose simvastatin pretreated animals.
In addition to simvastatin, some other statins like atorvastatin and lovastatin were also reported to increase the enzymatic activity of CYPs to different degrees, in which the capacity of this induction is CYP2C8>CYP3A4>CYP2C9>CYP2C19~CYP2D6.26 Schuetz and Kocarek also reported the induction of CYP3A4 by simvastatin. They found that the expression levels of CYP3A4 mRNA and protein were increased around 24–36 times and 6–20 times, respectively, after taking some statins like simvastatin 24 hours later.27,28 The explanation for the induction effect of simvastatin is that it can affect lipids metabolism and may trigger the activation of pregnane X receptor PXR, that is the main nuclear receptor mediating the effects of exogenous drugs on CYP3A4 expression.29,30
In addition to the hypolipidemic effect, simvastatin also exhibits certain anti-inflammatory effects by reducing the mRNA expression level and decreasing the production of the famous pro-inflammatory marker interleukin-6 (IL-6).31 Machavaram et al reported that upregulated IL-6 levels could decrease the metabolic activity of CYP3A4, whereas blocking the IL-6 signaling pathway could restore the enzyme activity.32 Accordingly, we believed that simvastatin may also induce the enhancement of CYP3A4 activity in this study, by inhibiting the production of IL-6 in simvastatin group.
It was reported that statins have different affinities for the membrane transport proteins that associated with intestinal absorption, hepatic metabolism, biliary excretion, and renal clearance. Whether this association can impact the extrahepatic metabolism of vonoprazan also needs further investigation, because vonoparazan can also be metabolized by some extrahepatic pathway, such as renal clearance.33–35
What is noteworthy is that, in clinic, both simvastatin and vonoprazan are well generally tolerated and most of the adverse reactions are mild and transient if take them solely. However, data in this study indicated that simvastatin exhibited strong inhibitory effect on the metabolism of vonoprazan, and some of the side effects of vonoprazan in digestive system might be stronger when co-administrated these two drugs together in clinic, such as diarrhea, constipation, nausea and abdominal distension. In addition, diseases of other systems such as headache, rash, edema, eosinophilia, and muscle injury may also be present rarely.
Conclusion
In vitro incubation experiments indicated that simvastatin had a metabolic inhibitory effect on vonoprazan. However, In vivo pharmacokinetic experiments in rats revealed that multiple doses of simvastatin could accelerate the metabolism of vonoprazan in rats. Considering for the wide usage of simvastatin and bright prospects of vonoprazan, our data suggest that the interactions between vonoprazan and simvastatin may need to be considered when they are combined clinically, so as to avoid or reduce the occurrence of some adverse reactions.
Acknowledgments
The authors are grateful to the members of Beijing Hospital and Lishui People’s Hospital for their advice and support.
Funding
This work was supported by the National Key R&D Program of China (2020YFC2008301), the National Natural Science Foundation of China (81971323, 81570307), Public Welfare Technology Research Funding Project of Lishui (2020GYX23), Public Welfare Technology Research Funding Project of Zhejiang (LGF21H310002), Key Research and Development Project of Lishui (2021ZDYF13 & 2021ZDYF15) and the CAMS Innovation Fund for Medical Sciences (2021-I2M-1-050).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tripathi DM, Vilaseca M, Lafoz E, et al. Simvastatin prevents progression of acute on chronic liver failure in rats with cirrhosis and portal hypertension. Gastroenterology. 2018;155(5):1564–1577. doi:10.1053/j.gastro.2018.07.022
2. Balasubramanian R, Maideen NMP. HMG-CoA reductase inhibitors (Statins) and their drug interactions involving CYP enzymes, P-glycoprotein and OATP transporters - an overview. Curr Drug Metab. 2021;22:328–341. doi:10.2174/1389200222666210114122729
3. Choi DH, Li C, Choi JS. Effects of simvastatin on the pharmacokinetics of verapamil and its main metabolite, norverapamil, in rats. Eur J Drug Metab Pharmacokinet. 2009;34(3–4):163–168. doi:10.1007/BF03191168
4. Gao J, Ren H, Feng Z, et al. Effects of multidose simvastatin co-administration on pharmacokinetic profile of apatinib in rats by UPLC-MS/MS. Xenobiotica. 2020;50(9):1115–1120. doi:10.1080/00498254.2020.1740952
5. Garnock-Jones KP. Vonoprazan: first global approval. Drugs. 2015;75(4):439–443. doi:10.1007/s40265-015-0368-z
6. Yoneyama T, Teshima K, Jinno F, et al. A validated simultaneous quantification method for vonoprazan (TAK-438F) and its 4 metabolites in human plasma by the liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1015–1016:42–49. doi:10.1016/j.jchromb.2016.01.051
7. Yamasaki H, Kawaguchi N, Nonaka M, et al. In vitro metabolism of TAK-438, vonoprazan fumarate, a novel potassium-competitive acid blocker. Xenobiotica. 2017;47:1027–1034. doi:10.1080/00498254.2016.1203505
8. Kong W-M, Sun B-B, Wang Z-J, et al. Physiologically based pharmacokinetic–pharmacodynamic modeling for prediction of vonoprazan pharmacokinetics and its inhibition on gastric acid secretion following intravenous/oral administration to rats, dogs and humans. Acta Pharmacol Sin. 2020;41(6):852–865. doi:10.1038/s41401-019-0353-2
9. Wang Y, Wang C, Wang S, et al. Cytochrome P450-based drug-drug interactions of vonoprazan in vitro and in vivo . Front Pharmacol. 2020;11:53. doi:10.3389/fphar.2020.00053
10. Shen J, Wang B, Wang S, et al. Effects of voriconazole on the pharmacokinetics of vonoprazan in rats. Drug Des Devel Ther. 2020;14:2199–2206. doi:10.2147/DDDT.S255427
11. Sevrioukova IF, Poulos TL. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci USA. 2017;114(3):486–491. doi:10.1073/pnas.1616198114
12. Bitencourt-Ferreira G, Pintro VO, de Azevedo WF. Docking with AutoDock4. In: de Azevedo WF Jr, editor. Docking Screens for Drug Discovery. New York: Springer New York; 2019:125–148.
13. Javaid S, Zafar H, Atia Tul W, et al. Identification of new lead molecules against anticancer drug target TFIIH subunit P8 using biophysical and molecular docking studies. Bioorg Chem. 2021;114:105021. doi:10.1016/j.bioorg.2021.105021
14. Schlessinger A, Welch MA, van Vlijmen H, Korzekwa K, Swaan PW, Matsson P. Molecular modeling of drug-transporter interactions-an international transporter consortium perspective. Clin Pharmacol Ther. 2018;104(5):818–835. doi:10.1002/cpt.1174
15. Patil R, Das S, Stanley A, Yadav L, Sudhakar A, Varma AK. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS One. 2010;5(8):e12029. doi:10.1371/journal.pone.0012029
16. Hu LM, Dai DP, Hu GX, et al. Genetic polymorphisms and novel allelic variants of CYP2C19 in the Chinese Han population. Pharmacogenomics. 2012;13:1571–1581. doi:10.2217/pgs.12.141
17. Desta Z, Zhao X, Shin JG, et al. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet. 2002;41(12):913–958. doi:10.2165/00003088-200241120-00002
18. Otake K, Sakurai Y, Nishida H, et al. Characteristics of the novel potassium-competitive acid blocker vonoprazan fumarate (TAK-438). Adv Ther. 2016;33(7):1140–1157. doi:10.1007/s12325-016-0345-2
19. Qiao Y, Zhao J, Yue X, et al. Study on pharmacokinetics and bioequivalence of Vonoprazan pyroglutamate in rats by liquid chromatography with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1059:56–65. doi:10.1016/j.jchromb.2017.05.013
20. Hori Y, Matsukawa J, Takeuchi T, et al. A study comparing the antisecretory effect of TAK-438, a novel potassium-competitive acid blocker, with lansoprazole in animals. J Pharmacol Exp Ther. 2011;337(3):797–804. doi:10.1124/jpet.111.179556
21. Martinucci I, Blandizzi C, Bodini G, et al. Vonoprazan fumarate for the management of acid-related diseases. Expert Opin Pharmacother. 2017;18(11):1145–1152. doi:10.1080/14656566.2017.1346087
22. Kogame A, Takeuchi T, Nonaka M, et al. Disposition and metabolism of TAK-438 (vonoprazan fumarate), a novel potassium-competitive acid blocker, in rats and dogs. Xenobiotica. 2017;47(3):255–266. doi:10.1080/00498254.2016.1182667
23. Ohno Y, Hisaka A, Ueno M, et al. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47(10):669–680. doi:10.2165/00003088-200847100-00004
24. Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–696. doi:10.2165/00003088-200746080-00005
25. Wang Y, Jin Y, Yun X, et al. Co-administration with simvastatin or lovastatin alters the pharmacokinetic profile of sinomenine in rats through cytochrome P450-mediated pathways. Life Sci. 2018;209:228–235. doi:10.1016/j.lfs.2018.08.012
26. Feidt DM, Klein K, Hofmann U, et al. Profiling induction of cytochrome p450 enzyme activity by statins using a new liquid chromatography-tandem mass spectrometry cocktail assay in human hepatocytes. Drug Metab Dispos. 2010;38(9):1589–1597. doi:10.1124/dmd.110.033886
27. Schuetz EG, Schuetz JD, Strom SC, et al. Regulation of human liver cytochromes P-450 in family 3A in primary and continuous culture of human hepatocytes. Hepatology. 1993;18(5):1254–1262. doi:10.1002/hep.1840180535
28. Kocarek TA, Dahn MS, Cai H, et al. Regulation of CYP2B6 and CYP3A expression by hydroxymethylglutaryl coenzyme A inhibitors in primary cultured human hepatocytes. Drug Metab Dispos. 2002;30(12):1400–1405. doi:10.1124/dmd.30.12.1400
29. Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98(6):3369–3374. doi:10.1073/pnas.051551698
30. Jones SA, Moore LB, Shenk JL, et al. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14(1):27–39. doi:10.1210/mend.14.1.0409
31. Berthold HK, Berneis K, Mantzoros CS, et al. Effects of simvastatin and ezetimibe on interleukin-6 and high-sensitivity C-reactive protein. Scand Cardiovasc J Suppl. 2013;47(1):20–27. doi:10.3109/14017431.2012.734635
32. Machavaram KK, Almond LM, Rostami-Hodjegan A, et al. A physiologically based pharmacokinetic modeling approach to predict disease-drug interactions: suppression of CYP3A by IL-6. Clin Pharmacol Ther. 2013;94(2):260–268. doi:10.1038/clpt.2013.79
33. Hirota T, Fujita Y, Ieiri I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin Drug Metab Toxicol. 2020;16(9):809–822. doi:10.1080/17425255.2020.1801634
34. Echizen H. The first-in-class potassium-competitive acid blocker, vonoprazan fumarate: pharmacokinetic and pharmacodynamic considerations. Clin Pharmacokinet. 2016;55(4):409–418. doi:10.1007/s40262-015-0326-7
35. Gertz M, Houston JB, Galetin A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos. 2011;39(9):1633–1642. doi:10.1124/dmd.111.039248
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.