")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Effects of resveratrol on P-glycoprotein and cytochrome P450 3A in vitro and on pharmacokinetics of oral saquinavir in rats
Authors Li JP, Liu Y, Zhang JR, Yu XT, Wang XL, Zhao LB
Received 2 August 2016
Accepted for publication 8 September 2016
Published 15 November 2016 Volume 2016:10 Pages 3699—3706
DOI https://doi.org/10.2147/DDDT.S118723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Jiapeng Li,1,2 Yang Liu,2 Jingru Zhang,1,2 Xiaotong Yu,1,2 Xiaoling Wang,1 Libo Zhao1
1Department of Pharmacy, Beijing Children’s Hospital, Capital Medical University, 2Department of Pharmacy Administration and Clinical Pharmacy, School of Pharmaceutical Sciences, Peking University, Beijing, People’s Republic of China
Background: The intestinal cytochrome P450 3A (CYP 3A) and P-glycoprotein (P-gp) present a barrier to the oral absorption of saquinavir (SQV). Resveratrol (RESV) has been indicated to have modulatory effects on P-gp and CYP 3A. Therefore, this study was to investigate the effects of RESV on P-gp and CYP 3A activities in vitro and in vivo on oral SQV pharmacokinetics in rats.
Methods: In vitro, intestinal microsomes were used to evaluate RESV effect on CYP 3A-mediated metabolism of SQV; MDR1-expressing Madin–Darby canine kidney (MDCKII-MDR1) cells were employed to assess the impact of RESV on P-gp-mediated efflux of SQV. In vivo effects were studied using 10 rats randomly assigned to receive oral SQV (30 mg/kg) with or without RESV (20 mg/kg). Serial blood samples were obtained over the following 24 h. Concentrations of SQV in samples were ascertained using high-performance liquid chromatography-tandem mass spectrometry analysis.
Results: RESV (1–100 µM) enhanced residual SQV (% of control) in a dose-dependent manner after incubation with intestinal microsomes. RESV (1–100 µM) reduced the accumulation of SQV in MDCKII-MDR1 cells in a concentration-dependent manner. A double peaking phenomenon was observed in the plasma SQV profiles in rats. The first peak of plasma SQV concentration was increased, but the second peak was reduced by coadministration with RESV. The mean AUC0–∞ of SQV was slightly decreased, with no statistical significance probably due to the high individual variation.
Conclusion: RESV can alter the plasma SQV concentration profiles, shorten the Tmax of SQV. RESV might also cause a slight decrease tendency in the SQV bioavailability in rats.
Keywords: resveratrol, saquinavir, P-glycoprotein, CYP 3A, pharmacokinetic
Introduction
The standard treatment for the human immunodeficiency virus (HIV)/acquired immunodeficiency disease recommended by the World Health Organization includes at least three drugs in combination: two nucleoside reverse transcriptase inhibitors and one protease inhibitor (PI) or non-nucleoside reverse transcriptase inhibitor. This treatment is commonly termed highly active antiretroviral therapy.1 Saquinavir (SQV), as with other PIs, exerts its pharmacological activities by binding to the active site of viral protease enzyme, leading to the formation of immature virus particles. Although SQV is highly effective in blocking HIV-1 infection, SQV efficacy in clinical practice is limited, mainly due to its low and highly variable oral bioavailability, which is ~4% with a coefficient of variation (CV) >100%.2,3 Multiple factors contribute to the low oral bioavailability of SQV, including poor solubility characteristics, extensive first pass metabolism and P-glycoprotein (P-gp) transport. After oral administration, SQV is extensively metabolized before systemic absorption by cytochrome P450 3A (CYP 3A) in the small intestine.4 In addition, SQV is a substrate for P-glycoprotein (P-gp) transporters with high affinity.5 The P-gp on intestine epithelia actively extrudes SQV from enterocytes back into the intestinal lumen, leading to a significant reduction in systematic plasma drug levels.5 Thus, coadministration of SQV with drug or diet that may inhibit or induce CYP 3A or/and P-gp activities should be viewed with caution.
Resveratrol (RESV) is a kind of stilbenes mainly found in red grapes, berries, and peanuts.6 RESV and its potential health benefits have attracted increasing interest since red wine consumption was found to be associated with low incidence of cardiovascular disease in the French despite a high-fat diet, which is popularly known as the “French paradox” phenomenon.7,8 During the past few years, RESV has been reported to exert various beneficial pharmacological effects, such as anti-inflammatory,9 anti-atherogenic,10 and anticarcinogenic11 activities. In addition, RESV exhibits anti-HIV-1 activity.12 Recent results suggest a synergistic inhibition of HIV-1 by the combination of RESV and decitabine.13
Given its multiple beneficial pharmacological effects, introducing RESV as a supplemental agent into HIV-1 therapy seems reasonable. However, RESV has been indicated to modulate P-gp and CYP 3A activities,14,15 suggesting it may alter the pharmacokinetics (PK) or bioavailability of concomitant drugs that are P-gp or/and CYP 3A substrates, such as SQV. Whether, and to what extent, RESV influences intestinal P-gp-mediated efflux or CYP 3A-mediated metabolism of SQV remains unclear. To our knowledge, no data are available concerning the impacts of RESV on the PK of SQV. Therefore, we conducted this study to investigate the effects of RESV on P-gp and CYP 3A activities and its impacts on the PK of SQV in rats.
Materials and methods
Chemicals and reagents
RESV (purity >98%) was purchased from FeiYu Biotechnology Corporation (Nantong, People’s Republic of China). SQV was purchased from USP (Rockville, MD, USA). Chemical structures of RESV and SQV are shown in Figure 1. Pooled Sprague-Dawley rat (male) intestine microsomes were obtained from Xenotech LLC (Kansas City, KS, USA). Verapamil (VER) fluid injection (50 mL/branch) was obtained from HeFeng Pharmaceutical Co. Ltd (Shanghai, People’s Republic of China). BCA Protein Assay Kit was purchased from Solarbio (Beijing, People’s Republic of China). Ketoconazole (KET) (purity >99%) was purchased from the National Institutes for Food and Drug Control (Beijing, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium was from Gibco (Shanghai, People’s Republic of China). Phosphate-buffered saline (PBS) and other reagents or solvents used were commercially available and of reagent grade.
 | Figure 1 Chemical structures of resveratrol (A) and saquinavir (B). |
Animals
Male Sprague-Dawley rats were purchased from the Academy of Military Medical Sciences (Beijing, People’s Republic of China). Rats were housed in well-ventilated cages at room temperature (24°C±2°C) and 40%–60% relative humidity. Rats were kept on a 12 h night cycle (lights on from 6 am to 6 pm). The animals were housed with free access to laboratory food and water ad libitum. Rats weighing 300–350 g (8–9 weeks old) were used in the PK study.
In vitro metabolism experiment
An in vitro experiment was conducted to assess the effect of RESV on intestinal CYP 3A-mediated metabolism of SQV. The procedures used are similar to previously reported methods.16,17 All incubations were performed in triplicate in 100 μL of 100 mM PBS (pH 7.4) containing 1 mM nicotinamide adenine dinucleotide phosphate, 0.8 mg/mL intestinal microsomal protein and modulator (10 μM KET, or 1, 10, or 100 μM RESV). The microsomes (replaced with equal volume of buffer in the control group) and KET or RESV were mixed and incubated at 37°C for 5 min. Nicotinamide adenine dinucleotide phosphate was then added to initiate the reaction, and the mixture further incubated at 37°C for 15 min. The reaction was terminated by the addition of 100 μL ice-cold acetonitrile. Residual SQV was measured with the high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) analysis method described in the drug assay section below.
In vitro cell accumulation experiment
Cell accumulation experiment to assess the effect of RESV on P-gp mediated efflux of SQV. MDR1-expressing Madin–Darby canine kidney (MDCKII-MDR1) cells were kindly provided by Prof Su Zeng. MDCKII-MDR1 cells were cultured in Dulbecco’s Modified Eagle’s culture Medium supplemented with 10% fetal bovine serum (Gibco, USA), 100 units/mL penicillin, and 100 mg/mL streptomycin. Cells were incubated at 37°C and 5% CO2 atmosphere.
MDCKII-MDR1 cells were incubated with 50 μΜ SQV in the absence or presence of various concentrations (1, 10, 33, and 100 μΜ) of RESV, the vehicle (0.5% dimethylsulfoxide), or the positive control (50 μΜ VER) at 37°C for 4 h. Cell viability was assessed through a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay.18 Results showed that agents at concentrations adopted in this study did not exert significant damage on MDCKII-MDR1 cell variability. After 4 h incubation, cells were washed with cold PBS, suspended in 1 mL distilled water, and then collected into a tube. Cells in each tube were broken using an ultrasonic cell disruption system at 0°C, and centrifuged (13,400× g for 5 min at 4°C). The supernatant fraction was then transferred into a tube and frozen at −20°C until analysis. The amount of protein in cells was measured using a BCA Protein Assay Kit (Solarbio). The intracellular accumulation of SQV was quantified as the concentration ratio (μg/mg protein), which was calculated by dividing the apparent uptake amount of SQV by protein content.
In vivo PK experiment
An in vivo experiment was conducted to assess the effects of RESV on SQV PK profiles in rats. The study protocol was approved, and adhered to the guidelines of the Animal Ethics Committee of Beijing Children’s Hospital. We designed the PK study based on previously reported SQV in vivo studies.19,20 Rats were fasted for 12 h prior to experiments with free access to water. They were then randomized into two groups: a control group (SQV 30 mg/kg, oral, aqueous suspension) and a RESV treatment group (SQV 30 mg/kg plus RESV 20 mg/kg, oral, aqueous suspension). SQV was suspended in solvent (20% ethanol, 30% propylene glycol, and 50% saline) at a concentration of 6 mg/mL; RESV was suspended in saline with 30% polyethylene glycol 400 at 20 mg/mL concentration. Each conscious animal was orally administrated an appropriate volume of suspension (30 mg/kg SQV or 30 mg/kg SQV +20 mg/kg RESV). Blood samples were taken from the posterior orbital venous plexus into heparinized Eppendorf tubes at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h after drug administration. Rats were given food 4 h after blood samples were obtained. Blood samples were immediately centrifuged and the obtained plasma samples were stored at −20°C until the time for LC-MS/MS analysis.
Drug assay
We used a validated HPLC-MS/MS analysis method21 to measure the concentrations of SQV in samples from the metabolism study, cell accumulation study, and PK study. Briefly, ritonavir was selected as the internal standard. The internal standard (200 μL, 20 ng/mL) was first added into the 20 μL buffer sample or 50 μL plasma sample prior to further extraction with 3 mL methyl tert-butyl ether. The upper layer (2.8 mL) was carefully transferred into another clean tube and then evaporated to dryness at 40°C under a gentle stream of nitrogen. The dry residue was reconstituted with 200 μL mobile phase (CH3CN/H2O, 76.65:23.35 [v/v], containing 0.2 mM NH4COOH). After vortex mixing, the extracted samples were centrifuged (13,400× g for 5 min at 4°C) and the supernatant (10 μL) was injected into the HPLC/MS-MS system.
The HPLC/MS-MS analysis was carried out on a Restek C18 (150×2.1 mm ID) column (Bellefonte, PA, USA) with the mobile phase at a flow rate of 300 μL per minute. SQV (retention time, 3.63 min) and ritonavir (retention time, 2.67 min) were analyzed by fragmentation of the parent compound and quantification of resulting fragments. The monitoring ions of SQV and ritonavir were m/z 671.4/570.4 and m/z 721.4/296.2, respectively. The lower limit of quantification of SQV was 1 ng/mL in extracted samples. The precision and accuracy of the quality control samples at three concentration levels (low, middle, and high: 3, 50, and 150 ng/mL, respectively) were within 15% relative standard deviation and 15% relative error, respectively.
PK analysis
The PK parameters of SQV in plasma were determined by noncompartmental method using WinNonlin version 6.4 (Pharsight Corporation, Mountain View, CA, USA). The area under the plasma concentration–time curves (AUC) from time 0 to 24 h (AUC0–t) was determined using the linear trapezoidal rule. The terminal elimination half-life (t1/2) was calculated as ln2/λz using the slope (λz) from a linear regression analysis of the terminal phase of the plasma concentration–time curve on a semilog scale. The AUC from time zero to infinity (AUC0–∞) was calculated using the linear trapezoidal rule, formulated as AUC0–∞ = AUC0–t + Ct/λz, where Ct was the last measured concentration in plasma. The apparent systemic clearance (CL/F) was calculated as: CL/F = dose/AUC0–∞.
Statistical analysis
The data for analysis of variance and Student’s t-test were normally distributed as assessed by Shapiro–Wilk test. One-way analysis of variance with Duncan’s multiple range test was used for statistical comparisons for the in vitro studies. For the in vivo study, AUC0–∞, Cmax, t1/2, CL/F, and MRT0–∞ were transformed into a logarithmic form and analyzed with independent-samples Student’s t-test; the time to Cmax, Tmax, was analyzed with Wilcoxon rank sum test. Statistical analyses were done with IBM SPSS Statistics for Windows, version 20.0 (IBM Corp, Armonk, NY, USA). A P-value of <0.05 was considered significant.
Results
RESV inhibits intestinal CYP 3A-mediated metabolism of SQV
The effects of RESV at various concentrations on the residual SQV presence after 15 min of metabolism are shown in Figure 2. The residual SQV in 0 μM RESV group (61.5% of control) was significantly lower than those in control (without CYP 3A) and KET (CYP 3A inhibitor) group (P<0.01). Residual SQV increased with RESV in a dose-dependent manner, with 67.5%, 70.7%, and 94.3% of control in the presence of 1, 10, and 100 μΜ RESV, respectively, suggesting a concentration-dependent inhibitory effect on intestinal CYP 3A-mediated SQV metabolism.
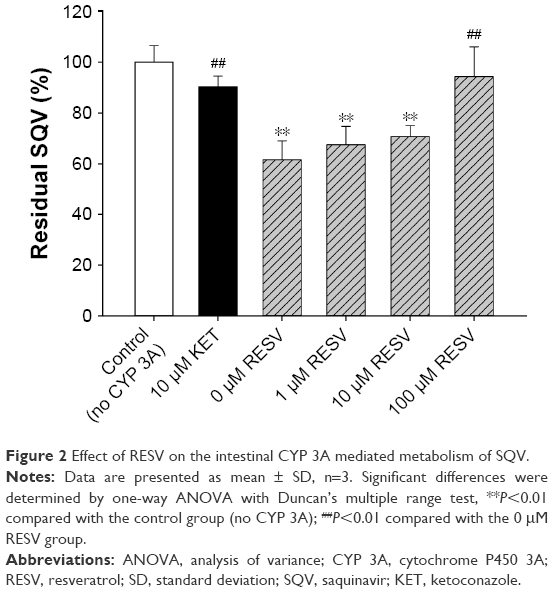 | Figure 2 Effect of RESV on the intestinal CYP 3A mediated metabolism of SQV. |
RESV stimulates P-gp mediated efflux of SQV
The effects of RESV at various concentrations on accumulations of SQV in MDCKII-MDR1 cells are shown in Figure 3. The treatment of 50 μΜ VER, a P-gp inhibitor served as a positive control; intracellular accumulation of SQV increased by nearly 200%, implying the experiment provides a reliable in vitro model for assessing P-gp regulatory effects. Significant decreases of SQV intracellular concentration were observed in the presence of 10 (P<0.05), 33 (P<0.01), and 100 μΜ RESV (P<0.01), a percent change with RESV dose of −58.1%, −71.2%, and −75.5%, respectively. This finding indicates that RESV stimulates P-gp-mediated efflux of SQV in a concentration-dependent manner.
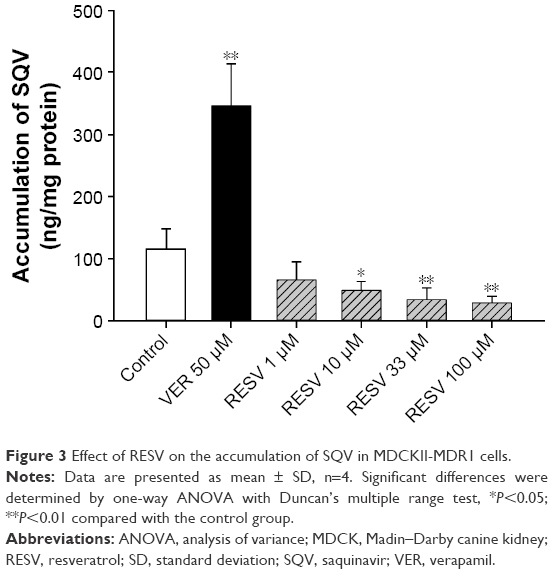 | Figure 3 Effect of RESV on the accumulation of SQV in MDCKII-MDR1 cells. |
The time profiles of the intracellular accumulation of SQV in the presence of RESV (33 μΜ) or VER (50 μΜ) are illustrated in Figure 4. The accumulation time profile of SQV reached steady state at 4 h after initiation of incubation. Compared with controls, intracellular concentrations of SQV decreased (P<0.01) in RESV treatments continuously after 2 h co-incubation, indicating a time-dependent stimulatory effect on the P-gp-mediated efflux of SQV.
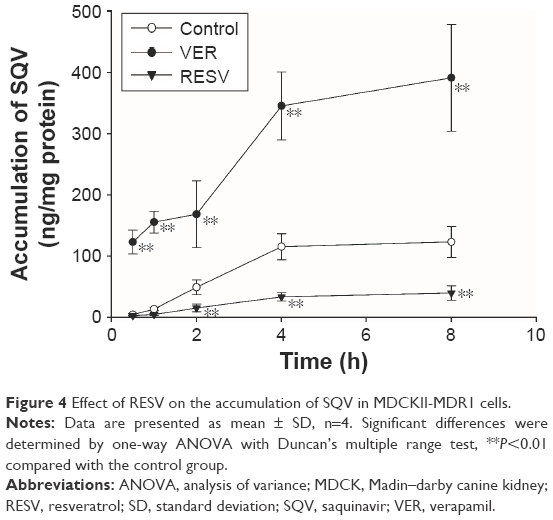 | Figure 4 Effect of RESV on the accumulation of SQV in MDCKII-MDR1 cells. |
RESV alters SQV PK profiles in rats
In vivo plasma concentration–time profiles of SQV in rats after receiving a single oral dose of SQV (30 mg/kg), with or without RESV (20 mg/kg), are shown in Figure 5. Wide interindividual variability was observed. In addition, a double peak phenomenon was observed, with the first peak in plasma levels reached at ~0.5 (control group) or 0.25 h (RESV treatment group), and the second peak at ~4 h (both groups). The profile of plasma SQV concentrations was significantly affected by coadministration of RESV. The first peak in the plasma drug profile in RESV treatment group was significantly higher than that in the control group, while the second one was significantly lower. The SQV plasma concentration curve declined more rapidly in the presence of RESV. (Composite curves graphed plasma SQV profiles for the control and treatment groups were displayed in Supplementary materials.)
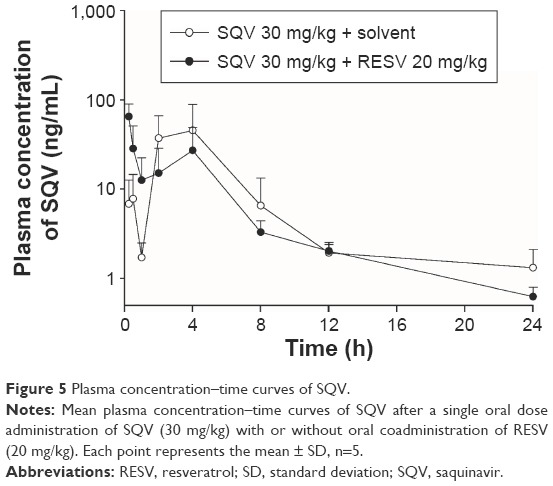 | Figure 5 Plasma concentration–time curves of SQV. |
The PK parameters of SQV are summarized in Table 1. Oral coadministration of RESV (20 mg/kg) decreased mean AUC0–∞ of SQV by ~31%, but this change had no statistical significance (P>0.05). The mean CL/F of SQV was increased by ~51% with no statistical significance (P>0.05). The Tmax of SQV was significantly shortened (from ~4 h to ~0.25 h, P<0.01) by coadministration with RESV, but Cmax was not significantly affected (P>0.05).
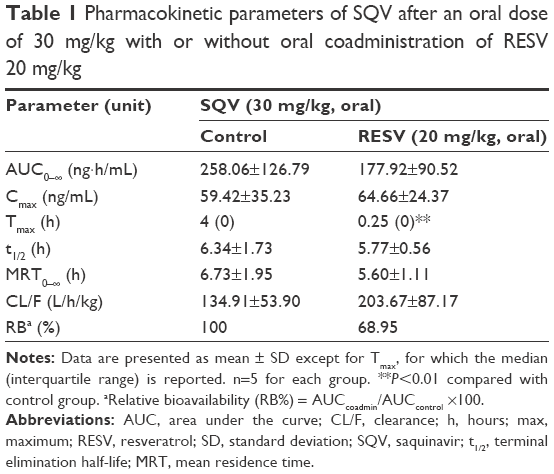 | Table 1 Pharmacokinetic parameters of SQV after an oral dose of 30 mg/kg with or without oral coadministration of RESV 20 mg/kg |
Discussion
This study includes both in vitro and in vivo experiments to explore the interaction between RESV and SQV. In vitro, RESV inhibited metabolism of SQV by rat intestinal CYP 3A, which is fairly consistent with the findings of previous studies.14,15,22,23 On the other hand, RESV showed stimulatory effects on the P-gp-mediated efflux of SQV. In vivo, RESV led to a greater peak in plasma SQV in the first hour after dosing, but a decreased level of SQV relative to the control after 2 h.
In order to investigate the impact of RESV on intestinal P-gp-mediated efflux, another major barrier for SQV absorption, we evaluated the accumulation of SQV in MDCKII-MDR1 cells in the presence or absence of RESV. MDCKII-MDR1 cells are MDCK cells transfected with the human MDR1 gene; they express P-gp but do not express other efflux transporters, such as BCRP and MRP2.24 MDCKII-MDR1 cells are commonly employed as an in vitro model for intestinal epithelium.25–27 Results in the accumulation study suggest that RESV stimulates the P-gp-mediated efflux of SQV.
Findings of the effect of RESV on P-gp from previous studies are controversial. For example, Choi et al found that RESV significantly reduced rhodamine123 efflux via P-gp in MCF-7/ADR cells that overexpress P-gp, and significantly increased the drug exposure of orally administered nicardipine in rats.28 In contrast, Yang et al reported that RESV exerted a stimulatory effect on P-gp, resulting in a reduction of cyclosporine oral bioavailability in rats.15 These findings suggest that RESV exerts stimulatory/inhibitory effects on P-gp depending on the substrate. It seems likely that the substrate-dependent modulation results from complicated interactions between substrates involving P-gp substrate binding sites. The binding sites for any particular P-gp substrate may be the same or distinct from those for another substrate.24,29–32 This may lead to competition between substrates that bind identical sites or to a positive cooperative effect between substrates that bind different sites.32,33 In this case, the latter could explain the stimulation of RESV on P-gp-mediated efflux of SQV.
In the PK study, rats were used as the animal model mainly due to the similarity in basic intestinal structures and physiology of rats and humans.34 We observed a high interindividual variability in the SQV plasma concentration–time profiles, which is generally consistent with previous PK studies in rats.35,36 Moreover, we noticed a double peak phenomenon in plasma SQV profiles in the presence and absence of RESV. Similar double peaking phenomena were also documented in multiple SQV PK studies involving rats35,37,38 and humans.39–41 Several physiological mechanisms can generate the double peaking phenomenon, including enterohepatic recycling, gastric emptying, small intestinal transit, and site-specific absorption.42 However, since no double peak has been observed after intravenous dosing of SQV,35,39,40 the second peak cannot be explained by enterohepatic recycling. Similarly, because the increase phase of SQV plasma profiles started earlier than food intake, it seems unlikely that gastric emptying plays an important role. Here, we speculate that the double peak phenomenon mainly results from site-specific intestinal absorption. SQV is one of the class II compounds categorized in Biopharmaceutics Classification System, with low solubility and high permeability.43 This property suggests that dissolution is a rate-limiting step for the oral absorption of SQV. Thus, the second peak in the SQV plasma profiles can be due to the increase of SQV dissolution in the more acidic environment of the colon compared with ileum.44 In addition, the regional differences in P-gp expression and activity along the intestine may play a significant role in causing the double peak.45 P-gp levels increase progressively from proximal to distal region in human or rodent small intestine.43,46 The ileum region shows significantly higher P-gp function than in other region.43 Therefore, we infer that after oral administration, SQV was absorbed rapidly in the proximal small intestine, and then it was transported back into the intestine lumen by P-gp at the ileum, resulting in a rapid rise of plasma SQV levels initially, followed by a decrease. After that, absorption increased again at the colon where P-gp expression is lower, leading to a second peak in the plasma SQV levels.
Overall, the mean AUC0–∞ of SQV was decreased by nearly 30% by coadministration with RESV, but this change had no statistical significance, which might be partially due to the high variation between individuals. The impact of RESV on SQV PK profiles can be attributed to the effects of RESV on intestinal CYP 3A and P-gp. The distribution of CYP 3A in the small intestine of rats is markedly different from that of P-gp. CYP 3A is one of the two most abundant P450 isoforms (the other one is CYP 2B1) in the duodenum and jejunum of rat intestines but it is not present in either the ileum or the colon.47 It is most likely that RESV inhibits the CYP 3A mediated metabolism of SQV, leading to an increase of SQV absorption in the proximal intestine. In contrast, RESV reduces the absorption of SQV in distal intestine (ileum and colon) by stimulating P-gp-mediated efflux of SQV. Overall, the positive effect of RESV on the absorption of SQV in the proximal intestine is offset by its negative effect in the distal intestine, eventually resulting in a nonsignificant change in the SQV oral bioavailability.
Conclusion
This study demonstrates that RESV can inhibit intestinal CYP 3A-mediated SQV metabolism and stimulate P-gp-mediated efflux of SQV. RESV can alter the SQV plasma concentration profiles and shorten the Tmax of SQV. RESV also leads to a decrease tendency in the SQV oral bioavailability, but this change has no statistical significance probably because of the high individual variation. Further clinical investigations are required to assess the benefit and risk of the concomitant use of SQV with RESV.
Acknowledgments
We gratefully acknowledge the assistance of Prof Su Zeng and Dr Xi Chen.
This study was funded by a grant from the National Natural Science Foundation of China (No 81102877).
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Consolidation Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection. Geneva, Switzerland. World Health Organization; 2013. | ||
Kitchen VS, Skinner C, Ariyoshi K, et al. Safety and activity of saquinavir in HIV infection. Lancet. 1995;345(8955):952–955. | ||
Noble S, Faulds D. Saquinavir. A review of its pharmacology and clinical potential in the management of HIV infection. Drugs. 1996;52(1):93–112. | ||
Fitzsimmons ME, Collins JM. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4 potential contribution to high first-pass metabolism. Drug Metab Dispos. 1997;25(2):256–266. | ||
Kim AE, Dintaman JM, Waddell DS, Silverman JA. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J Pharmacol Exp Ther. 1998;286(3):1439–1445. | ||
Giovinazzo G, Ingrosso I, Paradiso A, De Gara L, Santino A. Resveratrol biosynthesis: plant metabolic engineering for nutritional improvement of food. Plant Foods Hum Nutr. 2012;67(3):191–199. | ||
Sun AY, Simonyi A, Sun GY. The “French Paradox” and beyond: neuroprotective effects of polyphenols. Free Radic Biol Med. 2002;32(4):314–318. | ||
Criqui MH, Ringel BL. Does diet or alcohol explain the French paradox? Lancet. 1994;344(8939):1719–1723. | ||
Frémont L. Biological effects of resveratrol. Life Sci. 2000;66(8):663–673. | ||
Fan E, Zhang L, Jiang S, Bai Y. Beneficial effects of resveratrol on atherosclerosis. J Med Food. 2008;11(4):610–614. | ||
Jang M, Cai L, Udeani GO, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275(5297):218–220. | ||
Zhang H-S, Zhou Y, Wu M-R, Zhou H-S, Xu F. Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD(+)-dependent SIRT1 activity. Life Sci. 2009;85(13–14):484–489. | ||
Clouser CL, Chauhan J, Bess MA, et al. Anti-HIV-1 activity of resveratrol derivatives and synergistic inhibition of HIV-1 by the combination of resveratrol and decitabine. Bioorg Med Chem Lett. 2012;22(21):6642–6646. | ||
Chi Y-C, Lin S-P, Hou Y-C. A new herb–drug interaction of Polygonum cuspidatum, a resveratrol-rich nutraceutical, with carbamazepine in rats. Toxicol Appl Pharmacol. 2012;263(3):315–322. | ||
Yang S-Y, Tsai S-Y, Hou Y-C, Chao P-DL. Inductive modulation on P-glycoprotein and cytochrome 3A by resveratrol, a constituent of grapes. Food Chem. 2012;133(3):683–688. | ||
Zhuang X, Shen G, Yuan M, Li H. Investigation of the pharmacokinetic interaction between ritonavir and CMDCK, a new non-nucleoside reverse transcriptase inhibitor. Drug Res (Stuttg). 2013;63(5):237–242. | ||
Choi YH, Lee I, Lee MG. Effects of cysteine on metformin pharmacokinetics in rats with protein–calorie malnutrition: partial restoration of some parameters to control levels. J Pharm Pharmacol. 2008;60(2):153–161. | ||
Vellonen KS, Honkakoski P, Urtti A. Substrates and inhibitors of efflux proteins interfere with the MTT assay in cells and may lead to underestimation of drug toxicity. Eur J Pharm Sci. 2004;23(2):181–188. | ||
Kim S-A, Kim S-W, Choi H-K, Han H-K. Enhanced systemic exposure of saquinavir via the concomitant use of curcumin-loaded solid dispersion in rats. Eur J Pharm Sci. 2013;49(5):800–804. | ||
Vyas TK, Shahiwala A, Amiji MM. Improved oral bioavailability and brain transport of saquinavir upon administration in novel nanoemulsion formulations. Int J Pharm. 2008;347(1–2):93–101. | ||
Liang G, Li N, Ma L, et al. Effect of quercetin on the transport of ritonavir to the central nervous system in vitro and in vivo. Acta Pharm. 2016;66(1):97–107. | ||
Piver B, Berthou F, Dreano Y, Lucas D. Differential inhibition of human cytochrome P450 enzymes by ε-viniferin, the dimer of resveratrol: comparison with resveratrol and polyphenols from alcoholized beverages. Life Sci. 2003;73(9):1199–1213. | ||
Piver B, Berthou F, Dreano Y, Lucas D. Inhibition of CYP3A, CYP1A and CYP2E1 activities by resveratrol and other non volatile red wine components. Toxicol Lett. 2001;125(1):83–91. | ||
Taub ME, Podila L, Ely D, Almeida I. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: influence of cell line and modulator concentration on P-gp activity. Drug Metab Dispos. 2005;33(11):1679–1687. | ||
Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm Res. 2003;20(8):1210–1224. | ||
Soldner A, Benet LZ, Mutschler E, Christians U. Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and Caco-2 cell monolayers. Br J pharmacol. 2000;129(6):1235–1243. | ||
Liu Y, Zeng S. [Advances in the MDCK-MDR1 cell model and its applications to screen drug permeability]. Yao Xue Xue Bao. 2008;43(6):559–564. Chinese. | ||
Choi J-S, Choi B-C, Kang KW. Effect of resveratrol on the pharmacokinetics of oral and intravenous nicardipine in rats: possible role of P-glycoprotein inhibition by resveratrol. Pharmazie. 2009;64(1):49–52. | ||
Chufan EE, Kapoor K, Sim HM, et al. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PLoS One. 2013;8(12):e82463. | ||
Pajeva IK, Sterz K, Christlieb M, Steggemann K, Marighetti F, Wiese M. Interactions of the multidrug resistance modulators tariquidar and elacridar and their analogues with P-glycoprotein. ChemMedChem. 2013;8(10):1701–1713. | ||
Martinez L, Arnaud O, Henin E, et al. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014;281(3):673–682. | ||
Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250(1):130–137. | ||
Chufan EE, Sim H-M, Ambudkar SV. Chapter three – molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies. In: John DS, Toshihisa I, editors. Advances in Cancer Research. Academic Press; 2015:71–96. | ||
Jain R, Duvvuri S, Kansara V, Mandava NK, Mitra AK. Intestinal absorption of novel-dipeptide prodrugs of saquinavir in rats. Int J Pharm. 2007;336(2):233–240. | ||
Buchanan CM, Buchanan NL, Edgar KJ, et al. Pharmacokinetics of saquinavir after intravenous and oral dosing of saquinavir: hydroxybutenyl-beta-cyclodextrin formulations. Biomacromolecules. 2008;9(1):305–313. | ||
Hirunpanich V, Murakoso K, Sato H. Inhibitory effect of docosahexaenoic acid (DHA) on the intestinal metabolism of midazolam: in vitro and in vivo studies in rats. Int J Pharm. 2008;351(1–2):133–143. | ||
Pathak SM, Musmade P, Dengle S, Karthik A, Bhat K, Udupa N. Enhanced oral absorption of saquinavir with methyl-beta-cyclodextrin – preparation and in vitro and in vivo evaluation. Eur J Pharm Sci. 2010;41(3):440–451. | ||
Pathak SM, Kumar AR, Subramanian G, Udupa N. Development and validation of a reversed-phase liquid chromatographic method with fluorescence detection for the study of saquinavir pharmacokinetics in rat plasma. Anal Chim Acta. 2007;594(2):248–256. | ||
Kupferschmidt HH, Fattinger KE, Ha HR, Follath F, Krähenbühl S. Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br J Clin Pharmacol. 1998;45(4):355–359. | ||
Ha HR, Follath F, Bloemhard Y, Krähenbühl S. Determination of saquinavir in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1997;694(2):427–433. | ||
Fröhlich M, Burhenne J, Martin-Facklam M, et al. Oral contraception does not alter single dose saquinavir pharmacokinetics in women. Br J Clin Pharmacol. 2004;57(3):244–252. | ||
Davies NM, Takemoto JK, Brocks DR, Yanez JA. Multiple peaking phenomena in pharmacokinetic disposition. Clin Pharmacokinet. 2010;49(6):351–377. | ||
Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4(7):923–939. | ||
Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–S67. | ||
Wada S, Kano T, Mita S, et al. The role of inter-segmental differences in P-glycoprotein expression and activity along the rat small intestine in causing the double-peak phenomenon of substrate plasma concentration. Drug Metab Pharmacokinet. 2013;28(2):98–103. | ||
Mouly S, Paine MF. P-glycoprotein increases from proximal to distal regions of human small intestine. Pharm Res. 2003;20(10):1595–1599. | ||
Mitschke D, Reichel A, Fricker G, Moenning U. Characterization of cytochrome P450 protein expression along the entire length of the intestine of male and female rats. Drug Metab Dispos. 2008;36(6):1039–1045. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.