")
Back to Journals » Journal of Asthma and Allergy » Volume 16
Effects of Oxidative Stress on Airway Epithelium Permeability in Asthma and Potential Implications for Patients with Comorbid Obesity
Authors Kim HR , Ingram JL , Que LG
Received 22 December 2022
Accepted for publication 15 March 2023
Published 6 May 2023 Volume 2023:16 Pages 481—499
DOI https://doi.org/10.2147/JAA.S402340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Amrita Dosanjh
Haein R Kim, Jennifer L Ingram, Loretta G Que
Division of Pulmonary, Allergy, and Critical Care Medicine, Duke University Medical Center, Durham, NC, USA
Correspondence: Haein R Kim, Division of Pulmonary, Allergy and Critical Care Medicine, Duke University Medical Center, Durham, NC, USA, Tel +1 7063722118, Email [email protected]
Abstract: 20 million adults and 4.2 million children in the United States have asthma, a disease resulting in inflammation and airway obstruction in response to various factors, including allergens and pollutants and nonallergic triggers. Obesity, another highly prevalent disease in the US, is a major risk factor for asthma and a significant cause of oxidative stress throughout the body. People with asthma and comorbid obesity are susceptible to developing severe asthma that cannot be sufficiently controlled with current treatments. More research is needed to understand how asthma pathobiology is affected when the patient has comorbid obesity. Because the airway epithelium directly interacts with the outside environment and interacts closely with the immune system, understanding how the airway epithelium of patients with asthma and comorbid obesity is altered compared to that of lean asthma patients will be crucial for developing more effective treatments. In this review, we discuss how oxidative stress plays a role in two chronic inflammatory diseases, obesity and asthma, and propose a mechanism for how these conditions may compromise the airway epithelium.
Keywords: asthma, epithelium permeability, obesity, oxidative stress
Oxidative Stress
Oxidation and reduction of molecules is an important aspect of maintaining cellular homeostasis and engaging in host defense.1 Reactive oxygen species (ROS), such as superoxide and hydrogen peroxide (H2O2), are generated by mitochondria during the reduction of molecular oxygen in the electron transport chain and by immune cells activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) in response to pathogens.2 Cells also possess antioxidants, such as glutathione and catalase, to reduce both endogenous and exogenous sources of oxidizing molecules. Although normal cellular metabolic processes generate oxidants, during an inflammatory state or in response to chronic insults, excessive production of oxidants becomes harmful to host tissues as the presence of ROS and reactive nitrogen species (RNS) exceeds that of antioxidants, resulting in oxidative stress (Figure 1).1,3,4 Oxidants are capable of reacting with other biomolecules, such as DNA, lipids, and proteins, to form high quantities of highly reactive free radicals resulting in cell and tissue damage.3 Indeed, oxidative stress from mitochondrial dysfunction has been implicated in many diseases across several systems, including the cardiovascular (atherosclerosis), nervous (Alzheimer’s disease), digestive (ulcerative colitis), and respiratory systems.5
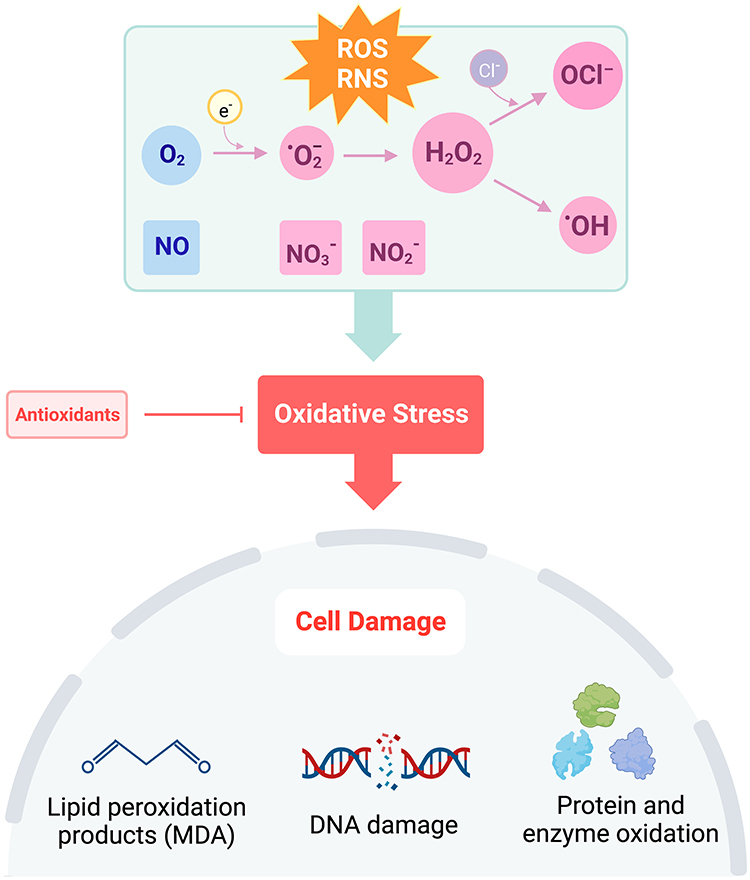 |
Figure 1 Disruption of the balance in ROS + RNS to antioxidants leads to oxidative stress. Under conditions of oxidative stress, oxygen (O2) is reduced to superoxide (·O2−), hydrogen peroxide (H2O2), or hydroxyl radical (·OH). Hypochlorite (OCl−)is formed through the reaction of H2O2 with chloride ions (Cl−). Nitric oxide (NO) is rapidly oxidized to form nitrite (NO2−) and in the presence of oxyhemoglobin, nitrate (NO3−). Increased production of ROS and RNS causes cell damage through lipid peroxidation, DNA damage, and oxidation of proteins and enzymes. Created with BioRender.com. Abbreviations: ROS, reactive oxygen species; RNS, reactive nitrogen species; O2, oxygen; ·O2−, superoxide ion; H2O2, hydrogen peroxide; OCl−, hypochlorite; ·OH, hydroxyl radical; NO, nitric oxide; NO3−, nitrate; NO2−, nitrite; DNA, deoxyribonucleic acid. |
Asthma
Increasing evidence demonstrates a significant role for oxidative stress in the pathogenesis of asthma, a chronic obstructive lung disease affecting over 20 million adults and 4.2 million children in the United States.6 Various allergens, such as house dust mites (HDM), mold, pollen, and pet dander, can trigger allergic asthma. However, asthma can also be exacerbated by factors other than allergens, including environmental pollutants, viral and bacterial infection, and cigarette smoke. Several other metabolic and cardiovascular conditions have been implicated in affecting the severity of asthma and quality of life, especially obesity, hypertension, diabetes, and dyslipidemia.7
Asthma is characterized by airway inflammation and airway hyperresponsiveness (AHR).8 Chronic inflammation in asthma is defined by Type-2-high and Type-2-low immune responses. Type-2-high asthma is typically provoked by allergies and characterized by elevated levels of eosinophils in the airways, while Type-2-low asthma is more often associated with non-allergic, neutrophilic asthma.9,10 Patients with Type-2-low asthma, who make up approximately 5–20% of patients with asthma,10,11 do not respond as well to current standard treatments for asthma such as inhaled corticosteroids. In the United States, uncontrolled asthma presents significant economic burdens on health care systems, reduces workforce productivity and decreases the health-related quality of life of adults and children living with uncontrolled asthma.12,13 Research is needed to understand the pathogenic processes governing Type-2-low asthma and to find specific interventions that best treat patients with this endotype.
Asthma and Obesity
Obesity is a major risk factor for asthma and a significant cause of oxidative stress in the body. Obesity is marked by adiposity defined by a body mass index (BMI) greater than 30 kg/m2.14 Adipose tissue, a source of cytokines and adipokines, can affect various physiological processes through cell signaling.15 As adipose tissue and free fatty acids in circulation increase, adipocyte production of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) increases.16 Leptin is also produced by adipose tissue and has been shown to generate lipid peroxidation products, isoprostanes, and protein carbonyls, biomarkers of oxidative stress.17 With release of adipokines and cytokines, immune cells that generate ROS are recruited to sites of inflammation and are also significant producers of ROS.18,19 Compared to adipocytes derived from lean patients, immune cell infiltration in adipose tissue promotes pro-inflammatory cytokine release, immune cell recruitment, and oxidant generation in adipocytes derived from patients in an obese and proinflammatory state.20 Excess ROS production exceeding the body’s antioxidant capabilities promotes tissue damage and oxidative stress.15 For people with obesity, ROS-induced inflammation is chronic and systemic and increases susceptibility to immune dysregulation.21
Obesity is associated with non-eosinophilic, Type-2-low asthma10 and decreases the effectiveness of inhaled corticosteroids, which is a main component of current treatment regimens.22 Children and adolescents with asthma and comorbid obesity are 24% more likely to be unresponsive to bronchodilators22 which suggests that BMI modifies asthma control.23 In addition to difficulties with controlling asthma, patients with obesity are more likely than lean and overweight patients to experience more severe asthma symptoms and miss more work days.24 The proportion of patients with asthma and comorbid obesity presenting to the emergency room is also significantly higher than other patients with asthma.24 Although the exact reasons for why and how asthma with comorbid obesity becomes difficult to control are unknown, there may be parallels between how both asthma and obesity progress. Studies show that a 5–10% decrease in body weight can reduce airway inflammation.25 However, more direct treatment options are needed for people with asthma and comorbid obesity to improve asthma control.
Oxidant stress could be an important factor to understand how asthma may be altered in the context of obesity, as among non-smoking patients with asthma, BMI is correlated with increased exhaled 8-isoprostanes, while this trend is not seen in comparisons of patients without asthma.26 In animal studies using wild-type and genetically obese mice lacking leptin signaling, exposures to ozone, which exacerbates asthma, versus filtered air (as a control) demonstrated increased methacholine-induced AHR following challenge with ozone, an exogenous source of oxidative stress.27 Interestingly, obese mice exposed to filtered air also exhibited greater AHR compared to wild-type mice, suggesting that obesity inherently contributes to worsening asthma symptoms.27
By 2030, the proportion of US adults who have obesity is expected to increase by 33%, underscoring the need for research on how obesity changes asthma progression.28 In 2010, a higher proportion of adults with obesity had asthma (38%) than did not have asthma (27%).29 With a significant proportion of the US already exhibiting obesity and the possibility of those who are overweight to develop obesity, understanding how obesity and its comorbidities interact to modify disease is becoming increasingly important.
The Airway Epithelium
As the primary protective barrier that defends the lung from agents such as allergens, pollutants, viruses and chemicals that trigger asthma, the airway epithelium plays a crucial role in helping to maintain barrier integrity. Thus, the epithelium is heavily affected by inflammation and oxidative stress caused by asthma and other diseases affecting the lungs (Figure 2).30
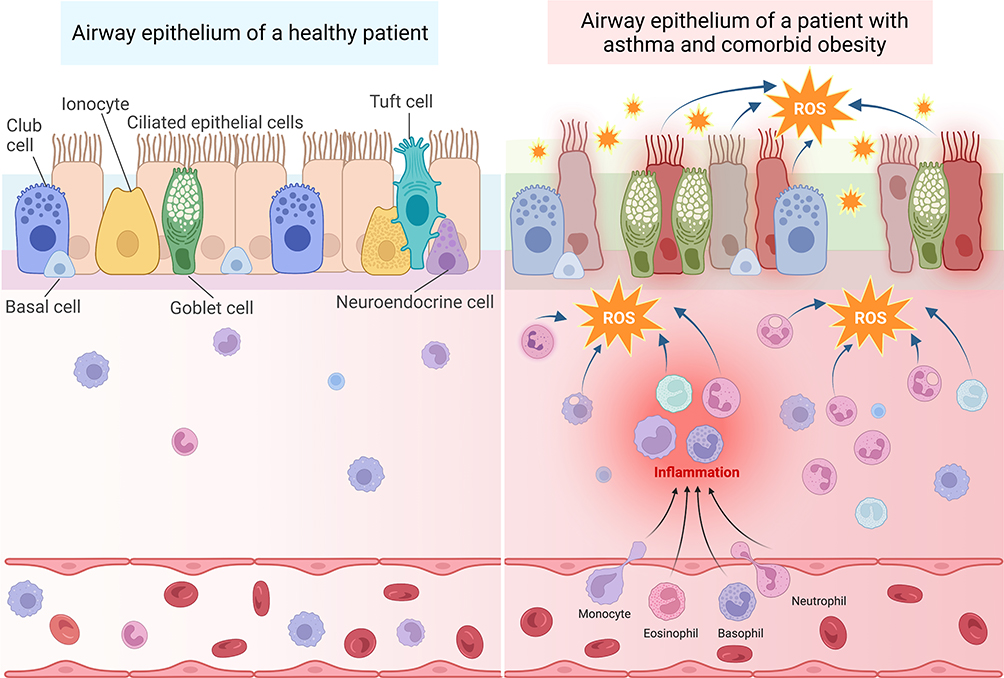 |
Figure 2 Oxidative stress in patients with asthma and comorbid obesity contributes to persistent immune cell recruitment and inflammation, leading to airway epithelium permeability. Left: The airway epithelium of healthy patients is comprised of several different cell types, including club cells, basal cells, ionocytes, ciliated epithelial cells, goblet cells, tuft cells, and neuroendocrine cells, which are attached to the basement membrane. The pseudostratified epithelium provides a sealed barrier to prevent environmental particles from infiltrating the lung tissue. The submucosal area lies between the basement membrane and blood vessels. Immune cells, such as macrophages, lymphocytes, eosinophils, and basophils are located within both the submucosal area and blood vessels. Right: In the presence of allergenic, viral, or air pollution particles that exacerbate asthma, immune cells are recruited from the blood stream into the submucosal area. These activated immune cells produce ROS in response to these provocative agents. Recurrent exacerbations and oxidative stress cause more inflammation and damage to the epithelial cells, resulting in epithelium permeability. In patients with obesity and asthma, inflamed adipose tissue serves as an additional source of oxidative stress, which may accelerate epithelial damage and barrier permeability. Created with BioRender.com. Abbreviation: ROS, Reactive oxygen species. |
Epithelium Structure and Apical Junctional Complexes
Various cell types comprise the airway epithelium (Figure 2), each having a role in maintaining the epithelial barrier. Goblet cells and ciliated cells work in tandem with immune cells to clear airway pathogens by producing mucus and mechanically moving particles out of the airway, respectively.31 Club cells become more numerous in the lower airway epithelium and produce club cell secretory protein (CCSP), which provides important anti-inflammatory properties for the lung.32,33 Basal cells are stem cells that can replenish the airway epithelial cells.34 Immune cells, including neutrophils, eosinophils, and macrophages, produce chemokines that recruit other immune cells, preventing additional damage to the epithelium and clearing invading pathogens.31 Once these pathogens have been cleared, macrophages remove remaining apoptotic neutrophils and produce anti-inflammatory cytokines to decrease the immune response once it is no longer needed.31
The epithelium creates a tight seal to regulate tissue selectivity for certain ions and water.35 The epithelium prevents pathogens from entering the lung, requiring uncompromised, close cell–cell contact throughout the epithelial sheet.35 This function is made possible by apical junctional complexes, consisting of tight junctions and adherens junctions, which work together to regulate ion permeability and maintain epithelial integrity (Figure 3).35
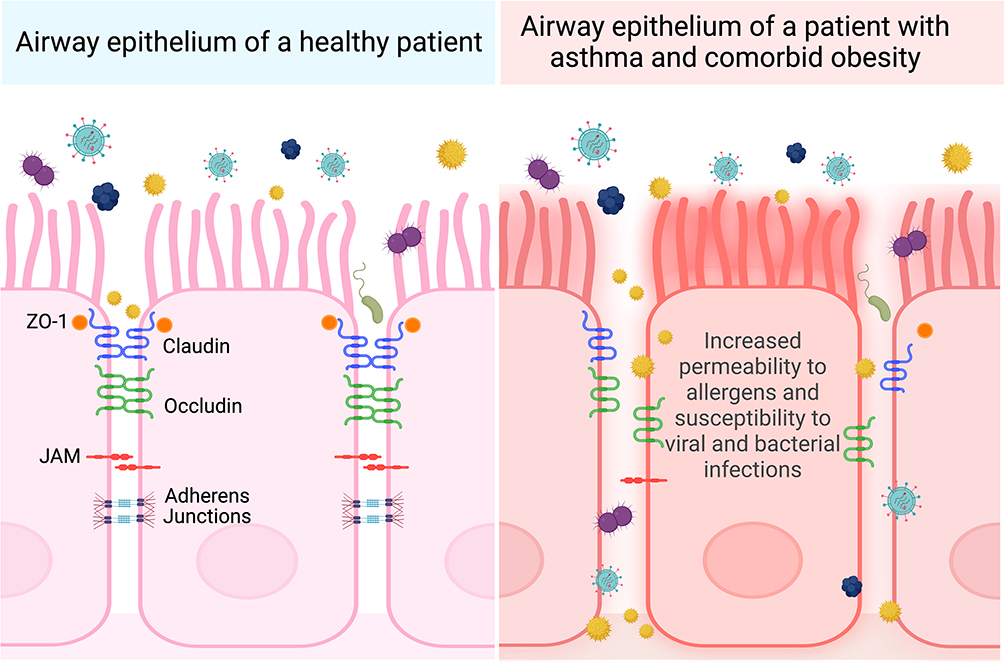 |
Figure 3 Comparison of postulated apical junctional protein organization in patients with asthma and comorbid obesity versus healthy patients. Airway epithelial cells are kept in close contact with adjacent epithelial cells through an organized arrangement of tight junction (ZO-1, claudin, occludin) and adherens junction proteins. People with asthma and comorbid obesity may have more severe asthma exacerbations caused by allergens, viral and bacterial pathogens, or pollutants, contributing to elevated oxidative stress. This may disrupt the organization expression levels of junction proteins, resulting in airway epithelium permeability. Created with Biorender.com. Abbreviations: ZO-1, Zona occludens 1; JAM, Junctional adhesion molecule. |
Tight Junctions
Tight junctions create cell polarity and distinguish the apical from basolateral membrane of the cell.35 Comprised of different types of claudins, these membrane-spanning proteins have varying abilities to regulate epithelium permeability by interacting with claudins on adjacent cells.36 Depending on the function and type of claudin, certain claudins are expressed in different regions of the lung. Sealing claudins (claudin-1, −3, −4, −5, −7, −18) maintain tight cell–cell contact, preventing ions and other molecules from passing through. Some claudins, such as claudin-2, −10, and −15, increase permeability of certain ions, while others maintain the seal with the help of occludins.36 Claudins are responsible for dictating selective permeability to molecules and ions, whereas occludins have been shown to be selective against certain macromolecules, such as inulin and dextran.37 Another protein that supports junction formation is the junctional adhesion molecule (JAM).38 Close in proximity to these tight junction proteins are Zona Occludins (ZO), which link the actin cytoskeleton to claudins and occludins.39
Adherens Junctions
Adherens junctions reside below tight junctions and provide adhesion between adjacent cells.39 Integral to the function of adherens junctions are catenins and E-cadherin, a homotypic adhesion molecule. The extracellular domain of E-cadherin forms the connection between two cells expressing the same E-cadherin, while the intracellular domain is in contact with the actin cytoskeleton. β-catenin and p120 catenin stabilize the adherens junction and the apical junctional complex as a whole, as the loss of one junctional protein can affect the formation and stability of the junction.39
Effects of Oxidative Stress on Airway Epithelium Permeability
In asthma, particles that infiltrate the lungs cause airway epithelial cells to initiate the inflammatory processes, including immune cell recruitment, against the stimuli.40 Chronic lung inflammation results in altered arrangement and expression of apical junctional complexes between airway epithelial cells, allowing the airway epithelial barrier to become permeable to molecules that exacerbate asthma through leaky tight junctions and adherens junctions.40 Dysfunction and damage to the airway epithelium may contribute to an increase in particles and pathogens reaching the submucosa of the airways, causing persistent inflammation, heightened immune responses, and compromising the barrier function from external pathogens.41 Breakdown in the barrier function activates more immune responses, generating oxidative stress, and may worsen AHR.8 Over time, recurrent exacerbations of asthma result in airway remodeling, a process that results in permanent changes to the airway wall through alterations in protein composition of the extracellular matrix and fibroblast and smooth muscle proliferation.42,43
In patients with asthma, the responsibility of the airway epithelial barrier to provide a tight seal also appears to be compromised (Table 1). Methods to study paracellular permeability involve a combination of measuring transepithelial electrical resistance (TEER) of epithelial cell monolayers and monitoring concentrations of various sizes of fluorescein isothiocyanate (FITC)-dextran or other labelled molecules, such as mannitol, bovine serum albumin (BSA), and pyranine, which do not pass between epithelial cells when junctions are intact. Immunofluorescent staining of junction proteins and quantification of gene and protein expression are also used to provide additional evidence of epithelial integrity or permeability.
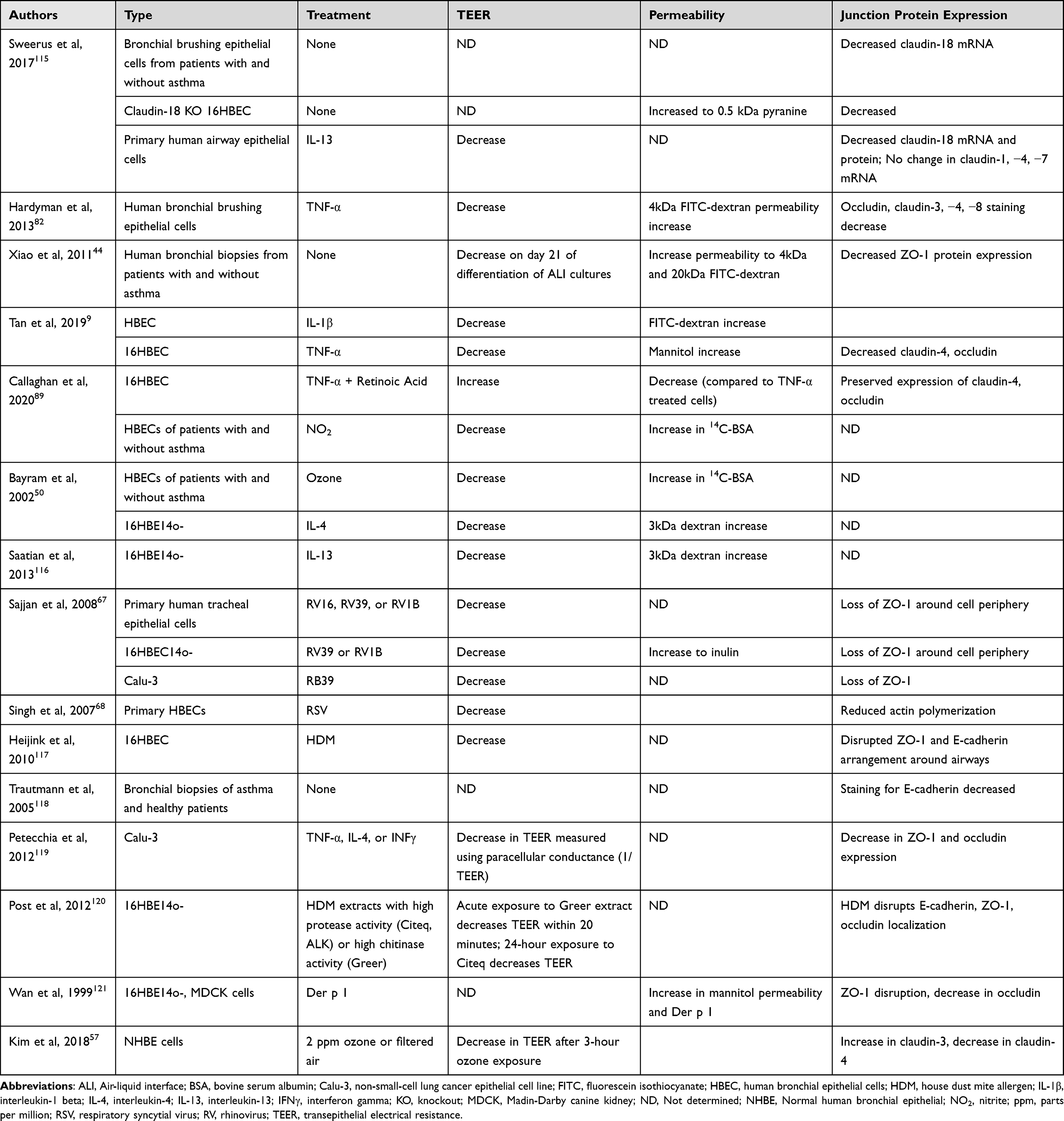 |
Table 1 Allergens, ROS, RNS, and Cytokines Affecting the Airway Epithelium in Human Cells |
When visualized through immunofluorescence, primary human bronchial epithelial cells (HBECs) isolated from patients with asthma, show an unorganized, uneven arrangement of occludin and ZO-1 compared to healthy patients, which show a more continuous arrangement of tight junction proteins.44 Furthermore, protein expression of ZO-1 is decreased in HBECs from patients with asthma compared to HBECs from healthy patients.44 Upon examination of airway biopsies, an uneven distribution of tight junctions is present, which is in agreement with the pattern seen in the cultured HBECs.44 Using primary airway epithelial cells cultured from healthy patients and those with asthma, the decrease in TEER changes in proportion to the severity of the disease. Cells with the lowest TEER also have low ZO-1 expression.44 Permeability assays demonstrate that FITC-dextran passage from the apical to the basolateral membrane is higher compared to airway epithelial cells cultured from healthy donor cells.44
In another study by Tan et al, mice challenged with different concentrations of house dust mite extract injected either intranasally or intraperitoneally acutely develop either eosinophilic, mixed, or neutrophilic phenotypes of experimental asthma.9 Structurally, tight junction protein expression in the lung changes among these three phenotypes of asthma following treatment with HDM extract. mRNA and protein expression of ZO-1 decreases, whereas mRNA and protein expression of Claudin-4 increases. Claudin-4 levels are highest in neutrophilic mice.9 Although Claudin-18 has only been found in the alveolar epithelium, its mRNA expression is decreased in all three asthma phenotypes compared to healthy mice.9
As a chronic inflammatory lung disease, asthma generates oxidative and nitrosative stress.7 While normal metabolic processes also generate oxidants, excessive production of oxidants becomes harmful to host tissues.4 Markers of oxidative stress, including 3-bromotyrosine, F2-isoprostanes, exhaled nitric oxide, superoxide, nitrites, nitrates, lipid peroxides, total protein carbonyls, total glutathione, aldehydes, and alkanes, are elevated in urine, exhaled breath condensate, and plasma samples of patients with asthma compared to healthy adult patients.45–47 In exhaled breath condensate samples, patients with persistent asthma show higher hydrogen peroxide levels than those with mild intermittent asthma48 and those who are healthy.49 Based on evidence demonstrating increased oxidative stress in asthma, oxidants may be contributing to the airway epithelium permeability that is observed in asthma models from primary human epithelial cells and mice (Table 1).
Several studies demonstrate the effect of strong oxidants on airway epithelium permeability. Of human bronchial epithelial cells (HBECs) isolated from healthy and allergic asthma patients, only cells from hosts with asthma demonstrate a significant decrease in TEER after 2 and 6 hours of 10 parts per billion (ppb) ozone exposure.50 Paracellular permeability to BSA also increases only in the HBEC cultures from patients with asthma following a 6-hour ozone exposure at concentrations between 10 and 100 ppb.50 While asthmatic epithelial cells respond more severely to ozone than cells from healthy patients, ozone still increases airway epithelium permeability in those who are healthy.51,52 Evidence of this ozone effect in healthy patients comes from studies measuring in vivo diethylenetriaminepentaacetic acid (DTPA) clearance rates in patients without asthma after ozone exposure.51,52 Even in healthy non-asthma patients, the clearance rate is much faster after ozone exposure, demonstrating causality between ozone and airway permeability.51,52 Since these acute exposures have been shown to affect the airways in healthy patients, chronic exposures to sources of oxidative stress would be expected to augment airway epithelium permeability in patients with asthma.
Surprisingly, however, lung clearance rates of DTPA do not consistently differ between healthy patients and those with asthma.53,54 A study conducted by Georas et al involving mannitol inhalation by ten healthy participants and eleven patients with mild asthma demonstrated that the average mannitol levels in serum did not differ significantly between subjects with and without asthma.55 While this finding suggests that epithelial permeability may not play a significant role in asthma, the study was conducted only in with patients with mild asthma that did not have any exacerbations within six months of the study.55 Patients who have both asthma and obesity often have worse asthma symptoms and, in this study, those with severe asthma were excluded.55 Whether patients with obesity and severe persistent asthma have a more permeable airway epithelium still remains to be determined.55 Further explorations of clearance rates among patients with asthma and comorbid obesity are needed to understand lung epithelium permeability in vivo (Box 1).
 |
Box 1 Future Areas of Study for Clinical and Basic Science Research on Asthma and Obesity |
Other studies demonstrate sensitivity of asthma patients to strong oxidants. In human lung epithelial cell lines (BEAS-2B), increasing the concentration of PM confirms elevation of ROS production after PM exposure.56 Following an infection with Pseudomonas aeruginosa, BEAS-2B cells challenged with PM are more susceptible to bacterial invasion than cells not challenged with PM. PM exposure reduces barrier integrity through decreased TEER and expression of occludin and claudin-1, suggesting that environmental pollutants can induce oxidative stress, which affects junction protein expression.56
Animal models also display epithelial changes in response to oxidants (Table 2). In a study conducted by Kim et al, wild-type mice subjected to a six-hour per day, short-term exposure of ozone57 had a significant increase in bronchoalveolar lavage (BAL) macrophage, eosinophil, and neutrophil cell number, indicating airway inflammation. Ozone exposure also resulted in an increase in ROS, as measured by the elevated lung carbonyl levels.57 Although the expression of sealing claudin proteins, Claudin-3 and Claudin-4, was significantly higher in ozone-exposed mice than FA-challenged mice, the circular distribution pattern appeared to be disrupted for claudins −3 and −4 in the ozone challenged airway.57
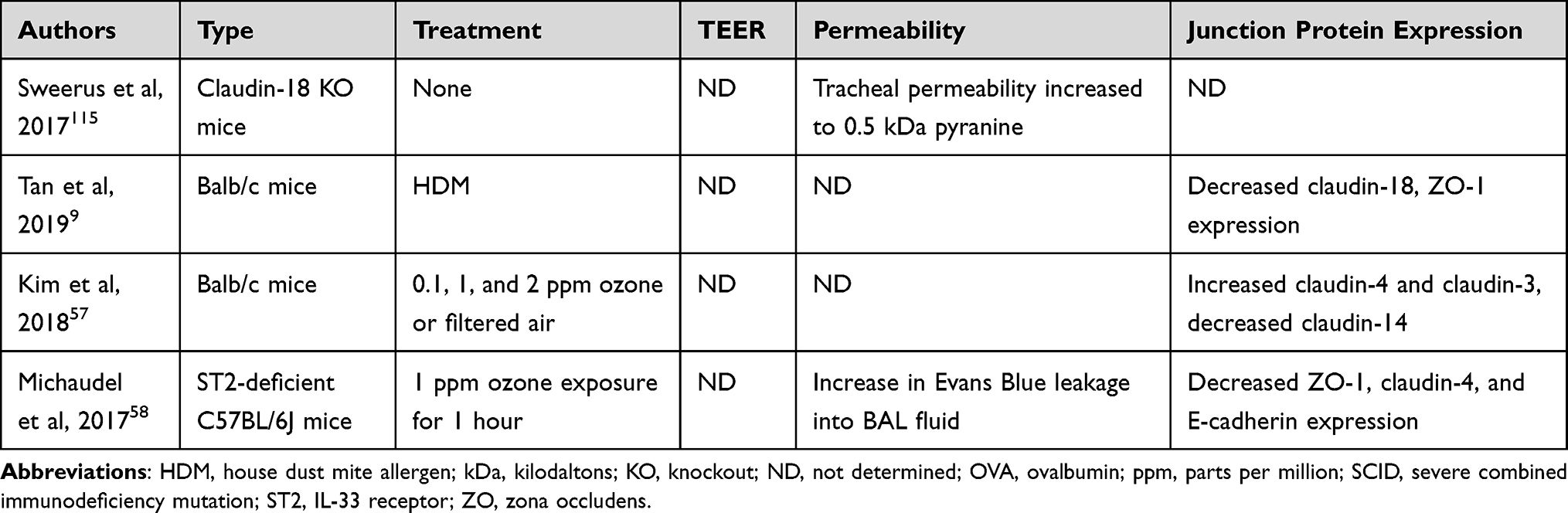 |
Table 2 Allergens and ROS Affecting the Airway Epithelium in Mouse Models |
However, claudin-4 expression is not consistently increased following ozone challenge. Michaudel et al determined that a 1 ppm ozone exposure for an hour resulted in decreased claudin-4 in ST2-deficient C57BL/6J mice,58 whereas Kim et al revealed an increase in claudin-4 levels in BALB/c mice after chronic exposure to 0.1, 1, or 2 ppm of ozone or ambient air for 6 h for 3 days.57 However, these discrepancies may be due to differences in the mouse models used, as well as the length of time ozone exposures were administered. Still, given that evidence suggests that oxidative stress-inducing agents, including ozone and PM, disrupt barrier proteins and increase airway epithelium permeability, the airway epithelium of patients with both asthma and obesity may be more severely affected, placing patients at higher risk for experiencing serious exacerbations and infections.56
Changes to the airway epithelium in asthma may be accelerated or worsened by comorbidities, specifically obesity, which contributes additional ROS as an endogenous source of oxidative stress. In fact, in patients with obesity, asthma is more severe and airway closure occurs to a greater extent than in lean patients with asthma.8 While asthma with comorbid obesity is associated with Type 2-low asthma, a subset of patients with obesity have allergic asthma. In response to allergens, such as HDM and fungi, cell culture models of human bronchial epithelial cells show decreases in tight junction proteins and epithelial resistance, along with increased permeability (Table 1).59 The processes by which obesity alters cell signaling and inflammatory pathways involved in atopic and nonatopic asthma have not yet been fully elucidated. We hypothesize that the airway epithelium of patients with asthma and comorbid obesity is affected by oxidative stress-induced disruptions in junctional protein dysfunction (Figures 2 and 3).
Sources of Oxidative Stress in Asthma and Obesity
In asthma, one potential source of oxidative stress in the lung is uncoupling of nitric oxide synthase (NOS), which results in reduced nitric oxide (NO) bioavailability and favors generation of superoxide over NO.60,61 The balance between l-arginine and NO is maintained by NOS, which forms NO using L-arginine as a substrate.60 L-arginine levels are also dependent on arginase, an enzyme that has been shown to be more active during asthma, causing the ratio of L-arginine to asymmetric dimethylarginine (ADMA) to decrease.60 Higher levels of ADMA have been shown to promote oxidative stress in lung epithelial cells of mice.61 Furthermore, ADMA inhibits NOS, reducing NO levels and resulting in oxidative stress.
Mitochondrial dysfunction is also seen in airway epithelial cells in patients with asthma and in patients with asthma and comorbid obesity. Among patients with asthma, those with obesity demonstrate higher rates of glycolysis in both platelets and airway epithelial cells, compared to those who are lean.62 Asthma also increases the oxygen consumption rate, ATP production, and activity of NOS in platelets and epithelial cells.62 Notably, platelet and airway epithelial cells from patients with asthma show an increase in mitochondrial superoxide and H2O2 production, two cellular sources of ROS.62 Furthermore, those with asthma and obesity demonstrate even higher levels of H2O2 production compared to those with asthma alone.62 Inhibiting NOS decreases H2O2 production in both airway epithelial cells and platelets, suggesting that oxidant production occurs during NOS uncoupling.62 Another potential mechanism for mitochondrial dysfunction in patients with asthma and comorbid obesity is the observation that in the airway epithelial cells of this subset of patients, levels of the antioxidant enzyme paraoxonase-2 (PON-2) are lower than in patients who are healthy, have asthma, or have obesity. PON-2 localizes to the inner mitochondrial membrane and plays a vital role in reducing oxidative stress from oxidants produced by the mitochondria.63,64
Increased oxidative stress is also seen with viral infections of the lung.1 The NOX family of NADPH oxidases are key producers of ROS in cells in response to microorganisms and pathogens.2 They are a seven-member family widely distributed in the body. Different NOX isoforms are expressed in different cell types and participate in host defense to inhibit viral propagation and bacterial infection. NOX2, NOX4, dual oxidase (DUOX)1, and DUOX2 are expressed in airway epithelial cells and become activated in asthma, increasing oxidative stress.1,65 In 16HBE14o-cells infected with rhinovirus, increased ROS was associated with a significant upregulation in NOX1 and DUOX2 activity.66 Respiratory infections, including rhinovirus, also result in decreased TEER of the airway epithelium and increased permeability to inulin (Table 1).67,68 This defect in the epithelial barrier function is associated with disruption to tight junction structure. Whereas ZO-1 is normally located in the cytoplasm of 15HBE14o-cells, ZO-1 was lost between cells, where the protein normally provides cell-to-cell connection.67 The increased permeability present after initial infection of primary human airway epithelial cells with rhinovirus, facilitates migration of bacteria from the apical end of the epithelium to the basolateral end.67
Viral infections also increase host susceptibility to bacterial infections.67 Patients with asthma are more likely to develop more severe and longer duration respiratory tract infections.69 Moreover, 80% of asthma exacerbations were found to be associated with colds in adults.70 Obesity is associated with higher risk for developing severe upper respiratory infections in adults. Adults with obesity also develop more frequent asthma exacerbations due to respiratory tract infections.71 Given the evidence demonstrating that viral respiratory infections disrupts the airway epithelium and is associated with higher oxidant burden, people with both obesity and asthma may be much more susceptible to airway epithelium damage and dysregulation especially after a viral or bacterial infection.
Environmental oxidants have also been shown to affect respiratory epithelial cells, especially in the context of viral infections, such as rhinovirus.72 Rhinovirus and environmental pollutant exposure (O3 or NO2) independently increase the release of interleukin-8 (IL-8), an inflammatory cytokine, in both BEAS-2B cells and human nasal epithelial cells (HNECs).72 Challenge of these cells to virus with and without O3 or NO2 significantly increases IL-8, which suggests that exogenous sources of oxidant stress can enhance the inflammatory response of epithelial cells in the respiratory system.72 This concept is further supported by the treatment of BEA-2B cells with the antioxidant N-acetylcysteine (NAC); NAC reduced IL-8 release in rhinovirus infected cells and in cells challenged with either O3 or NO2.72 However, because IL-8 release is not completely blocked by NAC, this finding indicates that although oxidant stress significantly increases cytokine production, it is not the only cause for cytokine secretion in airway epithelial cells.72
Effects of ROS, Cytokines, and Adipokines in Obesity: Lessons from the Intestinal Epithelium
Oxidative stress in obesity may result in low-grade, persistent systemic inflammation which can have a greater effect in people with asthma. While it has not been established whether patients with asthma and comorbid obesity exhibit greater epithelium permeability, adipose tissue signaling compromises epithelial integrity in the intestine.73 As a secretory organ, adipose cells are a source of adipokines, most notably leptin, and cytokines that are associated with asthma, including TNF-α, IL-6, and interleukin-1 beta (IL-1β).74 Importantly, oxidative stress contributes to the elevated levels of these cytokines and adipokines.75 To determine the effect of ROS on cytokine production, 3T3-L1 adipocytes were treated with glucose oxidase, which increases production of H2O2 within the cell. This treatment was found to increase lipid peroxidation and decrease adiponectin secretion in a dose-dependent manner. Similarly, in 3T3-L1 adipocytes treated with external H2O2, mRNA levels of adiponectin decreased, while IL-6 and TNF-α mRNA increased, demonstrating that ROS promotes production of inflammatory cytokines.16,76 Furthermore, in obese KKAy mice, a genetically modified mouse that becomes obese shortly after birth and serves as a model for type 2 diabetes, higher H2O2 levels are observed in plasma, as well as lipid peroxidation in white adipose tissue (WAT).16 As shown previously in the 3T3-L1 adipocytes, mRNA of adiponectin decreased in both plasma and WAT, while levels of TNF-α increased in obese KKAy mice, implicating the role of ROS in elevating cytokine levels.16
TNF-α
Patients with uncontrolled asthma are likely to have higher TNF-α and TNF-α receptor levels in BAL fluid and on peripheral blood monocytes.77,78 To determine the role of TNF-α in asthma, TNF-α was neutralized with antibodies in mice after challenged with ovalbumin. When T-helper 17 cells, which produce interleukin-17A (IL-17A), a cytokine that may recruit neutrophils into the airways, were transferred to the trachea of ovalbumin-challenged mice, increased neutrophil and macrophage migration, as well as increased concentrations of TNF-α and IL-17A levels, were observed in BAL fluid.79 Stained lung sections confirmed an increase in airway inflammation. Mice treated with anti-TNF-α antibodies had decreased Il6 transcription and reduced airway inflammation, although the amount of inflammation remained higher than in control mice.79 Targeting TNF-α has had mixed results in patients with moderate to severe asthma suggesting that there are other complex effects of TNF-α that need to be better understood.80,81 Studies show that TNF-α has the capacity to reduce the strength of the airway epithelial barrier by decreasing epithelial resistance and permeability (Table 1).82 Challenging human bronchial epithelial cells at air-liquid interface (ALI) with basolateral TNF-α increases TNF-α, IL-1β, IL-6 and decreases expression of occludin and claudin expression indicating disturbance of tight junctions. Treatment with a Src-family kinase (SFK) inhibitor reverses the increase in airway epithelium permeability. TNF-α decreases TEER and increases the passage of FITC-dextran molecules.82 Because SFK activation is dependent on TNF-α, the results obtained from inhibiting its activity provide evidence for TNF-α’s role in affecting tight junction structures.83 TNF-α also disrupts apical junction complexes.82 E-cadherin levels decrease and p120 catenin increase after TNF-α treatment, both of which are components of adherens junctions. This disruption of the adherens junction may contribute to other changes in the tight junctions and barrier function.82 It has yet to be determined whether targeting specific molecules involved in cytokine release or maintenance of junction proteins can be a potential therapeutic for people with severe asthma.
IL-6
IL-6 is another example of a proinflammatory cytokine known to be increased in populations of patients with obesity.84 In addition, IL-6 and IL-6 mRNA are elevated in the BAL fluid of patients with Type 2-low asthma.85,86 Among those with asthma, patients with high circulating IL-6 levels are more likely to have higher BMIs, hypertension, and lower measures of lung function (forced expiratory volume (FEV1) and forced vital capacity (FVC)).87 Peters et al conducted a study of 93 healthy subjects, 249 subjects with non-severe asthma, and 387 subjects with severe asthma.87 The threshold for “IL-6 high” classification was based on the 95th percentile for plasma IL-6 in healthy subjects, which was ~3.1 pg/mL. Patients with high IL-6 levels had higher BMI, worse lung function (lower FEV1) and more exacerbations. Although 75% of “IL-6 high” patients had obesity, 37% of patients with obesity were part of the “IL-6 high” category. Among those with severe asthma, a history of diabetes was more common for those with high plasma IL-6 levels. Asthma control test scores demonstrated poorer asthma control in IL-6 high groups. The likelihood of a patient having had a visit to the emergency department and a hospitalization in the last year, in addition to exacerbations in the past 1 and 2 years, increased for those having high plasma IL-6 levels.87
In patients with high levels of plasma IL-6 trans-signaling (IL-6 TS) demonstrate downregulation of genes involved in apical junctional complexes, including CTNNB1 (β-catenin), CLDN1, CLDN8, CLDN18 (claudins-1, −8, −18), and TJP1 (ZO-1).88 No change is observed in OCLN (occludin) mRNA expression between IL-6 TS-high and IL-6 TS-low patients.88 In human bronchial epithelial cells, treatment with IL-6 and soluble IL-6 receptor show the greatest decrease in TEER, compared to IL-6 and its receptor alone.88 However, other groups report that IL-6 treatment significantly increases epithelial permeability to mannitol without a decrease in TEER.89
The effect of IL-6 has also been studied in intestinal Caco-2 cells.90 IL-6 decreases TEER in these cells, and resistance is most affected by IL-6 added to the basolateral membrane more than the apical membrane.90 Epithelial permeability to urea, but not mannitol, inulin, or dextran, which are larger molecules, also increases in Caco-2 cells.90 Supporting the permeability results, protein expression of claudin-2, a leaky claudin expressed in the intestines, increases, which may be causing the decrease in TEER.90 A separate study also determined that larger molecules, like dextran, were impermeable to Caco-2 monolayers treated with IL-6.91 Permeability for small ions, however, was found to increase, and TEER was reduced after 48 hours of IL-6 treatment to the basal end of the cells. Claudin-2 protein also increased in a dose-dependent manner in response to increasing IL-6 concentrations.91
IL-1β
Interleukin-1 beta (IL-1β) is a cytokine that may also affect obesity and airway inflammation. TNF-α, common to both asthma and obesity, increases IL-1β release in primary bronchial epithelial cells grown at air–liquid interface (ALI).82 IL-1β mRNA expression was significantly elevated in mouse models of eosinophilic, neutrophilic, and mixed asthma types compared to control mice, with the highest levels of IL-1β mRNA and pro-IL-1β protein in neutrophilic asthma.9 ALI-cultured HBECs were treated with IL-1β, which decreased TEER after 12 hours. In addition to a decrease in resistance, FITC-dextran levels were higher in IL-1β-treated HBEC cultures, suggesting that in asthma, especially neutrophilic asthma, the cytokines produced contribute to increased epithelial permeability.9 However, the mRNA expression of occludins and CLDN1 and CLDN4 (claudin-1 and −4) is unchanged by either IL-1β or IL-17A.9 This study also determined that IL-1β and IL-17 may be acting together to disrupt barrier function. Future research efforts may be directed towards understanding which cytokines act synergistically to promote epithelial barrier dysfunction (Box 1).
The effects of IL-1β have also been studied in the intestinal epithelium.92 IL-1β reduces epithelial resistance and as TEER decreases, paracellular permeability of the Caco-2 cells measured by inulin flux increases. This increase in permeability appears to be NF-κB dependent. NF-κB p65, a subunit of the NF-κB transcription factor, is normally present in the cytoplasm. However, more p65 is found in the nucleus when cells are stimulated with IL-1β. Inhibiting NF-κB p65 prevents IL-1β-induced permeability.92
Adipose Tissue
Although, to our knowledge, the effects of comorbid obesity and asthma on lung epithelial permeability have not yet been studied, evidence suggests that obesity induces changes to intestinal tight junctions and epithelial permeability.93,94 Genser et al compared intestinal permeability in patients with severe obesity and non-obese patients.93 The tight junction protein, occludin, was decreased in jejunal samples of patients with obesity. In investigating the effect of lipid micelles on Caco-2 cells, lipids did not change epithelial resistance, but did increase permeability to macromolecules. Importantly, micelles with digested triglycerides had a significant effect on permeability to sulfonic acid, 4kDa dextran, and 10kDa dextran, whereas micelles containing bile did not. However, upon further examination, the increase in permeability may be more likely to be evident in jejunum samples from diabetic patients with obesity, compared to nondiabetic patients with obesity.93
In mice, high fat diet (HFD)-induced obesity significantly increased conductance and intestinal permeability to FITC-dextran and reduced TEER, compared to mice on a normal chow diet.95 Stained sections of intestine and colon reveal the large presence of immune cells, indicating inflammation.95 As in the previous studies, claudin-2 levels in both the small intestine and colon are significantly higher in obese mice than control mice. Occludin and β-catenin, a component of the adherens junction, are unchanged. Expression of several claudins (−1, −3, −4, −7, −15) and E-cadherin decreases in obese mice in the small intestine. Interestingly, claudin-7 expression in the colon increases in HFD-fed mice, suggesting that changes to the epithelium are not uniform, but are unique to the type of tissue in which they reside.95
Work by Suzuki and Hara provides insight into the possible causes of intestinal epithelial permeability in obese states, which may be through dietary fat intake and the presence of bile. In Otsuka Long Evans Tokushima Fatty (OLETF) obese rats and their lean strain, LETO, fed high fat diets, intestinal permeability, measured by the amount of phenolsulfonphthalein and chromium ethylenediaminetetraacetate (Cr-EDTA) excreted in urine, increases compared to rats fed a normal diet.96 After consuming their respective diets for 15 weeks, HFD-fed OLETF rats had the greatest degree of intestinal permeability. All HFD-fed rats had increased leptin levels, but leptin was significantly higher specifically in OLETF rats, suggesting that obese phenotype modifies hormone expression. Plasma TNF-α levels were also significantly higher in OLETF fed HFD, compared to standard diet OLETF and LETO rats.96 Diet also influences the composition of junction proteins. HFD-fed OLETF rats had decreased expression of claudin-3, claudin-1, junctional adhesion molecule 1 (JAM-1), and occludin, compared to standard diet-fed OLETF rats. Consistent with findings of Genser et al, the presence of emulsified fat affects permeability and epithelial resistance of Caco-2 cell cultures. However, while Genser et al did not find bile micelles to affect permeability significantly, Suzuki and Hara observed that a 20% rat bile solution increased permeability to luciferase even more than a 1% fat emulsion solution. While decreases in the expression of JAM-1, claudin-1, and claudin-3 were indirectly proportional to the concentration of rat bile and fat emulsion used, occludin expression was significantly elevated at the highest treatment concentration. Whether high dietary fat intake or other aspects of obesity affect the lung epithelium during the pathogenesis of asthma has yet to be determined (Box 1).96
Leptin and Adiponectin Balance
Adipocytes secrete adipokines, most notably leptin and adiponectin, which affect feeding habits, weight, and interactions with the immune system.97 Leptin is a peptide hormone and levels of leptin are directly proportional to the amount of adipose tissue present.97 In relationship to other cytokines involved in inflammation, leptin is able to increase secretion of TNF-α and IL-6.98 This stimulation establishes a feed-forward mechanism where TNF-α and IL-1β can further increase the leptin production.98 Leptin is pro-inflammatory, while adiponectin acts as an anti-inflammatory factor.97 However, both TNF-α and IL-6 suppress adiponectin expression,97 indicating that when leptin levels are high in the body, adiponectin may be significantly inhibited, promoting more inflammation.
Leptin and its receptor are expressed by 16HBE cells, normal HBECs, and primary bronchial epithelial cells from airway brushings, and may be a potential link between obesity and asthma.99 In obese, leptin receptor-deficient mice (db/db), BAL fluid shows elevated leptin and reduced adiponectin concentrations compared to lean wildtype mice.100 This difference in the leptin-adiponectin balance may promote oxidative stress in the lung, exacerbating asthma symptoms and inflammation.101 Leptin augments the sensitivity of ovalbumin-challenged mice to methacholine compared to control mice, suggesting that leptin can worsen asthma symptoms.102
Again, evidence from studies of the intestinal epithelium may shed light on potential pathways in which obesity alters asthma pathogenesis, and especially the airway epithelium. Using ob/ob mice lacking the leptin protein and db/db mice lacking a functional leptin receptor, both of which have an obese phenotype, Brun et al, showed that in obese mice, TEER was reduced, the intestinal epithelium was more permeable to horse radish peroxidase (HRP), and the IL-1β, IL-6, and TNF-α cytokines were elevated, compared to wildtype lean mice.103 Consistent with these changes, ZO-1 and occludin distribution in the epithelium were also significantly disrupted.103 Furthermore, Le Dréan et al sought to determine the mechanisms by which obesity contributes to intestinal permeability and focused on the potential effects of visceral adipose tissue (VAT), leptin, and adiponectin.73 Adult rats born from intrauterine growth restriction (IUGR) conditions are more susceptible to developing higher amounts of VAT and have significantly increased leptin mRNA expression, as expected. Although ZO-1 protein expression in the colon did not differ between control and IUGR rats, barrier properties of the epithelium did change, with TEER decreasing. Interestingly, the decrease in resistance was not due to any increases in TNF-α and IL-6. In a separate experiment, VAT from HFD-fed rats was isolated and transplanted into lean rats. This procedure resulted in increased epithelial permeability of FITC-dextran, as well as a decrease in ZO-1 protein expression. No change was observed in TNF-α and IL-6 levels, which points to an inflammation-independent mechanism for obesity reducing the integrity of the epithelial barrier. Confirming this finding, Caco-2 cells cultured with adipocytes demonstrate a decrease in TEER and significantly increased levels of leptin and adiponectin.73
To determine the effect of leptin on intestinal permeability, leptin was injected into normal rats and Zucker rats, which lack functional leptin receptors.73 In normal rats, leptin increased intestinal permeability as demonstrated by increased Cr-EDTA in urine. No change in permeability was detected in Zucker rats, indicating that leptin interacting with its receptor is necessary for permeability to increase. Furthermore, claudin-1 and occludin arrangements in the colon were disrupted. In cultured colonic epithelial cells, leptin treatment results in a decrease in TEER, but adiponectin does not. The study also determined that leptin influences cytoskeleton rearrangement through the ras homolog family member A (RhoA)/Rho-kinase pathway, which participates in airway remodeling and hyperresponsiveness, especially in severe asthma.73,104 In cells treated with leptin, RhoA, a GTPase which activates the Rho-kinase ROCK, activation is increased, and the F-actin cytoskeleton changes form. This activation is reversed by the addition of a ROCK inhibitor.73 These findings suggest that leptin contributes to epithelium paracellular permeability, and the potential exists for this phenomenon to also occur in patients with asthma and comorbid obesity. More studies must be conducted to understand the effects of leptin on the airway epithelium specifically (Box 1).
Future Directions and Clinical Implications
Several clinical trials have been conducted to determine the efficacy of antioxidant molecules on reducing oxidative stress and improving asthma control. However, potential treatments such as Vitamin D3 supplementation did not significantly reduce asthma exacerbations nor did Vitamin D3 improve asthma control.105 In this study, 29% of patients enrolled in the control group experienced the primary outcome of treatment failure within 28 weeks, most often through needing additional steroid treatment or experiencing an exacerbation. Because the event rate was lower than expected, a greater sample size may have been needed to detect differences between the control and Vitamin D3 supplementation group. However, a beneficial effect was found in the percentage of participants in the Vitamin D3 group who were able to reduce their dose of ciclesonide, a corticosteroid used to treat asthma symptoms, by 75% compared to the placebo group.105
Additional clinical trials targeting patients with asthma and comorbid obesity are ongoing. One potential avenue for treatment is adjusting diet. A pilot study is being conducted to determine the effect of three different nutrition plans (diet following Dietary Guidelines for Americans (DGA), diet following DGA with medium-chain triglyceride supplementation, and a ketogenic diet) on asthma control and lung function in adults with obesity and asthma (NCT05222451). Weight loss has also generated interest as an intervention and a 5% reduction in weight was found to have positive effects on asthma control in patients with asthma and obesity.106 Currently, Semaglutide, a glucagon-like peptide (GLP-1) receptor agonist used to achieve weight loss in patients with obesity and improve glycemic control in patients with Type 2 Diabetes Mellitus, is being studied in a randomized control trial to determine if the drug improves asthma control in adult patients with obesity and persisting asthma symptoms (NCT05254314).107 GLP-1 is a hormone secreted by the intestine and acts to promote insulin secretion and inhibit glucagon secretion.108 Epithelial cells in the lung express the GLP-1 receptor, stimulating investigations in the potential role for GLP-1 agonists in treating asthma symptoms in patients with obesity or metabolic dysregulation.108,109 Other studies are targeting genes involved in both asthma and obesity, such as the β2-adrenergic receptor (ADRB2).110 β2-agonists are bronchodilators used to manage asthma.111 A randomized clinical trial involving patients with type-2 low asthma is being conducted to determine if responses to the β2-agonist albuterol is improved when used in conjunction with Roflumilast, a phosphodiesterase inhibitor which blocks the degradation of cyclic adenosine monophosphate, resulting in airway smooth muscle cell relaxation (NCT04108377).111 It will be interesting to see if this is effective, as a recent double-blinded placebo-controlled trial with roflumilast in people with obesity and late-onset, poorly controlled asthma did not improve asthma control and actually appeared to increase risk of exacerbation.112
Another pilot study in patients with asthma and comorbid obesity is currently underway (NCT04026711) with the antioxidant, Mitoquinone (MitoQ), which targets mitochondrial ROS. In mouse studies, mitoquinone decreased lung cytokine release, eosinophil infiltration, and AHR to methacholine in an obese, allergic asthma mouse model.113 In a separate open-label pilot study involving adult patients with asthma and obesity, L-citrulline supplementation for 2 weeks improved asthma control and lung function.114 L-citrulline is postulated to protect airway epithelial cells from NOS uncoupling, thereby reducing oxidant stress.114 L-citrulline supplementation for obese asthma is now being studied in a double blinded placebo controlled trial (NCT03885245). While treatments involving antioxidants are promising potential avenues for treating asthma in patients who also have obesity, more research is needed to understand the mechanism of redox signaling and how ROS can specifically be targeted to improve nonallergic asthma in people with obesity (Box 1).
Conclusion
While asthma is more likely to be severe and difficult to control among patients with asthma and comorbid obesity, the manner in which obesity changes asthma outcomes is not well understood. We hypothesize that oxidative stress is playing an important role in altering the structure and organization of the airway epithelium. Because obesity profoundly changes intestinal epithelial tight junction and junctional protein expression, there is a significant opportunity for parallel research in patients with asthma and comorbid obesity. Important research has already been conducted to demonstrate the effects of exogenous oxidative stress on airway epithelial tight junction complexes, permeability, and cytokine secretion in asthma. Damage to the airway epithelium arising from oxidative stress may be contributing to airway inflammation and airway permeability and subsequent AHR and airway remodeling in patients with asthma and obesity. With the projected increase in obesity in the US and the high prevalence of asthma, research of asthma with comorbid obesity is especially relevant today and will continue to be in the future.
Acknowledgments
Drs. Que and Ingram are supported by the NIH grants R01HL153641, R01HL136917, R01HL146542 (Que) and R01HL130234 (Ingram).
Disclosure
Drs. Que and Ingram report grants from National Institutes of Health during the conduct of the study, and report grants from Sanofi/Regeneron, outside the submitted work. The authors report no conflicts of interest in this work.
References
1. Michaeloudes C, Abubakar-Waziri H, Lakhdar R, et al. Molecular mechanisms of oxidative stress in asthma. Mol Aspects Med. 2022;85:101026. doi:10.1016/j.mam.2021.101026
2. van der Vliet A, Janssen-Heininger YMW, Anathy V. Oxidative stress in chronic lung disease: from mitochondrial dysfunction to dysregulated redox signaling. Mol Aspects Med. 2018;63:59–69. doi:10.1016/j.mam.2018.08.001
3. Betteridge DJ. What is oxidative stress? Metabolism. 2000;49(2Suppl 1):3–8. doi:10.1016/s0026-0495(00)80077-3
4. Sahiner UM, Birben E, Erzurum S, Sackesen C, Kalayci O. Oxidative stress in asthma: part of the puzzle. Pediatr Allergy Immunol. 2018;29(8):789–800. doi:10.1111/pai.12965
5. Liu Z, Ren Z, Zhang J, et al. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. 2018;9:477. doi:10.3389/fphys.2018.00477
6. CDC. Most recent national asthma data. Available from: https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm.
7. Su X, Ren Y, Li M, Zhao X, Kong L, Kang J. Prevalence of comorbidities in asthma and nonasthma patients: a meta-analysis. Medicine. 2016;95(22):e3459. doi:10.1097/MD.0000000000003459
8. Chapman DG, Irvin CG. Mechanisms of airway hyper-responsiveness in asthma: the past, present and yet to come. Clin Exp Allergy. 2015;45(4):706–719. doi:10.1111/cea.12506
9. Tan HT, Hagner S, Ruchti F, et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. 2019;74(2):294–307. doi:10.1111/all.13619
10. Hinks TSC, Levine SJ, Brusselle GG. Treatment options in type-2 low asthma. Eur Respir J. 2021;57(1):2000528. doi:10.1183/13993003.00528-2020
11. Ricciardolo FLM, Sprio AE, Baroso A, et al. Characterization of T2-low and T2-high asthma phenotypes in real-life. Biomedicines. 2021;9(11):1684. doi:10.3390/biomedicines9111684
12. Yaghoubi M, Adibi A, Safari A, FitzGerald JM, Sadatsafavi M. The projected economic and health burden of uncontrolled asthma in the United States. Am J Respir Crit Care Med. 2019;200(9):1102–1112. doi:10.1164/rccm.201901-0016OC
13. Guilbert TW, Garris C, Jhingran P, et al. Asthma that is not well-controlled is associated with increased healthcare utilization and decreased quality of life. J Asthma. 2011;48(2):126–132. doi:10.3109/02770903.2010.535879
14. NHLBI Obesity Education Initiative Expert Panel on the Identification E, and Treatment of Obesity in Adults (US). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults. National Heart, Lung, and Blood Institute; 1998:98–4083.
15. Fernandez-Sanchez A, Madrigal-Santillan E, Bautista M, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12(5):3117–3132. doi:10.3390/ijms12053117
16. Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114(12):1752–1761. doi:10.1172/JCI21625
17. Beltowski J, Wojcicka G, Jamroz A. Leptin decreases plasma paraoxonase 1 (PON1) activity and induces oxidative stress: the possible novel mechanism for proatherogenic effect of chronic hyperleptinemia. Atherosclerosis. 2003;170(1):21–29. doi:10.1016/s0021-9150(03)00236-3
18. Cinkajzlova A, Mraz M, Haluzik M. Adipose tissue immune cells in obesity, type 2 diabetes mellitus and cardiovascular diseases. J Endocrinol. 2021;252(1):R1–R22. doi:10.1530/JOE-21-0159
19. Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol. 2019;10:1587. doi:10.3389/fimmu.2019.01587
20. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. 2021;320(3):C375–C391. doi:10.1152/ajpcell.00379.2020
21. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi:10.1038/nri2921
22. McGarry ME, Castellanos E, Thakur N, et al. Obesity and bronchodilator response in black and Hispanic children and adolescents with asthma. Chest. 2015;147(6):1591–1598. doi:10.1378/chest.14-2689
23. Farah CS, Kermode JA, Downie SR, et al. Obesity is a determinant of asthma control independent of inflammation and lung mechanics. Chest. 2011;140(3):659–666. doi:10.1378/chest.11-0027
24. Taylor B, Mannino D, Brown C, Crocker D, Twum-Baah N, Holguin F. Body mass index and asthma severity in the national asthma survey. Thorax. 2008;63(1):14–20. doi:10.1136/thx.2007.082784
25. Scott HA, Gibson PG, Garg ML, et al. Dietary restriction and exercise improve airway inflammation and clinical outcomes in overweight and obese asthma: a randomized trial. Clin Exp Allergy. 2013;43(1):36–49. doi:10.1111/cea.12004
26. Komakula S, Khatri S, Mermis J, et al. Body mass index is associated with reduced exhaled nitric oxide and higher exhaled 8-isoprostanes in asthmatics. Respir Res. 2007;8:32. doi:10.1186/1465-9921-8-32
27. Shore SA, Rivera-Sanchez YM, Schwartzman IN, Johnston RA. Responses to ozone are increased in obese mice. J Appl Physiol. 2003;95(3):938–945. doi:10.1152/japplphysiol.00336.2003
28. Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42(6):563–570. doi:10.1016/j.amepre.2011.10.026
29. CDC. Asthma and obesity. Available from: https://www.cdc.gov/asthma/asthma_stats/asthma_obesity.htm.
30. Holguin F. Oxidative stress in airway diseases. Ann Am Thorac Soc. 2013;10(Supplement):S150–S157. doi:10.1513/AnnalsATS.201305-116AW
31. David WH, Riches TRM. Overview of innate lung immunity and inflammation. In: Lung Innate Immunity and Inflammation Methods in Molecular Biology. Vol. 1809. Springer; 2018.
32. Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. 2008;5(7):772–777. doi:10.1513/pats.200805-041HR
33. Wang SZ, Rosenberger CL, Bao YX, Stark JM, Harrod KS. Clara cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J Immunol. 2003;171(2):1051–1060. doi:10.1183/13993003.00528-2020
34. Hiemstra PS, McCray PB
35. Campbell HK, Maiers JL, DeMali KA. Interplay between tight junctions & adherens junctions. Exp Cell Res. 2017;358(1):39–44. doi:10.1016/j.yexcr.2017.03.061
36. Schlingmann B, Molina SA, Koval M. Claudins: gatekeepers of lung epithelial function. Semin Cell Dev Biol. 2015;42:47. doi:10.1016/J.SEMCDB.2015.04.009
37. Loxham M, Davies DE. Phenotypic and genetic aspects of epithelial barrier function in asthmatic patients. J Allergy Clin Immunol. 2017;139(6):1736–1751. doi:10.1016/j.jaci.2017.04.005
38. Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13(9):3218–3234. doi:10.1091/mbc.e02-03-0134
39. Yuksel H, Ocalan M, Yilmaz O. E-cadherin: an important functional molecule at respiratory barrier between defence and dysfunction. Front Physiol. 2021;12:720227. doi:10.3389/fphys.2021.720227
40. Erle DJ, Sheppard D. The cell biology of asthma. J Cell Biol. 2014;205(5):621–631. doi:10.1083/jcb.201401050
41. Gon Y, Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int. 2018;67(1):12–17. doi:10.1016/j.alit.2017.08.011
42. Hough KP, Curtiss ML, Blain TJ, et al. Airway remodeling in asthma. Front Med. 2020;7:191. doi:10.3389/fmed.2020.00191
43. Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. 2010;17(4):e85–e93. doi:10.1155/2010/318029
44. Xiao C, Puddicombe SM, Field S, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128(3):549–612. doi:10.1016/j.jaci.2011.05.038
45. Wedes SH, Khatri SB, Zhang R, et al. Noninvasive markers of airway inflammation in asthma. Clin Transl Sci. 2009;2(2):112–117. doi:10.1111/j.1752-8062.2009.00095.x
46. Nadeem A, Chhabra SK, Masood A, Raj HG. Increased oxidative stress and altered levels of antioxidants in asthma. J Allergy Clin Immunol. 2003;111(1):72–78. doi:10.1067/mai.2003.17
47. Zhao JJ, Shimizu Y, Dobashi K, et al. The relationship between oxidative stress and acid stress in adult patients with mild asthma. J Investig Allergol Clin Immunol. 2008;18(1):41–45.
48. Ganas K, Loukides S, Papatheodorou G, Panagou P, Kalogeropoulos N. Total nitrite/nitrate in expired breath condensate of patients with asthma. Respir Med. 2001;95(8):649–654. doi:10.1053/rmed.2001.1117
49. Svensson S, Olin AC, Larstad M, Ljungkvist G, Toren K. Determination of hydrogen peroxide in exhaled breath condensate by flow injection analysis with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;809(2):199–203. doi:10.1016/j.jchromb.2004.06.027
50. Bayram H, Rusznak C, Khair OA, Sapsford RJ, Abdelaziz MM. Effect of ozone and nitrogen dioxide on the permeability of bronchial epithelial cell cultures of non-asthmatic and asthmatic subjects. Clin Exp Allergy. 2002;32(9):1285–1292. doi:10.1046/j.1365-2745.2002.01435.x
51. Que LG, Stiles JV, Sundy JS, Foster WM. Pulmonary function, bronchial reactivity, and epithelial permeability are response phenotypes to ozone and develop differentially in healthy humans. J Appl Physiol. 2011;111(3):679–687. doi:10.1152/japplphysiol.00337.2011
52. Kehrl HR, Vincent LM, Kowalsky RJ, et al. Ozone exposure increases respiratory epithelial permeability in humans. Am Rev Respir Dis. 1987;135(5):1124–1128. doi:10.1164/arrd.1987.135.5.1124
53. Del Donno M, Chetta A, Foresi A, Olivieri D, Gavaruzzi G, Ugolotti G. Lung epithelial permeability and bronchial responsiveness in subjects with stable asthma. Chest. 1997;111(5):1255–1260. doi:10.1378/chest.111.5.1255
54. Elwood RK, Kennedy S, Belzberg A, Hogg JC, Pare PD. Respiratory mucosal permeability in asthma. Am Rev Respir Dis. 1983;128(3):523–527. doi:10.1164/arrd.1983.128.3.523
55. Georas S, Ransom N, Hillman S, et al. The leaky lung test: a pilot study using inhaled mannitol to measure airway barrier function in asthma. J Asthma. 2019;56(12):1257–1265. doi:10.1080/02770903.2018.1536145
56. Liu J, Chen X, Dou M, et al. Particulate matter disrupts airway epithelial barrier via oxidative stress to promote Pseudomonas aeruginosa infection. J Thorac Dis. 2019;11(6):2617–2627. doi:10.21037/jtd.2019.05.77
57. Kim BG, Lee PH, Lee SH, Park CS, Jang AS. Impact of ozone on claudins and tight junctions in the lungs. Environ Toxicol. 2018;33(7):798–806. doi:10.1002/tox.22566
58. Michaudel C, Mackowiak C, Maillet I, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol. 2018;142(3):942–958. doi:10.1016/j.jaci.2017.11.044
59. Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134(3):509–520. doi:10.1016/j.jaci.2014.05.049
60. Grasemann H, Holguin F. Oxidative stress and obesity-related asthma. Paediatr Respir Rev. 2021;37:18–21. doi:10.1016/j.prrv.2020.05.004
61. Wells SM, Holian A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol. 2007;36(5):520–528. doi:10.1165/rcmb.2006-0302SM
62. Winnica D, Corey C, Mullett S, et al. Bioenergetic differences in the airway epithelium of lean versus obese asthmatics are driven by nitric oxide and reflected in circulating platelets. Antioxid Redox Signal. 2019;31(10):673–686. doi:10.1089/ars.2018.7627
63. Devarajan A, Bourquard N, Hama S, et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid Redox Signal. 2011;14(3):341–351. doi:10.1089/ars.2010.3430
64. Winnica DE, Monzon A, Ye S, et al. Airway epithelial paraoxonase-2 in obese asthma. PLoS One. 2022;17(3):e0261504. doi:10.1371/journal.pone.0261504
65. Schiffers C, Reynaert NL, Wouters EFM, van der Vliet A. Redox dysregulation in aging and COPD: role of NOX enzymes and implications for antioxidant strategies. Antioxidants. 2021;10(11):1799. doi:10.3390/antiox10111799
66. Comstock AT, Ganesan S, Chattoraj A, et al. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. 2011;85(13):6795–6808. doi:10.1128/JVI.02074-10
67. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178(12):1271–1281. doi:10.1164/rccm.200801-136OC
68. Singh D, McCann KL, Imani F. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L436–L445. doi:10.1152/ajplung.00097.2007
69. Corne JM, Marshall C, Smith S, et al. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359(9309):831–834. doi:10.1016/S0140-6736(02)07953-9
70. Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307(6910):982–986. doi:10.1136/bmj.307.6910.982
71. Tang M, Henderson RJ, Holbrook JT, et al. Does obesity increase respiratory tract infections in patients with asthma? J Allergy Clin Immunol Pract. 2019;7(3):954–961e6. doi:10.1016/j.jaip.2018.09.033
72. Spannhake EW, Reddy SP, Jacoby DB, Yu XY, Saatian B, Tian J. Synergism between rhinovirus infection and oxidant pollutant exposure enhances airway epithelial cell cytokine production. Environ Health Perspect. 2002;110(7):665–670. doi:10.1289/ehp.02110665
73. Le Dréan G, Haure-Mirande V, Ferrier L, et al. Visceral adipose tissue and leptin increase colonic epithelial tight junction permeability via a RhoA-ROCK-dependent pathway. FASEB J. 2014;28(3):1059–1070. doi:10.1096/fj.13-234203
74. Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. 2006;74:443–477. doi:10.1016/S0083-6729(06
75. Maslov LN, Naryzhnaya NV, Boshchenko AA, Popov SV, Ivanov VV, Oeltgen PR. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J Clin Transl Endocrinol. 2019;15:1–5. doi:10.1016/j.jcte.2018.11.001
76. Monickaraj F, Aravind S, Nandhini P, et al. Accelerated fat cell aging links oxidative stress and insulin resistance in adipocytes. J Biosci. 2013;38(1):113–122. doi:10.1007/s12038-012-9289-0
77. Berry MA, Hargadon B, Shelley M, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354(7):697–708. doi:10.1056/NEJMoa050580
78. Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. 1992;89(5):958–967. doi:10.1016/0091-6749(92)90218-q
79. Manni ML, Trudeau JB, Scheller EV, et al. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol. 2014;7(5):1186–1198. doi:10.1038/mi.2014.8
80. Holgate ST, Noonan M, Chanez P, et al. Efficacy and safety of etanercept in moderate-to-severe asthma: a randomised, controlled trial. Eur Respir J. 2011;37(6):1352–1359. doi:10.1183/09031936.00063510
81. Taille C, Poulet C, Marchand-Adam S, et al. Monoclonal anti-TNF-alpha antibodies for severe steroid-dependent asthma: a case series. Open Respir Med J. 2013;7:21–25. doi:10.2174/1874306401307010021
82. Hardyman MA, Wilkinson E, Martin E, et al. TNF-alpha-mediated bronchial barrier disruption and regulation by src-family kinase activation. J Allergy Clin Immunol. 2013;132(3):665–675 e8. doi:10.1016/j.jaci.2013.03.005
83. Huang S, Dudez T, Scerri I, et al. Defective activation of c-Src in cystic fibrosis airway epithelial cells results in loss of tumor necrosis factor-alpha-induced gap junction regulation. J Biol Chem. 2003;278(10):8326–8332. doi:10.1074/jbc.M208264200
84. Tashiro H, Shore SA. Obesity and severe asthma. Allergol Int. 2019;68(2):135–142. doi:10.1016/j.alit.2018.10.004
85. Marini M, Avoni E, Hollemborg J, Mattoli S. Cytokine mRNA profile and cell activation in bronchoalveolar lavage fluid from nonatopic patients with symptomatic asthma. Chest. 1992;102(3):661–669. doi:10.1378/chest.102.3.661
86. Virchow JC
87. Peters MC, McGrath KW, Hawkins GA, et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med. 2016;4(7):574–584. doi:10.1016/S2213-2600(16)30048-0
88. Jevnikar Z, Ostling J, Ax E, et al. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. 2019;143(2):577–590. doi:10.1016/j.jaci.2018.05.026
89. Callaghan PJ, Rybakovsky E, Ferrick B, Thomas S, Mullin JM. Retinoic acid improves baseline barrier function and attenuates TNF-alpha-induced barrier leak in human bronchial epithelial cell culture model, 16HBE 14o. PLoS One. 2020;15(12):e0242536. doi:10.1371/journal.pone.0242536
90. Al-Sadi R, Ye D, Boivin M, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One. 2014;9(3):e85345. doi:10.1371/journal.pone.0085345
91. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. 2011;286(36):31263–31271. doi:10.1074/jbc.M111.238147
92. Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178(7):4641–4649. doi:10.4049/jimmunol.178.7.4641
93. Genser L, Aguanno D, Soula HA, et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J Pathol. 2018;246(2):217–230. doi:10.1002/path.5134
94. Shore SA. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol. 2010;108(3):735–743. doi:10.1152/japplphysiol.00749.2009
95. Ahmad R, Rah B, Bastola D, Dhawan P, Singh AB. Obesity-induces organ and tissue specific tight junction restructuring and barrier deregulation by claudin switching. Sci Rep. 2017;7(1):5125. doi:10.1038/s41598-017-04989-8
96. Suzuki T, Hara H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr Metab. 2010;7(1):19. doi:10.1186/1743-7075-7-19
97. Marseglia L, Manti S, D’Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2014;16(1):378–400. doi:10.3390/ijms16010378
98. Iikuni N, Lam QL, Lu L, Matarese G, La Cava A. Leptin and inflammation. Curr Immunol Rev. 2008;4(2):70–79. doi:10.2174/157339508784325046
99. Bruno A, Pace E, Chanez P, et al. Leptin and leptin receptor expression in asthma. J Allergy Clin Immunol. 2009;124(2):230–7, 237 e1–4. doi:10.1016/j.jaci.2009.04.032
100. Holguin F, Rojas M, Hart CM. The peroxisome proliferator activated receptor gamma (PPARgamma) ligand rosiglitazone modulates bronchoalveolar lavage levels of leptin, adiponectin, and inflammatory cytokines in lean and obese mice. Lung. 2007;185(6):367–372. doi:10.1007/s00408-007-9035-9
101. Holguin F, Fitzpatrick A. Obesity, asthma, and oxidative stress. J Appl Physiol. 2010;108(3):754–759. doi:10.1152/japplphysiol.00702.2009
102. Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, Johnston RA. Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol. 2005;115(1):103–109. doi:10.1016/j.jaci.2004.10.007
103. Brun P, Castagliuolo I, Di Leo V, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292(2):G518–G525. doi:10.1152/ajpgi.00024.2006
104. Zhang Y, Saradna A, Ratan R, et al. RhoA/Rho-kinases in asthma: from pathogenesis to therapeutic targets. Clin Transl Immunol. 2020;9(5):e01134. doi:10.1002/cti2.1134
105. Castro M, King TS, Kunselman SJ, et al. Effect of vitamin D3 on asthma treatment failures in adults with symptomatic asthma and lower vitamin D levels: the VIDA randomized clinical trial. JAMA. 2014;311(20):2083–2091. doi:10.1001/jama.2014.5052
106. Johnson O, Gerald LB, Harvey J, et al. An online weight loss intervention for people with obesity and poorly controlled asthma. J Allergy Clin Immunol Pract. 2022;10(6):1577–1586 e3. doi:10.1016/j.jaip.2022.02.040
107. Hinnen D. Glucagon-like peptide 1 receptor agonists for type 2 diabetes. diabetes spectr. 2017;30(3):202–210. doi:10.2337/ds16-0026
108. Foer D, Beeler PE, Cui J, Karlson EW, Bates DW, Cahill KN. Asthma exacerbations in patients with type 2 diabetes and asthma on glucagon-like peptide-1 receptor agonists. Am J Respir Crit Care Med. 2021;203(7):831–840. doi:10.1164/rccm.202004-0993OC
109. Rogliani P, Calzetta L, Capuani B, et al. Glucagon-like peptide 1 receptor: a novel pharmacological target for treating human bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2016;55(6):804–814. doi:10.1165/rcmb.2015-0311OC
110. Danielewicz H. What the genetic background of individuals with asthma and obesity can reveal: is beta2-adrenergic receptor gene polymorphism important? Pediatr Allergy Immunol Pulmonol. 2014;27(3):104–110. doi:10.1089/ped.2014.0360
111. Cazzola M, Page CP, Rogliani P, Matera MG. Beta 2-agonist therapy in lung disease. Am J Respir Crit Care Med. 2013;187(7):690–696. doi:10.1164/rccm.201209-1739PP
112. Dixon AE, Que LG, Kalhan R, et al. Roflumilast may increase risk of exacerbations when used to treat poorly controlled asthma in people with obesity. Ann Am Thorac Soc. 2023;20(2):206–214. doi:10.1513/AnnalsATS.202204-368OC
113. Chandrasekaran R, Bruno SR, Mark ZF, et al. Mitoquinone mesylate attenuates pathological features of lean and obese allergic asthma in mice. Am J Physiol Lung Cell Mol Physiol. 2023;324(2):L141–L153. doi:10.1152/ajplung.00249.2022
114. Holguin F, Grasemann H, Sharma S, et al. L-Citrulline increases nitric oxide and improves control in obese asthmatics. JCI Insight. 2019;4(24). doi:10.1172/jci.insight.131733
115. Sweerus K, Lachowicz-Scroggins M, Gordon E, et al. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J Allergy Clin Immunol. 2017;139(1):72–81e1. doi:10.1016/j.jaci.2016.02.035
116. Saatian B, Rezaee F, Desando S, et al. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1(2):e24333. doi:10.4161/tisb.24333
117. Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J. 2010;36(5):1016–1026. doi:10.1183/09031936.00125809
118. Trautmann A, Kruger K, Akdis M, et al. Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol. 2005;138(2):142–150. doi:10.1159/000088436
119. Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Invest. 2012;92(8):1140–1148. doi:10.1038/labinvest.2012.67
120. Post S, Nawijn MC, Hackett TL, et al. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax. 2012;67(6):488–495. doi:10.1136/thoraxjnl-2011-200606
121. Wan H, Winton HL, Soeller C, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104(1):123–133. doi:10.1172/JCI5844
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.