")
Back to Journals » Cancer Management and Research » Volume 12
Effects of Nicotinamide on Cervical Cancer-Derived Fibroblasts: Evidence for Therapeutic Potential
Authors Hassan RN , Luo H , Jiang W
Received 31 August 2019
Accepted for publication 16 January 2020
Published 12 February 2020 Volume 2020:12 Pages 1089—1100
DOI https://doi.org/10.2147/CMAR.S229395
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Reem N Hassan,* Hualei Luo,* Weiying Jiang
Department of Medical Genetics, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou, Guangdong 510080, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weiying Jiang
Department of Medical Genetics, Zhongshan School of Medicine, Sun Yat-Sen University, No. 74, Zhongshan Second Road, Yuexiu District, Guangzhou, Guangdong 510080, People’s Republic of China
Tel +86-20-87331928
Email [email protected]
Purpose: The present study aimed to examine the effects of nicotinamide (NAM) on cervical cancer-associated fibroblasts (CAF) for its in vitro efficacy, gross inhibition, and mechanism of inhibition.
Methods: The fibroblasts were treated with pre-specified concentrations of NAM followed by measurement of the cell proliferation using CCK-8 assay. The production of reactive oxygen species (ROS) was measured by 2ʹ,7ʹ-Dichlorofluorescin diacetate. We further investigated the apoptosis by flow cytometry using Annexin-V. We employed JC-1 assay to detect changes in the potential of the mitochondrial membrane. We further determined the expression of apoptotic genes was measured using qRT-PCR. And lastly, cell cycle experiments were conducted to determine the influence of NAM on arresting the growth of CAF in a cell cycle.
Results: Our study showed that NAM was able to reduce fibroblasts viability. We specifically observed a significantly increased intracellular ROS with resultant exhaustion of cellular antioxidant defense machinery, including reduced glutathione (GSH). We further observed the involvement of mitochondrial pathway in the NAM induced apoptosis of fibroblasts.
Conclusion: Our study supports the therapeutic potential of NAM for the treatment of cervical cancer and necessitates a further investigation of the reported findings.
Keywords: apoptosis, cervical cancer, fibroblasts, mitochondrial pathway, nicotinamide, therapeutic potential
Introduction
Cervical cancer is a spectrum of disease with a gradual progression from pre-malignant condition to aggressive disease.1 The onset of the disease begins in the cervix, which later spreads towards the lower uterine segment, vagina, para-cervical space, along with the broad and uterosacral ligaments. Current treatment approaches involve mainly the use of intensive chemotherapy, aided by radiotherapy and surgery. Cervical cancer patients have an overall five-year survival of 84% across all the stages.2 However, the survival rate is further reduced to 17%, if cancer manages to metastasis to distant organs in the body, which emphasizes the importance of early diagnosis and treatment of cancer.3 Unfortunately, drug resistance is inevitable in this cancer, which further makes it very difficult to contain and prevent metastasis. For instance, resistance to cisplatin, one of the most commonly used drug for the treatment of cervical cancer, is a frequently reported observation.4–6 Henceforth, alternative therapeutic strategies are urgently needed to not only to improve the treatment outcome but also to reduce the associated cost of treatment.
Nicotinamide (NAM), the amide form of niacin (i.e., vitamin B3) is known to play a major role in the metabolic pathway involved in the production of nicotinamide adenine dinucleotide (NAD+).7,8 It has been long used to treat pellagra, a diseased condition characterized by severe dermatitis, diarrhea, dementia, and multi-organ dysfunction.9,10 Furthermore, the therapeutic benefits of NAM has also been observed in the treatment of actinic keratosis11 and melanoma.12,13 NAM primarily exerts its effect on cellular energy metabolism through its precursor nicotinamide adenine dinucleotide (NAD+), a component of the electron transport chain machinery in the mitochondria. NAM henceforth plays a crucial role in cell metabolism and regulates cell survival and cell death.14 NAD+ is henceforth, also known as a universal pro-survival factor, which plays a vital role in protecting the cell against extrinsic and intrinsic risk factors.15 NAM, as a backbone of phosphorylated NAD+ (i.e., NADP+), is also known to participate in the synthesis of fatty acids and thereby maintains the oxidative balance in the cells.16
In consent to its central role on several intracellular pathways in the cell, the potential therapeutic role of NAM has been well documented in the literature. Its therapeutic efficacy has been widely studied on various cancer cell models, namely pancreatic cancer,17 chronic lymphocytic leukaemia,18 prostate,19 and breast cancers.20 For instance, Feng et al recently investigated the effects of NAM on cervical cancer cell lines and demonstrated significant gross inhibition of the cell lines primarily mediated by oxidative stress.20 In addition, CAFs have been recently shown to modulate components of the immune system, leading to evasion immune response to cancer immunotherapy.21 Another study revealed the potential of CAFs to induce drug resistance in cancer cells.22 Furthermore, animal model studies have shown that anti-cancer agents such as N-acetyl L-cysteine (NAC) can modulate the interaction between stromal cells and cancer cells by influencing the NAM mediated oxidative damage.23 NAC is thereby a known antioxidant that prevents cells from damage by free oxygen radicals and other cytotoxic substances in vitro as well as in vivo. However, NAC potential combination with NAM in cervical cancers has not been explored to date.
Recently, cancer-associated fibroblasts (CAFs), one of the stromal cell types in cancer mesenchyme, have evolved as a potential target of anti-cancer immunotherapy. The CAFs are known to be one of the most critical component of the tumor microenvironment (TME) that not only promote tumorigenesis but may also contribute towards therapeutic resistance.24–26 These recent findings further emphasize the enormous potential of studying the effects of novel therapeutic agents on microenvironment components of cervical cancer lines for developing novel anticancer drugs. Henceforth, the present study aims to examine the effects of NAM as well NAM in combination with NAC on the cervical cancer-associated fibroblasts for its in vitro efficacy, gross inhibition, and mechanism of inhibition.
Patient Consent and Research Ethics
The present study was obtained from a primary cervical cancer patient through surgical biopsy of the tissue. The patient provided written informed consent for the tissue to be used for inclusion in this study. Furthermore, this study was approved by Zhongshan school of medicine local ethics committee, Sun Yat-sen University and conducted our study according to the Ethical Guidelines for Human Genome/Gene Research enacted by the Helsinki Declaration.
Materials and Methods
Materials
Following reagents ere used: PBS (Thermo Fisher Scientific, China), Trypsin (Sigma-Aldrich (Vermont, USA), 6-carboxy-2ʹ,7ʹ-dichlorofluorescein diacetate (DCFH-DA, Molecular Probes), DMEM medium (GEMINI Company, USA), Fetal bovine serum (GEMINI Company, USA)., collagenase (Sigma Chemical Co., St. Louis, MO, USA), Anti-fibroblast microbeads (MiltenyiBiotec, Auburn, CA, USA), reagents for flow cytometry (Becton, Dickinson and Company, USA), NAM and NAC (Sigma Aldrich, China), antibodies (Abcam, Cambridge, UK)), In addition, Sterile plastic materials were used for cell culture (Corning, Inc.).
Methods
Isolation, Cell Sorting and Culture
The fibroblast cells (CAF) for the experiments in the present study were obtained from a primary cervical cancer patient through surgical biopsy of the tissue.
The specimen was washed three times with PBS, then minced into approximately 1-2mm2 sized pieces followed by digestion using 10mL of a 0.1% trypsin solution containing 0.75 µg/µL collagenase I. After variable digestion, the homogenate was collected and passed through a 30mm strainer. The CAFs were then purified using the BD influx (BD Medical Technology, New Jersey, USA) method. The resulting CAFs were incubated for 30 mins with anti-fibroblast microbeads. After passing through a MiniMACS separator, positive cells were resuspended and plated in a DMEM (Dulbecco’s Modified Eagle Medium) containing 1% penicillin/streptomycin, and 10% FBS (fetal bovine serum). After 24 hrs culture, the medium was replaced to remove floating cells to obtain purified fibroblasts for the present study. The CAFs were suspended in DMEM containing 10% fetal bovine serum (FBS) with 1% penicillin/streptomycin and cultured in a constant temperature incubator at 37ºC with 5% carbon dioxide.29
Treatment with Nicotinamide
At the beginning of each experiment, the cells were seeded to well plates (6 or 96 as per the experimental procedure) for 24 hrs with each well comprising of 3×105 cells in 6 well plate experiments and 3×105 cells in 96 well plate experiments. After 24 hrs of incubating the fibroblast, different dilutions of NAM were prepared in varying concentrations of 0 (negative control), 1, 2 and 4 mg/mL, for pretreatment of the fibroblasts. Also, a group of cells were pretreated with NAC with a final concentration of 5 Mm followed by incubation for 1 hr at 37ºC and then finally treated with 2 mg/mL of NAM then the cells are incubated at 37ºC for 24 hrs’ time period. Post-treatment and incubation for 24 hrs, we conducted cell-viability assay, Intracellular ROS, GSH and MDA measurements, mitochondrial Membrane Potential, targeted gene expression and cell-cycle experiments.
Cell Viability Assay
The cell viability was evaluated using the cell counting kit (CCK-8, Dojindo Molecular Technologies, Japan), as per the manufacturer’s instructions to detect proliferation. The fibroblasts were seeded in 96-well plates with 3×103 cells per well, and incubated for 24 hrs at 37ºC with varying concentrations of NAM: 0 mg/mL (control), 0.25 mg/mL, 0.5 mg/mL, 1 mg/mL, 2 mg/mL, and 4 mg/mL. The cells were then incubated at 37ºC. Post-incubations, the plates were read using the microplate reader at 450 nm. The cell proliferation growth curves were constructed at 14, 24, 36, 56, and 72 hrs. The mean optical density (OD) or absorbance were calculated for each time point. Graphical representation of Individual OD curves was compared for assessing cellular viability. Different doses of NAM were selected to be used for later experiments. All the experiments were conducted in triplicates.
Intracellular ROS, GSH and MDA Measurement
We used the DCFH-DA assay (Sigma-Aldrich, Vermont, USA) for measuring the ROS level. In this experiment, an extra well of fibroblasts (untreated with NAM) was added to be used as positive control cells. Post incubation i.e., after treatment with varying concentrations of NAM and collecting the cells in tubes, DCFH-DA dye with a final concentration of 10µM was added to each tube (except for the negative control cells tube) and incubated at 37ºC for 15 mins. The tubes were vigorously shaken during the incubation. In the positive control tube, 1µL of Rosup buffer (50mg/mL) was added to the cells and incubated at 37ºC for 20 mins. Post incubation, the dye solution was removed by centrifugation followed by washing twice with cold PBS. Finally, ROS was detected in the cells by CytoFlex (Beckman Coulter, USA) after transferring to the flow tubes. All the experiments were conducted in triplicates.
Glutathione and lipid peroxidation were measured using the Reduced glutathione (GSH) assay kit (Nanjing Jiancheng Bioengineering Institute, China) and Malondialdehyde (MDA) assay kit (Nanjing Jiancheng Bioengineering Institute, China), respectively. Post incubation, cells were harvested followed by sonication and homogenization in ice PBS buffer. This was followed by centrifugation of cell lysates for 15 mins with 12,000 g content at 4°C. The supernatant was collected to measure the protein levels. We used total protein BCA assay kit (A045–3, Nanjing Jiancheng Bioengineering Institute) to measure total protein concentrations, before subjecting the cells to GSH assay and MDA assay as elaborated in the manufacturers’ guidelines. Afterwards, the levels of GSH and MDA were detected using a microplate reader at 405 nm and 532 nm, respectively. All experiments were performed in triplicates.
Mitochondrial Membrane Potential
We used the Mitochondrial membrane potential assay kit with JC-1 (Beyotime, China) to measure the mitochondrial membrane potential after treatment of CAFs with various concentrations of nicotinamide treatment. Post 24 hrs incubation and collecting the cells in tubes, two buffers were used for the JC-1 assay kit. The JC-1 assay kit primarily employs two buffers. The first buffer is JC-1 dye buffer, which was prepared by diluting 4mL of the JC-1 (5X) reagent with 16mL of cold ddH2O. The second buffer was prepared by diluting 100µL of JC-1 Buffer (light-sensitive buffer) with 16 mL of ddH2O. As a first step, 1µL of cccp buffer was added to the positive control group cells well and then incubated for 20 mins at 37ºC. Next, the cells were collected then washed twice with PBS and centrifuged twice at 1000 rpm for 5 mins. This was followed by addition of 500µL of the light-sensitive buffer to all group of cells in the Eppendorf tubes except for the JC-1 negative control tube, in which only 500µL of the medium was added. The tubes were then shaken and incubated for 20 mins at 37ºC. After incubation, the tubes were centrifuged at 600xg for 3 mins followed by discarding of supernatants. At this stage, 1mL of the prepared Jc-1 dye buffer was added to all tubes, except for the negative control tube and tubes were then centrifuged for 3 mins at 4ºC at 600Xg. The last stage was repeated. Finally, 300µL of the JC-1 dye buffer was added to all tubes except negative controls tubes. The cells were transferred in CytoFlex (Beckman Coulter, USA). All experiments were performed in triplicates.
Targeted Gene Expression Using qPCR
Post incubation i.e., after treatment with varying concentrations of NAM, we evaluated the expression of apoptosis and redox-related genes. From the treated cells, total RNA was isolated using TRIzol followed by reverse transcription to cDNA on a PrimeScript RT reagent KIT (Takara Biomedical Technology, Beijing, China) according to the manufacturer’s guidelines. The purity and concentration of the total RNA for every sample was analyzed using OD260:OD280 ratio on Nanodrop 2000 (ThermoFisher Scientific, USA). The real-time SYBR Green PCR was undertaken using quantitative real-time PCR on CFX96 touch (Bio-Rad, California, USA). Each of the 20μL reaction volume well contained 7μL H2O, 1μL of each primer, 1μL of the template, and 10μL of 2×RealStar Power SYBR Mixture. The amplification reaction conditions included an initial denaturation for 10 mins at 95°C followed by 40 cycles each of denaturation for 15 seconds at 95°C, annealing for 25 seconds at 60°C, extension for 35 seconds at 72°C, and a final extension for 5 seconds at 72°C. All Threshold cycle (CT) values were repeated in triplicates. β-actin was used as an internal standard to normalize the relative gene expression levels, which were calculated with the 2−ΔΔCt method. All the primers have been further provided in Table 1.
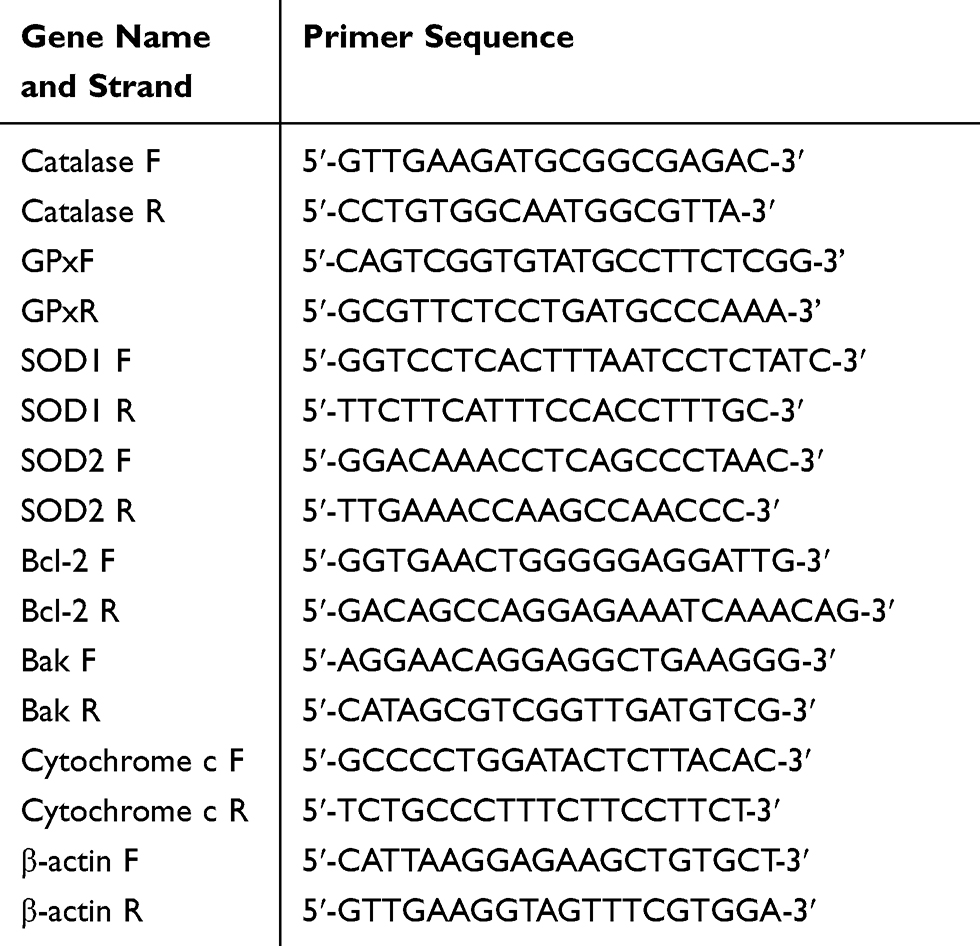 |
Table 1 PCR Primers Used in the Study |
Cell Cycle Experiments
Post incubation, i.e., after treatment with varying concentrations of NAMs, the cell cycle experiment was conducted using the Cell Cycle kit (Yeasen, China). The kit comprises of a mix-buffer of 1800µL of staining solution, 36µL of RNA solution (7µLx5), and 36µL of Propidium iodide (PI) buffer which was later mixed in a 15 mL falcon tube. All treated cells were transferred to five tubes of 15mL and centrifuged at 1000 RPM for 5 mins. After centrifugation, the supernatants were discarded and 500 mL of PBS was added to each tube. The tubes were again centrifuged at 1000 RPM for 5 mins, after which, the supernatants were discarded, and 1mL of ethanol was added to each tube followed by incubation at 4ºC for 2 hrs. At the end of incubation, the tubes were centrifuged again at 1000 RPM for 5 mins followed by discarding of ethanol. As a next step, 1mL of cold PBS was added to each tube followed by centrifugation at 1000 RPM for 5 mins. Finally, 300µL of the mix-buffer was added to each tube followed by incubation at 37°C for 30 mins for use in CytoFlex (Beckman Coulter, USA) for flow cytometric analysis. All the experiments were conducted in triplicates.
Statistical Analysis
Continuous variables were expressed in mean and standard deviations (sd). The distribution of continuous variables was compared using Student’s t-tests and one way ANOVA. A p-value of less than 0.05 was considered to be significant. All the statistical analysis was performed using GraphPad Prism (Version 8). The data generated in Flow cytometry was further analyzed using FlowJo®, version 10 (Ashland, Oregon, USA).
Results
Cell Viability Assay
The cell viability results at different concentrations of NAM are shown in Figure 1. Treatment with both 1 and 2 mg/mL NAM closely mimic the control with mean OD ranging from 0.5 to 1.5 at 24 hrs, 1.0 to 1.5 at 36 hrs, and 1.5 to 2.5 at 56 hrs. On the contrary, 4 mg/mL NAM treated cells showed a significant reduction in the viability as compared to the negative control (p<0.01), at 36 hrs’ time point (OD of 4 mg/mL: 0.5). Both, 2 mg/mL and 4 mg/mL induced a significant decrease in cell viability at 56 hrs as compared to negative control (p<0.01). The significant difference was further retained on comparison of 4 mg/mL NAM treated cells with other treatment groups (4 vs 2 mg/mL, p<0.04, 4 vs 1 mg/mL, p<0.009). The decline in viability continued with time and reached its lowest at 72 hrs corresponding to an OD of less than 0.4.
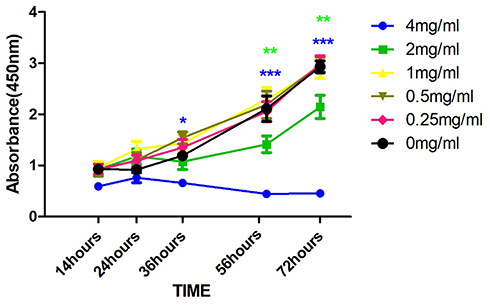 |
Figure 1 Cellular proliferation using CCK8 after drug treatment with varying concentration of Nicotinamide (NAM) at the absorbance of 450 nm (***p≤0.001, **p≤0.01, *p≤0.05). |
In summary, we observed a reduction in cell viability in a dose-dependent manner at different concentrations of NAM. Our results henceforth suggest that NAM could serve as a potent viability inhibitor of cervical cancer cell-derived fibroblasts.
Intracellular ROS, GSH and MDA Measurement
The concentrations of MDA and ROS for each treatment group are shown in Figure 2A and B. The 4 mg/mL NAM treated group showed the highest level of MDA and ROS (4 nmol/mg) which was significantly higher in comparison to all treatment groups including negative controls (p<0.01). Our results henceforth suggest that NAM induced oxidative stress that could lead to increased lipid peroxidation. The GSH assay further showed a significant reduction in glutathione levels in 4 mg/mL NAM treated cells compared to negative controls (p<0.01). We also observed a lower level of GSH in 2 mg/mL NAM treated cells in comparison to cells treated with NAC+ 2 mg/mL NAM (Figure 2C). Thus, pretreating the cells with NAC confers them the ability to be more effective against GSH reduction. We further observed a two-fold increase in mRNA expression levels of intracellular catalase (p<0.001) in the 1 mg/mL NAM treated group, approximately 3-fold increase (p<0.001) in the 2 mg/mL NAM treated group, and a 6 fold increase in the 4 mg/mL NAM treated group (p<0.001). A similar trend was observed in expression levels of SOD1 and SOD2, suggesting that the expression levels of the antioxidant enzymes in the cells are affected by NAM, resulting in an imbalance of redox balance (Figure 2D–F). In summary, our results show that NAM treatment induces an increase in ROS levels and a decrease in GSH levels in CAF, leading to oxidative stress in the cells.
 |
Figure 2 Nicotinamide induces oxidation of cervical cancer-associated fibroblasts (***p≤0.001, **p≤0.01, *p≤0.05). (A) MDA; (B) Mean fluorescent intensity of ROS; (C) GSH; (D) Relative mRNA level of Catalase; (E) Relative mRNA level of SOD1; (F) Relative mRNA level of SOD2. |
Cell Apoptosis
The two-dimensional dot plot showing apoptosis rate of CAF is shown in Figure 3A. The figure has been divided into four quadrants. The lower right quadrant represented by FITC+/PI- corresponds to early apoptosis of cells, and the upper right quadrant represented by FITC+/PI+ corresponds to late cell apoptosis. The results show that a greater number of cells enter the apoptotic state as a result of a higher concentration of NAM. Figure 3B and C further show that the early apoptosis rate of cells gradually increases with NAM concentration with a statistically significant difference at a concentration of 4 mg/mL. Concerning late apoptosis, we observed the most significant increase in the 2 mg/mL treated group (p<0.05). Our results further suggest that 1 mg/mL NAM treated cells begin to undergo apoptosis after 24 hrs of treatment. We also observed that NAC can partially reverse the apoptotic rate of CAF (Figure 3B and C), thereby preventing early apoptosis and reducing the late apoptotic rate of cells. In summary, our results suggest that NAM induces apoptosis of CAF by oxidative stress.
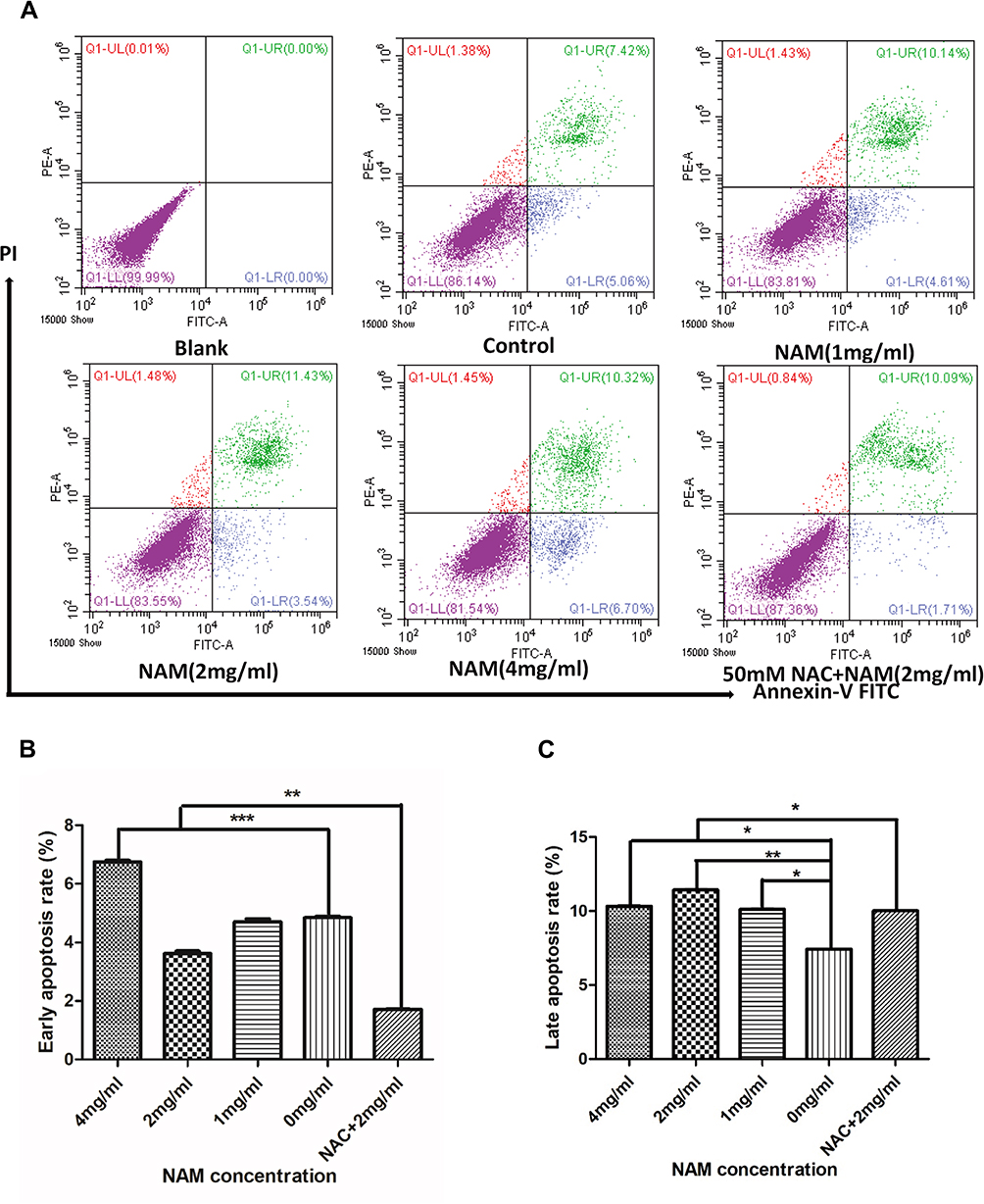 |
Figure 3 Nicotinamide (NAM) can induce apoptosis in cervical cancer-associated fibroblasts (***p≤0.001, **p≤0.01, *p≤0.05). (A) flow cytometry of quantitative detection of apoptosis by Annexin V method. Blank group: cells without NAM treatment, without FITC Annexin V buffer, and without Propidium Iodide staining solution (PI), while Control group: cells without NAM treatment, but with FITC buffer and PI solution.; (B) Early apoptosis; (C) Late apoptosis. |
Mitochondrial Membrane Potential
Compared with negative control, the 1 mg/mL NAM treated cells showed loss of mitochondrial membrane potential as indicated by an increased cell population in the lower right quadrant (26.7%) (Figure 4). We further observed a similar loss of mitochondrial membrane potential for all concentrations of NAM and NAC+NAM (2 mg/mL) treated cells. The NAC+NAM (2 mg/mL) treated cells, however, demonstrated a slightly higher proportion of cells showing a decline in membrane potential compared to cells treated with 2 mg/mL of NAM alone. In summary, NAM alone seemed to be sufficient to induce mitochondrial membrane potential decline in cervical cancer-associated fibroblasts.
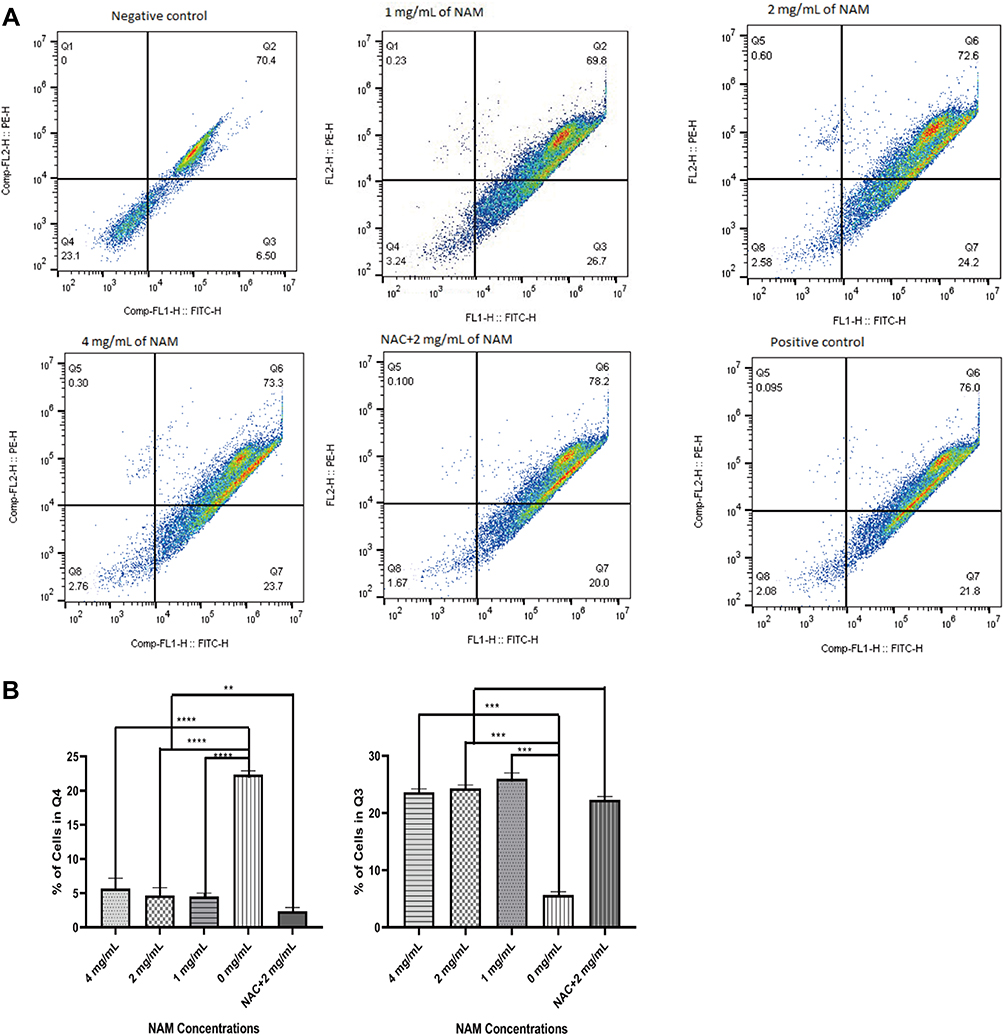 |
Figure 4 Mitochondrial membrane potential for nicotinamide (NAM) treated cells at varying concentrations along with N-acetyl-L-cysteine (NAC) pre-treated cells (****p≤0.0001, ***p≤0.001, **p≤0.01). (A) Representative scatterplot showing cells in quarters with NAM treatment. (B) Charts showing the proportions of cells in two quarters (Q4) and Q3 (higher proportion indicates loss of mitochondrial membrane potential). |
Targeted Gene Expression
The gene expression of apoptosis-related genes has been shown in Figure 5A–C. The studied genes have been further listed in Table 1. We observed higher mRNA expression in the cells treated with 4 mg/mL NAM compared to untreated cells and other treatment groups. Among all the genes studied, the most pronounced change was observed in the expression levels of cytochrome C with more than 80 log fold change (LFC) in the cells treated with 4 mg/mL NAM compared to untreated cells. In the same comparison, another prominent gene of interest was anti-apoptotic BAK gene, which showed more than 5 LFC increase in its expression levels. However, we observed a much larger increase in the expression of pro-apoptotic gene BCL-2, which demonstrated more than 35 LFC in cells treated with 4 mg/mL NAM compared to untreated cells. We further observed significant differences in the mRNA expression of antioxidant genes such as catalase, SOD1 and SOD2, on comparison of 2 mg/mL NAM treated cells with 2 mg/mL NAM+NAC treatment. In summary, our study suggests the ability of high concentration of NAM (4 mg/mL) to induce oxidative stress in CAFs since NAM treatment prompted the cells to recruit the anti-oxidative stress mechanisms at a molecular level. In addition, our study detected cleaved caspase-9 by Western blot and used GAPDH as an internal reference. Our findings further confirm the involvement of endogenous mitochondrial pathway in NAM induced CAF apoptosis.
 |
Figure 5 Nicotinamide-induced cervical cancer-associated fibroblast apoptosis depends on the mitochondrial pathway (***p≤0.001, *p≤0.05). (A) Bak; (B) bcl-2; (C) Cytochrome C mRNA levels; (D) Western Blot detection of cleaved caspase 9 protein expression with GAPDH as an internal reference; (E) Cleaved caspase 9 protein level quantitative analysis. |
Cell Cycle Experiments
We further conducted cell cycle experiments to assess if NAM treatment, in association with NAC arrests the growth of the CAFs (Figure 6). We observed a substantial but a non-significant reduction in the proportion of G1 cells in 4 mg/mL NAM treated cells in comparison to untreated cells (78.9% (4 mg/mL of NAM) vs 92.2% (control cells), p=0.017). However, we did observe a significant increase in the proportion of G2/M cells (9.56% (4 mg/mL of NAM) vs 2.74% (control cells), p=0.04). A similar significant increase in the proportion of G2/M cells was also observed when comparing NAC+2 mg/mL NAM treated cells to 2 mg/mL NAM treated cells (8.87% (NAC+ 2 mg/mL) vs 4.01% (2mg/mL of NAM), p=0.03), suggesting that NAC drives the cells from G1 to G2/M phase of cell cycle. In summary, our data suggest that both high concentration of NAM (4 mg/mL) or lower concentration of NAM (2 mg/mL) in combination with NAC promotes growth arrest in CAFs, indicative of the lethal effect of NAM as well as NAC.
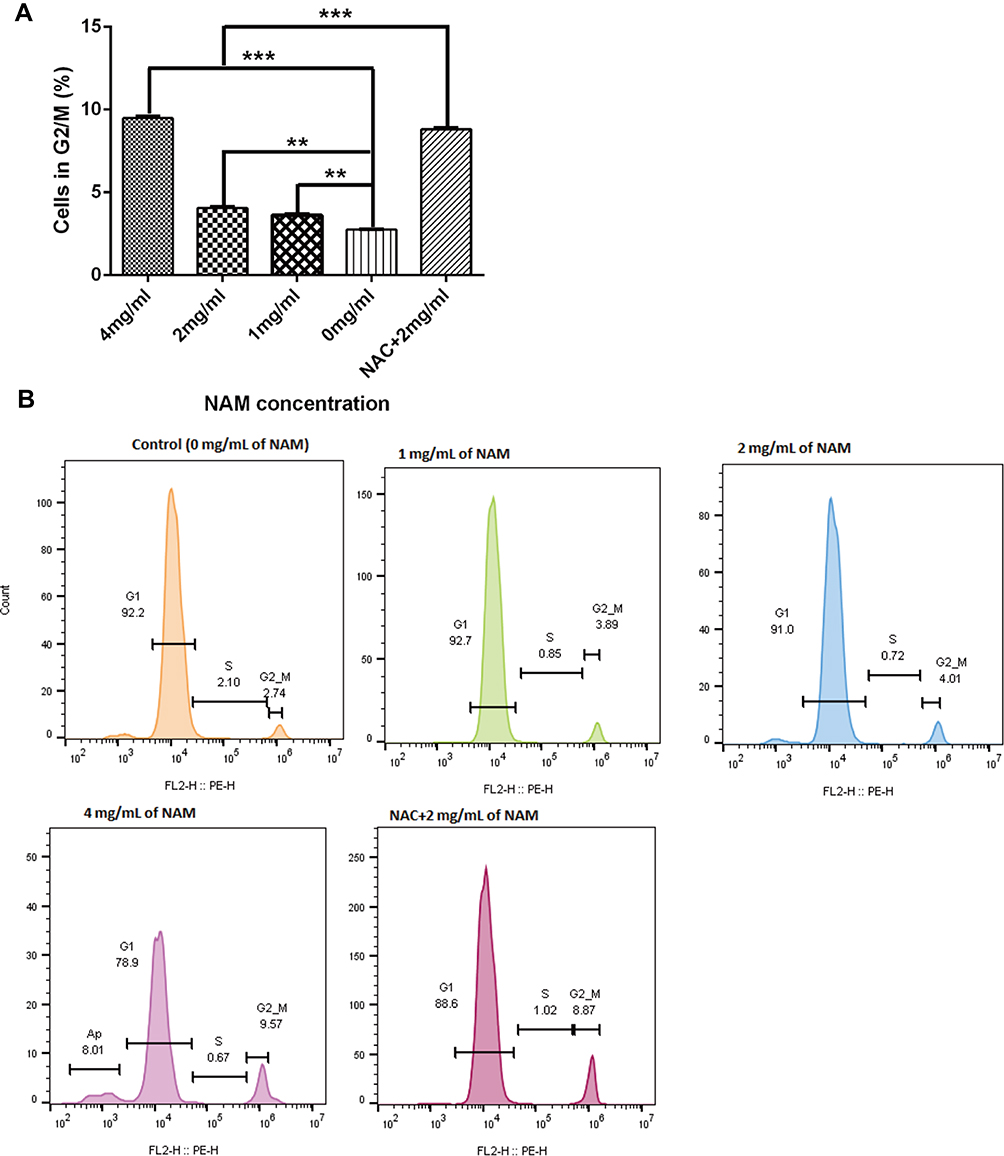 |
Figure 6 Nicotinamide treatment induces cell cycle arrest at G2/M for fibroblasts (***p≤0.001, **p≤0.01). (A) The proportion of cells in G2M phase after NAM treatment and (B) Represents flow cytometric analysis results of cellular phases with the indicated concentrations of nicotinamide. |
Discussion
The treatment of cervical cancer, one of the most common malignancies of the female reproductive system, relies heavily on intensive chemotherapy. Henceforth, there is an urgent need to develop new therapeutic agents for the treatment of cervical cancer, specifically in the advanced and recurrent disease setting.27 The present study, therefore, investigated the effects of NAM on CAFs to uncover its efficacy and mechanisms of action. Our results showed a significant reduction in cell viability in a time and dose-dependent manner with the highest reduction in viability observed at 4 mg/mL of NAM concentration. Our findings, therefore, suggest that 4 mg/mL of NAM could be an effective dose in vitro. We also observed that NAM treatment induced early and late-onset apoptosis in the cells in a dose-dependent manner, suggesting the therapeutic potential of NAM.
Our findings of significant apoptosis of CAF with 4 mg/mL NAM treatment and arresting of CAF in a G2/M arrest of the fibroblasts are in consent with the previous findings report in the literature. The study by Wang et al 2018 showed apoptotic potential and cell growth arrest inducing properties of NAM in intrahepatic cholangiocarcinoma cells.28 A high concentration of NAM at 4 mg/mL was able to cause oxidative stress to the cells by elevating the MDA levels and reducing the level of GSH, known to be an important component of the antioxidant defense system of the cells. Furthermore, NAM was able to generate intracellular ROS in a dose-dependent manner. NAM has been also reported in previous studies for its ability to induce increase in ROS and MDA levels in cervical cancer cell line (ie, HeLa cells).24 Another study reported with antioxidant effects of NAM in activated macrophages30,31 and brain cells in mice.31 Furthermore, it is worth addressing that the dose-specific effect of NAM observed in the present study suggest that NAM can act as an oxidative stressor only after a certain threshold (i.e., 2 mg/mL) is reached.
NAM treatment was further observed to increase oxidative stress in fibroblasts responds by modulating the expressions of anti-oxidative enzymes and anti-apoptotic genes. Besides, altered metabolism resulting from mutations in pro-apoptotic genes has long been considered as a hallmark characteristics of cancer cells.32,33 Free oxygen radicals, induced by the higher rate of metabolism, can cause altered fuel consumption in the cancer cells aided by the higher availability of NADPH.34 Glucose-6-phosphate dehydrogenase (G6PD) enzyme is specifically known to play a critical role in supplying the cells with NADPH and ribose, which in turn, strengthens the antioxidant defense system of the cells. NAM is also well-known to be a non-competitive inhibitor of the G6PD enzyme, and therefore inhibition of G6PD activity could starve the cells from its essential fuel. Consistent with our study, Fang et al35 had previously demonstrated that NAM treatment of HeLa cells could result in G6PD deficiency, ultimately leading to an increased ROS induced apoptosis and thereby decreased HeLa cell migration and proliferation. Our study, therefore, provides a proof-of-principle for G6PD inhibition through NAM in cervical cancer cell lines. The NAM treatment of the adipocytes has been earlier shown to alter redox balance, where NAM acted as an anti-oxidant and maintained NADPH level.36 This is consistent with the observation that NAM lethality is dose-specific being cytoprotective at lower doses and pro-lethal at higher doses. Thus, the next step could be to uncover the role of NAM through inhibiting G6PD in fibroblasts as well as primary cervical cancer cells both in vitro and in vivo studies, and explore the evidence of the effect of nicotinamide through some animal experiments.
Conclusion
The present study has shown the importance of an intriguing component of the cervical cancer tumor microenvironment; the fibroblasts cells demonstrated that NAM treatment could be lethal for these cells through similar mechanisms as observed earlier in HeLa cell lines. The present study has examined the effects of NAM on CAF concerning its in vitro efficacy, gross inhibition, and mechanism of inhibition. Our study showed that NAM at a higher concentration results in higher oxidative stress in CAF through ROS and inhibits the proliferation of fibroblasts. The lethality is contributed by reducing the mitochondrial membrane potential and inducing cell cycle arrest and apoptosis. Our study supports the therapeutic potential of NAM for the treatment of cervical cancer. However, the results of the study should be treated with caution as the findings need to be confirmed with cells from at least two more patients because the heterogeneity of cell populations between patients could confound the study results.
Acknowledgments
We highly appreciate the financial support of the National Natural Science Foundation of China grants U1132606 and No.31171214. The authors thank Jin Yan, Peng Du, Qiuyue Hu and Yue Zhu for their technical and general support in this study.
Author Contributions
All authors contributed equally to data analysis, drafting and revising the article. All authors gave final approval of the version to be published and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ryan BM, Faupel-Badger JM. The hallmarks of premalignant conditions: a molecular basis for cancer prevention. Semin Oncol. 2016;43:22–35. doi:10.1053/j.seminoncol.2015.09.007
2. Carneiro SR, de Araújo Fagundes M, Do Rosário PD, et al. Five-year survival and associated factors in women treated for cervical cancer at a reference hospital in the Brazilian Amazon. PLoS One. 2017;12:e0187579. doi:10.1371/journal.pone.0187579
3. Li H, Wu X, Cheng X. Advances in diagnosis and treatment of metastatic cervical cancer. J Gynecol Oncol. 2016;27. doi:10.3802/jgo.2016.27.e43
4. Zhu H, Luo H, Zhang W, Shen Z, Hu X, Zhu X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des Devel Ther. 2016;10:1885. doi:10.2147/dddt.s106412
5. Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64:706–721. doi:10.1124/pr.111.005637
6. Wang Y, Gao Y, Cheng H, Yang G, Tan W. Stanniocalcin 2 promotes cell proliferation and cisplatin resistance in cervical cancer. Biochem Biophys Res Commun. 2015;466:362–368. doi:10.1016/j.bbrc.2015.09.029
7. Fricker RA, Green EL, Jenkins SI, Griffin SM. The influence of nicotinamide on health and disease in the central nervous system. Int J Tryptophan Res. 2018;11:1178646918776658. doi:10.1177/1178646918776658
8. Gasperi V, Sibilano M, Savini I, Catani MV. Niacin in the central nervous system: an update of biological aspects and clinical applications. Int J Mol Sci. 2019;20:974. doi:10.3390/ijms20040974
9. Fouts PJ, Helmer OM, Lepkovsky S, Jukes TH. Treatment of human pellagra with nicotinic acid. Proc Soc Exp Biol Med. 1937;37:405–407. doi:10.3181/00379727-37-9589p
10. Trammell SA, Yu L, Redpath P, Migaud ME, Brenner C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. J Nutr. 2016;146(5):957–963. doi:10.3945/jn.116.230078
11. Moloney F, Vestergaard M, Radojkovic B, Damian D. Randomized, double-blinded, placebo controlled study to assess the effect of topical 1% nicotinamide on actinic keratoses. Br J Dermatol. 2010;162:1138–1139. doi:10.1111/j.1365-2133.2010.09659.x
12. Chen AC, Martin AJ, Choy B, et al. A Phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373:1618–1626. doi:10.1056/nejmoa1506197
13. Itzhaki O, Greenberg E, Shalmon B, et al. Nicotinamide inhibits vasculogenic mimicry, an alternative vascularization pathway observed in highly aggressive melanoma. PLoS One. 2013;8:e57160. doi:10.1371/journal.pone.0057160
14. Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med. 2012;53:421–436. doi:10.1016/j.freeradbiomed.2012.05.006
15. Preyat N, Rossi M, Kers J, et al. Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner. Cell Death Differ. 2016;23:29. doi:10.1038/cdd.2015.60
16. Zhang JG, Zhao G, Qin Q, et al. Nicotinamide prohibits proliferation and enhances chemosensitivity of pancreatic cancer cells through deregulating SIRT1 and Ras/Akt pathways. Pancreatology. 2013;13:140–146. doi:10.1016/j.pan.2013.01.001
17. Audrito V, Vaisitti T, Rossi D, et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res. 2011;71:4473–4483. doi:10.1158/0008-5472.can-10-4452
18. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–1785. doi:10.1158/0008-5472.can-07-5259
19. Beall HD, Murphy AM, Siegel D, Hargreaves RH, Butler J, Ross D. Nicotinamide adenine dinucleotide (phosphate): quinone oxidoreductase (DT-diaphorase) as a target for bioreductive antitumor quinones: quinone cytotoxicity and selectivity in human lung and breast cancer cell lines. Mol Pharmacol. 1995;48:499–504.
20. Feng Y, Wang Y, Jiang C, et al. Nicotinamide induces mitochondrial-mediated apoptosis through oxidative stress in human cervical cancer HeLa cells. Life Sci. 2017;181:62–69. doi:10.1016/j.lfs.2017.06.003
21. Liu T, Han C, Wang S, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12:86. doi:10.1186/s13045-019-0770-1
22. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel). 2015;2443–2458. doi:10.3390/cancers7040902
23. Lei S, Liu Y, Liu H, Yu H, Wang H, Xia Z. Effects of N-acetylcysteine on nicotinamide dinucleotide phosphate oxidase activation and antioxidant status in heart, lung, liver and kidney in streptozotocin-induced diabetic rats. Yonsei Med J. 2012;53:294–303. doi:10.3349/ymj.2012.53.2.294
24. Pavlides S, Whitaker-Menezes D, Castello-Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi:10.4161/cc.8.23.10238
25. Erez N, Truitt M, Olson P, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell. 2010;17:135–147. doi:10.1016/j.ccr.2009.12.041
26. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi:10.3322/caac.21254
27. Duska LR. Cervical cancer: the next generation of prevention, detection, and treatment. Clin Ther. 2016;38:446–448. doi:10.1016/j.clinthera.2016.02.013
28. Wang Y, Ryu HS, Jang JJ. Nicotinamide suppresses cell growth by G1-phase arrest and induces apoptosis in intrahepatic cholangiocarcinoma. Mol Cell Toxicol. 2018;14(1):43–51. doi:10.1007/s13273-018-0006-4
29. Rashid M-U, Coombs KM. Serum-reduced media impacts on cell viability and protein expression in human lung epithelial cells. J Cell Physiol. 2019;234:7718–7724. doi:10.1002/jcp.27890
30. Kamat JP, Devasagayam TP. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Report. 1999;4:179–184. doi:10.1179/135100099101534882
31. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi:10.1016/j.cmet.2015.12.006
32. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi:10.1016/j.ccr.2012.02.014
33. Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–3658. doi:10.1172/jci67228
34. Stanton RC. Glucose‐6‐phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64:362–369. doi:10.1002/iub.1017
35. Fang Z, Jiang C, Feng Y, et al. Effects of G6PD activity inhibition on the viability, ROS generation and mechanical properties of cervical cancer cells. Biochim Biophys Acta. 2016;1863:2245–2254. doi:10.1016/j.bbamcr.2016.05.016
36. Torres-Ramírez N, Baiza-Gutman LA, García-Macedo R, et al. Nicotinamide, a glucose-6-phosphate dehydrogenase non-competitive mixed inhibitor, modifies redox balance and lipid accumulation in 3T3-L1 cells. Life Sci. 2013;93:975–985. doi:10.1016/j.lfs.2013.10.023
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.