")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 7
Effects of LRP1B Regulated by HSF1 on Lipid Metabolism in Hepatocellular Carcinoma
Authors Li M, Hu J, Jin R, Cheng H, Chen H, Li L, Guo K
Received 27 August 2020
Accepted for publication 18 November 2020
Published 8 December 2020 Volume 2020:7 Pages 361—376
DOI https://doi.org/10.2147/JHC.S279123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Miaomiao Li,1,* Juntao Hu,2,* Riming Jin,3,* Hongxia Cheng,1 Huaping Chen,4 Limin Li,4 Kun Guo1
1Liver Cancer Institute, Zhongshan Hospital, Fudan University, Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Shanghai, People’s Republic of China; 2Department of Anatomy, Histology and Embryology, School of Basic Medical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China; 3Department of First Surgery, the Third Affiliated Hospital, NAVY Medical University, Shanghai, People’s Republic of China; 4Department of Clinical Laboratory, First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kun Guo
Liver Cancer Institute, Zhongshan Hospital, Fudan University, Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Building 19, No. 180, Fenglin Road, Shanghai 20032, People’s Republic of China
Tel +86-21-54237962
Fax +86-21-54237959
Email [email protected]
Background: To date, aberrated lipid metabolism has been recognized as an important feature of hepatocellular carcinoma (HCC); however, it remains poorly defined. As a large member of the low-density lipoprotein receptor family, LRP1B plays a pivotal role in maintaining lipid homeostasis. Here we investigated the expression feature of LRP1B in HCC and elucidated its effects on lipid metabolism of HCC cells.
Materials and Methods: LRP1B expression in HCC cells and tumor tissues was respectively examined by quantitative PCR, Western blotting and immunohistochemistry. Crispr-cas9 RNA inference and CRISPRa transcription activation system were used to downregulate and upregulate LRP1B expression, respectively. Oil red O staining, DiD staining combined with flow cytometry and transmission electron microscopy were used to evaluate the lipid content in HCC cells. Overall survival (OS) and time to recurrence (TTR) were calculated; meanwhile, Kaplan–Meier and the Cox proportional hazards model were used to assess the prognosis of HCC patients.
Results: In contrast to inactivation expression in a majority of cancers, LRP1B showed predominantly strong expression in HCC. LRP1B knockdown induced the decrease of intracellular lipid content, downregulated expressions of lipid synthesis-related enzymes and upregulated expressions of β-oxidation-related enzymes as well as activated the AMPK signaling. Moreover, HSF1 directly regulated the transcription of LRP1B and was involved in LRP1B-mediated lipid metabolism in HCC; meanwhile, the combination of LRP1B knockdown and HSF1 inhibition suppressed synergistically the proliferation of HCC cells. In addition, simultaneous expression of HSF1 and LRP1B was an independent prognostic factor for HCC patients.
Conclusion: Altogether, the study reveals a novel unique role of LRP1B in HCC by serving as a mediator in lipid metabolism, which provides an insight for making explorable therapeutic strategies for HCC.
Keywords: hepatocellular carcinoma, lipid metabolism, LRP1B, HSF1, prognosis
Introduction
The updated cancer epidemiological statistics data illustrates the continuous improvement of global cancer diagnosis and management. However, the situation of prevention and treatment of liver cancer is not optimistic.1 Hepatocellular carcinoma (HCC) accounts for about 90% of liver cancer and represents the second leading cause of cancer-related mortality and morbidity worldwide.2 Unfortunately, the aggressive and refractory nature of HCC often makes currently available therapeutic options and multi-modal strategies unsatisfying.3 Data from multi-omics and pharmacogenomics suggest that the refractory characterization of HCC results from its highly heterogeneous feature, associated with a variety of complex altered signaling pathways.4–6 The heterogeneity of HCC comes from complex multi-levels including cellular behaviors, molecular diversity, genetic mutation and metabolism within the tumor and among individuals.7 Particularly, data accumulated over recent years demonstrate that metabolic heterogeneity has been recognized as a significant feature of tumor, closely related to tumorigenesis and development.8 Moreover, two recent key studies in our institute revealed the proteogenomic characteristics of HBV-related HCC in China and identified liver-specific metabolism alterations (such as enhanced glucose metabolism, overall activation of lipid biosynthesis, etc.) as a distinct feature in the Chinese population with HBV-related HCC.9,10 Of note, some key proteins related to cholesterol homeostasis are found to be significantly up-regulated in HBV-related HCC.9 Hence, the multi-omics data suggest that targeting against liver-specific lipid metabolism might provide a potential opportunity for precise therapies of HBV-related HCC in China.
As a central organ for lipid metabolism, the liver is in charge of lipid profiling and fatty acid composition in body; this process requires the participation of various molecules, associated with lipid synthesis, trafficking, and storage.11 The low-density lipoprotein (LDL) receptor-related protein 1B (LRP1B) is a novel member of the LDL receptor family and fulfills a variety of different physiological functions, ranging from cell signaling, lipoprotein trafficking, transportation of nutrients and vitamins to developmental processes.12 In recent years, LRP1B has gained more and more attention in the cancer research field and has been found to be involved in malignant proliferation, angiogenesis, differentiation and metastases in multiple types of cancers including NSCLC,13 prostate cancer,14 cervical squamous cell cancer,15 renal cell cancer16 and glioma.17 In these cancers, LRP1B has been identified to be downregulated and serve as a potential tumor suppressor. In addition, in a genomic context, LRP1B mutations are often related to tumor mutation burden (TMB) and can be regarded as an useful predictor for treatment efficacy and outcomes in patients with cancer such as lung cancer, melanoma, renal cell cancer, and myeloid leukemia.13,18,19 More recently, it is also reported that LRP1B mutations were associated with TMB and survival in patients with liver cancer.20 Nevertheless, in contrast to other cancers, expression status and possible functions of LRP1B in HCC are largely unknown. Therefore, in the study, we investigated the expression feature of LRP1B in HCC and elucidated its effects on lipid metabolism of HCC cells; mechanistically, revealed the involvement of HSF1 in LRP1B-mediated lipid metabolism through regulating AMPK signaling in HCC. These findings might provide an insight for making explorable therapeutic strategies for HCC in the future.
Materials and Methods
Reagents
Anti-HSF1 (#12972), β-actin (#4970), anti-AMPKα (#5831), anti-phospho-AMPKα (50081), anti-rabbit anti-mouse HRP-linked secondary antibody IgG (#7074 and #7076) were purchased from Cell Signaling Technology. Anti-LRP1B (DF9609) was purchased from Affinity Bioscience. For lipid content measurement, a lipophilic fluorescent dye (DiD) (# V22887, ThermoFisher) and Oil Red O Stain kit (#YB0843, ScienCell) were used. Human FAS ELISA Kit (MS1108), ACC ELISA Kit (MS2411) and CPT-1A ELISA Kit (CSB-E17408h) were respectively from Shanghai Suobio Technology and Ximei Biotech.
Cell Culture
The human HCC cell lines MHCC97L, MHCC97H and HCCLM3 (Liver Cancer Institute, Zhongshan Hospital), and Huh7, Hep3B and HepG2 (American Type Culture Collection) with high/low metastatic potential, and the rat HCC cell line RH-35 (American Type Culture Collection) and the murine HCC cell line Hep1-6 (American Type Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Genom, GNM12800) supplemented with 10% fetal bovine serum (FBS) (BI, 04–001-1acs) and 1% penicillin-streptomycin (Gibco, 15140–122). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 and treated according to experiment design. The use of the cell lines was approved by the Ethics Committee of Zhongshan Hospital, Fudan University.
Patients and Samples
To analyze the expression feature of LRP1B and HSF1, eighty formalin-fixed paraffin-embedded (FFPE) HCCs with a matched peritumoral tissue and 305 FFPE HCCs without a matched peritumoral tissue were collected, which were diagnosed at the Eastern Hepatobiliary Surgery Hospital between January 2003 and March 2012. Follow-up in these patients was conducted every 3 months during the 1st year after surgery and every 6 months thereafter until December 2014. The overall survival (OS) and the time to recurrence (TTR) were respectively calculated as the interval between surgery and death or the last observation taken and from the date of hepatectomy until the detection of tumor recurrence, death or last observation. Moreover, 30 fresh resected HCCs with a matched peritumoral tissue were gained between January and July 2017 for quantitative methylation-specific PCR (Q-MSP). Written informed consent was obtained from all patients. The study protocol was approved by the Ethics Committee of Eastern Hepatobiliary Surgery Hospital (EHBHKY2015-01-001) and was conducted in accordance with the Declaration of Helsinki.
Quantitative PCR
Trizol Reagent (Invitrogen, 15596026) was used to extract total RNA of cultured cells and tumor/adjacent tissues. The reverse transcription was performed by using PrimeScript™ RT Reagent kit (TaKaRa, M0402-1061). SYBR Premix Ex Taq Ⅱ (TaKaRa, DRR420A) was used to preform quantitative PCR at ABI 7500 instrument (Applied Biosystems). The cycle threshold (Ct) of β-actin gene was set up as a control, and relative expression levels of target genes were calculated as 2−ΔCt [ΔCt = Ct - Ct (control)]. The sequences of primers used in the study can be found in supplementary Table.
Immunofluorescence Assay
For cultured HCC cells, 0.1% Triton X-100 was used to permeabilize for 15 min at room temperature; after rinsing twice with 1×PBS solution for 5 min, cells were incubated with blocking buffer (1×PBS containing 5% BSA) for 30 min at room temperature. Then, cells were rinsed twice with 1×PBS and incubated with specific primary anti-LRP1B antibody (1:50) overnight at 4 °C. After being twice washed with 1×PBS solution, cells were incubated with goat anti-rabbit secondary antibody for 1 h at room temperature. The slices were rinsed twice with 1×PBS and counterstained with diamidino phenylindole (DAPI); finally observed by using fluorescence microscopy (Leica Microsystems Imaging Solutions) for photograph.
Western Blotting
Harvested cells were washed by the ice-cold 1×PBS, and lysed by radio immunoprecipitation assay (RIPA) lysis buffer containing protease cocktail inhibitor (Thermo Scientific, 89900). The cell lysates were centrifuged × 12,000 rpm at 4 °C for 30 minutes. The concentration of supernatant was determined by using BCA protein assay kit (Bio-Rad, 500–0001). After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, ISEQ00010). After being blocked with 5% BSA buffer, membranes were incubated with specific primary antibodies (dilution 1:1000) at 4 °C overnight. After washing three times with 1×TBST buffer, membranes were incubated with HRP-conjugated secondary antibodies for another 1 h at room temperature. Membranes were washed three times with 1×TBST buffer and then visualized by using the enhanced chemiluminescence (ECL) reagents (Thermo Scientific, 34577). Protein bands were quantified by ImageJ Software (Bio-Rad).
Dual Luciferase Reporter Gene Assay
HEK 293T cells were cultured in 12-well plates with 1×105 cells per well overnight. The HSF1 vector, wild-type LRP1B promoter, mutant LRP1B promoters (MT1 and MT2) were cloned into pGL3-basic plasmid and pRL-TK. The recombines were transfected into the cells with the confluence of 60–70% per well. According to protocols of manufacturer from Dual-Luciferases Reporter Assay kit (Promega, E1980), cells were lysed by lysis buffer for 20 min at room temperature after the transfection 48h; then lysates were centrifuged and supernatants were extracted; finally, the luciferase activity was determined.
Crispr-Cas9 and RNA Inference
Recombinant lentiviruses were constructed by lentiviral vectors Lenti-CRISPR-V2, pLKO.1 TRC and pWPI.1. Three single guide RNAs (sgRNAs) specific to LRP1B and one control sgRNA were designed using the CRISPR Design software (http://crispr.mit.edu) (supplementary Table). The oligonucleotides were ligated into the lentiviral vector Lenti-CRISPR-V2 for construction of lentivirus expression plasmids. For RNA interference, short interference RNA (shRNA) constructs were generated by using oligonucleotides encoding hairpin precursors for sh-HSF1 (supplementary Table); meanwhile a scrambled non-targeting sequence was used as a control. Recombinant lentivirus was amplified in HEK293T cells. The cells (2×105) were planted in 6-well plates. The lentiviral vector and plasmids were transfected into cells by Lipofectamine 3000 (Invitrogen, L3000001). After indicated timepoints, the cells were lysed and knockdown efficacy was defined by Western blotting.
Immunohistochemistry
Tissue microarrays were constructed according to our previous studies.21 Paraffin sections were sliced to 4 μm thickness and placed on slides coated with 3-aminopropyltriethoxysilane. After deparaffinization in xylene and rehydration in serial concentrations of ethanol (100%, 95%, 85% and 75% for 5 min each), antigen retrieval was performed in pH 6.0 citric buffer by microwave irradiation for 3 min. Endogenous peroxidase activity and nonspecific binding sites were respectively blocked by 3% H2O2/phosphate-buffered saline and goat serum. Immunohistochemistry (IHC) was performed according to the instructions of the manufacturer (The UltraSensitive SP Kit; Maixin-Bio, KiT-97). Tissue sections were counterstained with hematoxylin for 5 min; meanwhile slides without primary antibody was set up as negative control. These stained slides were scanned and photographed on a KFBIO KF-PRO-005-EX digital section scanner (Konfoong Biotech International Co., Ltd., Yuyao, China) and the staining IODs in images was calculated as previous studies.21 The category of low vs high HSF1 or LRP1B expression in IHC staining was determined by using X-tile analysis. In brief, to assess HSF1 or LRP1B expression which was represented as IOD/area (μm2), the X-tile plots were used and cut-points was optimized based on outcome. Detailed analysis process was performed according to Camp’s method.22 Then, by a standard log-rank method, statistical significance was assessed through using the cut-off score derived from included HCC cases; meanwhile the associated p-values were obtained from a lookup table.
Oil Red O Staining Assay
Stock solution for 0.5% Oil Red O was prepared by dissolving the staining powder with isopropanol in the dark. HCC cells were washed three times with 1×PBS; then fixed with 4% paraformaldehyde solution for 30 min at room temperature. After fixation, cells were stained with filtered Oil red O staining working solution (with 60% deionized water) for 30 min at room temperature. Images of cells were photographed by using a fluorescent microscope (Lecia, DM6 M). Moreover, after 100% isopropyl alcohol was used to dilute the stain, the eluted Oil Red O staining was quantified through detecting the absorbance at 510 nm.
DiD Staining Assay and Flow Cytometry Analyses
HCC cells were plated for DiD staining assay. After being washed three times in 1×PBS buffer, these cells were incubated with DiD solution at a 1:200 dilution at room temperature for 30 min with gentle shaking in the dark. After incubation, cells were washed three times with 1×PBS buffer. The labelled cells were photographed using a fluorescent microscope (Lecia, DM6 M) at 633 nm absorbance. Moreover, these cells were collected through centrifuge at 1000 rpm for 3 min and then were resuspended in FACS flow buffer for analysis by the flow cytometry (BD Biosciences, BD FACSCanto).
ELISA Assay
The levels of FAS, ACC, and CPT-1A activities in cell extracts were determined respectively by ELISA Kits according to the protocols of the manufacturer. The average value of three independent repeats was implemented for statistical analyses.
Statistical Analyses
For the statistical analysis, GraphPad Prism 7 (GraphPad Software), and SPSS version v.24.0 (IBM Corporation) were used. The tests used included two-tailed Student’s t-test, chi-squared test, one-way and two-way ANOVA, Kaplan-Meier’s method and Log rank test. P-values < 0.05 or <0.01 were considered a statistically significant difference or extremely significant difference. Preliminary data or previous reports were the source of the sample size. No data was removed from the analyses. As shown in figure legends, the experiments were repeated independently many times and the results were similar.
Results
LRP1B is High Expressed in HCC
Firstly, the pan-cancer view of LRP1B expression was explored by using Human Protein Atlas (HPA; http://www.proteinatlas.org) as well as UALCAN (http://ualcan.path.uab.edu/) databases. It was observed that LRP1B was predominantly high-expressed in HCC, while low-expressed or negatively expressed in the majority of remaining cancers. The finding encouraged us to investigate the feature of LRP1B expression in various liver cancer cell lines and clinical tissue samples. As Figure 1A shows, the mRNA level of LRP1B was detectable in various liver cancer cell lines including Hep3B, HepG2, Huh7, MHCC97L, MHCC97H, HCCLM3, RH-35, and Hep1-6, displaying a possible increased tendency as the increase of metastatic potential in these cells. Moreover, the levels of LRP1B protein in these cell lines were consistent with mRNA levels, higher levels in HCCLM3, MHCC97H, MHCC97L and Huh7 cells while a lower level in Hep3B cells (Figure 1B-C). Moreover, higher expression of LRP1B in liver cancer cells was observed, as compared to non-cancerous cells (sFigure 1). In addition, the result from cellular immunofluorescence staining showed the typical cytoplasmic positivity of LRP1B in HCC cells (Figure 1D). Furthermore, by using tissue chip construction followed by immunohistochemical staining and IPP software analysis, we detected LRP1B expression in 80 pairs of HCC tissues and adjacent tissues and found that the IOD/μm2 value of LRP1B staining in HCC tissues was 0.62±0.12, significantly higher than that in matched adjacent tissues (0.39±0.01; P=0.004) (Figure 1E-F).
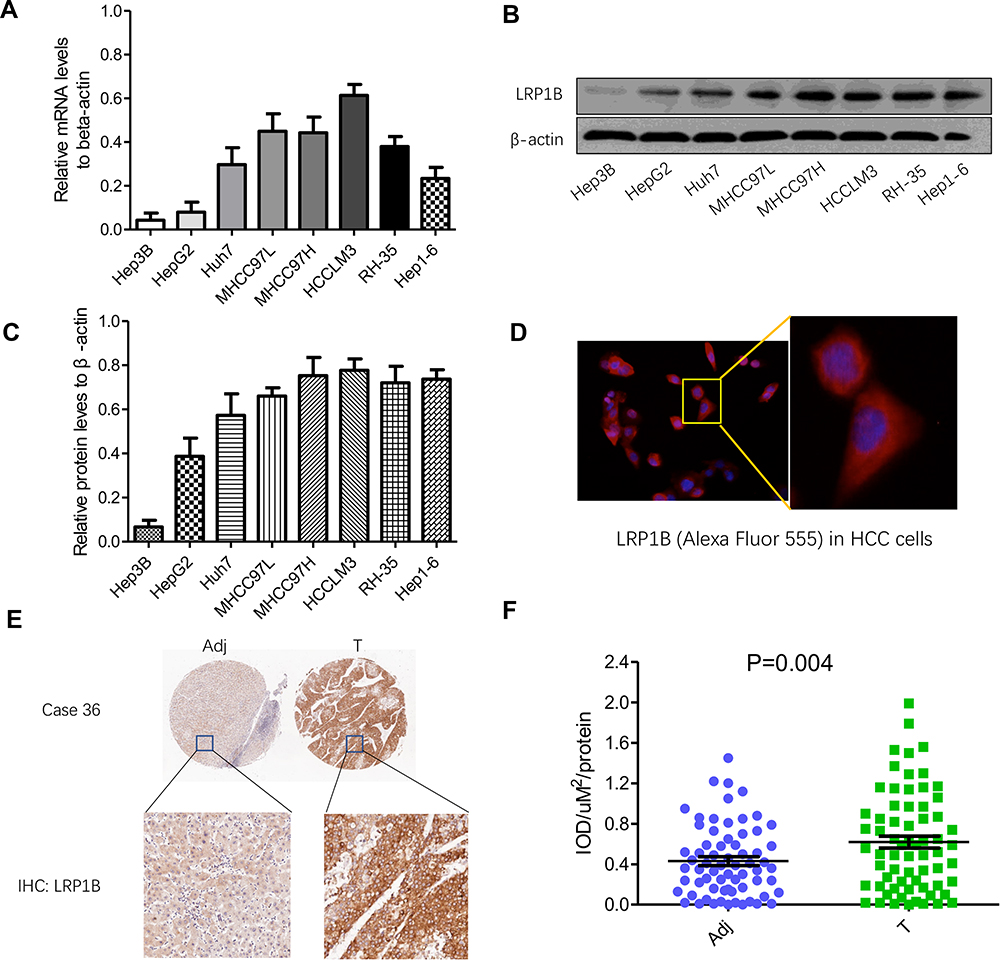 |
Figure 1 Expression of LRP1B in HCC cell lines and tissues is different. (A) Relative mRNA levels of LRP1B in different HCC cell lines including Hep3B, HepG2, Huh7, MHCC97L, MHCC97H, HCCLM3, RH-35 and Hep1-6. (B) Expression of LRP1B protein in liver cancer cell lines mentioned above by Western blotting. β-actin was regarded as an internal control. (C) Relative levels of LRP1B protein to β-actin in liver cancer cell lines. (D) Representative images of LRP1B in HCC cells by immunofluorescence staining. Immunofluorescence assay result of HCC cells (from MHCC97H) expressing the primary antibody of LRP1B, followed by Alexa Fluor 555-conjugated secondary antibody (red) is shown. Cell nuclei were counterstained with diamidino phenylindole DAPI (blue). Scale bar = 100 μm, magnification×200 (left). Scale bar = 200 μm, magnification × 400 (right). (E) Representative IHC staining of LRP1B in 80 pairs of HCC tissues (T) and matched adjacent liver tissues (Adj). Immunoreactivity of LRP1B was mainly located in the cytoplasm. (F) Integrated optical density (IOD) for LRP1B staining was obtained. The measurement values represented the mean±standard deviation (SD). Statistical significance was analyzed by two-tailed unpaired Student’s t-test (A, C, F). Triplicate experiments independently with similar results. |
Effects of LRP1B on Lipid Metabolism of HCC Cells
In normal cells, LRP1B is involved in lipid transport and synthesis;23 however, its effect on lipid metabolism in HCC cells is still unclear. To investigate effects of LRP1B on lipid accumulation and storage in HCC cells, we performed an efficacious knockdown of LRP1B expression by using the lentivirus-induced CRISPR/Cas9 system in HCC cells (sFigure 2). After LRP1B knockdown, HCC cells presented a significantly attenuated growth from the 2nd day (sFigure 3). Next, we observed the cellular lipid accumulation after LRP1B knockdown by using Oil Red O staining and found that lipid accumulation was significantly reduced in HCC cells with LRP1B knockdown, as compared to the controls and the untreated cells (Figure 2A). Moreover, we reconfirmed the phenomena by DiD staining followed by flow cytometry and further verified the decrease of lipid content after LRP1B knockdown (Figure 2B). In addition, the result from transmission electron microscopy also showed that lipid droplets in HCC cells with LRP1B knockdown were significantly shrunk or reduced as compared to the control and the untreated cells (Figure 2C).
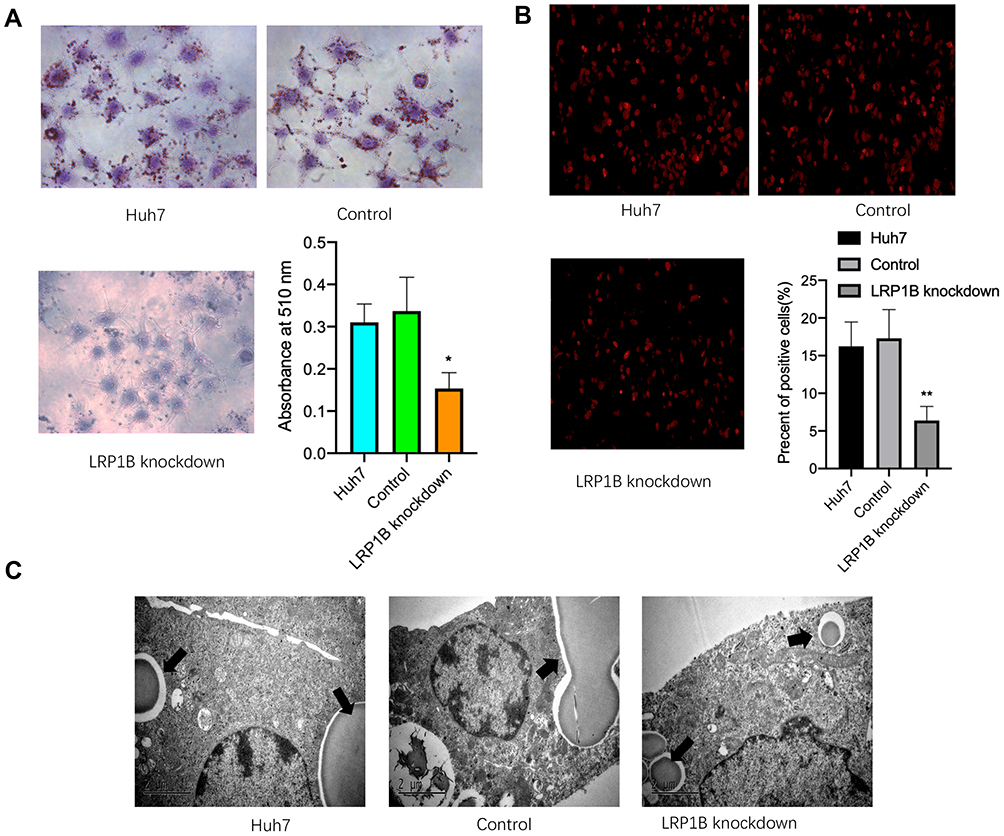 |
Figure 2 Knockdown of LRP1B suppresses the lipid metabolism of HCC cells. (A) Oil Red O staining assay was used to determine the effect of LRP1B knockdown on intracellular lipid deposits in Huh7 cells (×200). Histogram indicated that OD values measured at the 510 nm wavelength and represented the mean±SD. **p<0.01 vs control. (B) Representative images of DiD staining in Huh7 cells with or without LRP1B knockdown. The red indicated the positive lipophilic cells (×100). Histogram indicated the proportion of lipophilic cells in various Huh7 cells by using flow cytometry. (C) Transmission electron microscopy revealed representative manifestation of intracellular lipid droplets in different groups. |
Abnormal lipid metabolism of cancer cells mainly manifested in de novo fatty acid synthesis and enhanced lipid synthesis, and reduced fatty acid breakdown.24 Hence, we detected the mRNA levels of several key enzymes for fatty acid synthesis-fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), sterol regulatory element binding protein 1c (SREBP1c), ATP-citrate lyase (ACLY), stearoyl-CoA desaturase-1 (SCD1) as well as important enzymes for β-oxidation of fatty acids-carnitine palmitoyltransterase-1 (CPT-1), CPT-2, long-chain acyl-CoA dehydrogenase (LCAD), and medium-chain Acyl-CoA dehydrogenase (MCAD). As Figure 3A-B shows, the mRNA levels of FAS, ACC, SREBP1c, and ACLY significantly decreased while the mRNA levels of MCAD, LCAD and CPT-1 obviously increased after LRP1B knockdown in HCC cells, as compared to the other two groups. Meanwhile, further measures of FAS, ACC and CPT-1 activity were performed by using enzyme activity detection kits and the result showed that the activity of FAS and ACC decreased while the activity of CPT-1 increased after LRP1B knockdown (Figure 3C-E). These results suggested that LRP1B might promote lipid synthesis, inhibit lipolysis and finally regulate the lipid metabolism reprogramming in HCC cells. Of note, in recent years, AMPK signaling has been recognized as a main regulator of cellular lipid metabolism.25 Herein, we detected the levels of p-AMPK and AMPK proteins in HCC cells and found that p-AMPK/AMPK ratio was significantly increased after LRP1B knockdown (Figure 3F), suggesting the involvement of LRP1B in lipid metabolism in HCC cells might be related to AMPK signaling.
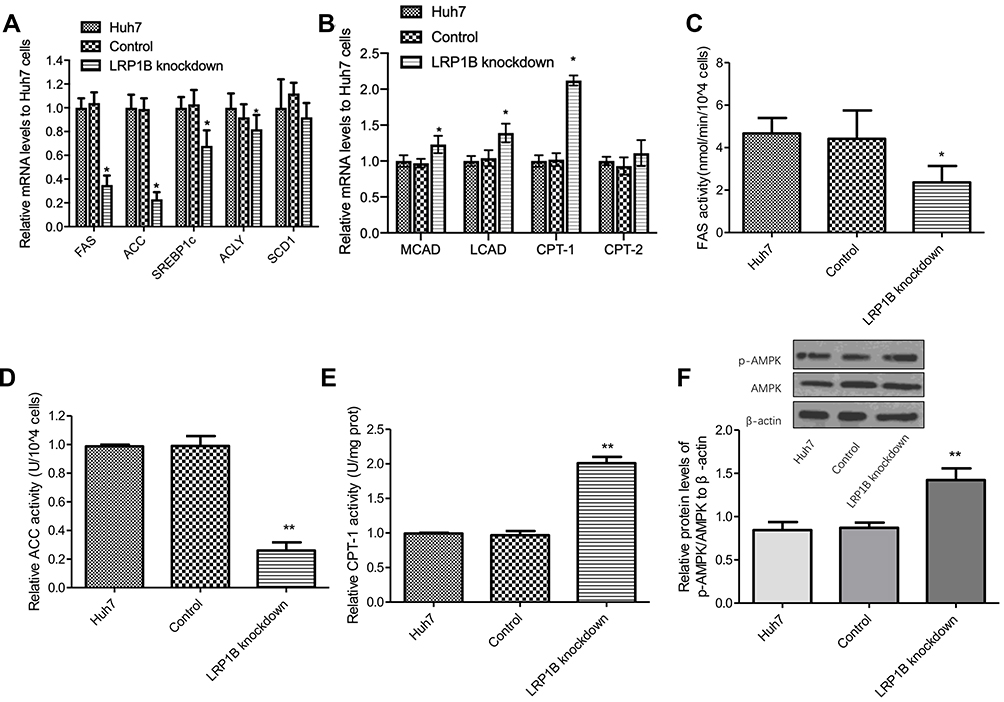 |
Figure 3 LRP1B regulated expression and activity of lipid metabolism-related enzymes and signaling. (A) Relative mRNA levels of several key enzymes for fatty acid synthesis-FAS, ACC, SREBP1c, ACLY, SCD1 in Huh7 cells with or without LRP1B knockdown were assessed by quantitative PCR. (B) Relative mRNA levels of important enzymes for β-oxidation of fatty acids-CPT-1, CPT-2, LCAD, MCAD in Huh7 cells with or without LRP1B knockdown were assessed by quantitative PCR. (C–E) Relative activity of FAS (C), ACC (D), CPT-1 (E) in Huh7 cells after LRP1B knockdown was determined by relevant Kit. (F) Expressions of p-AMPK and AMPK in Huh7 cells with or without LRP1B knockdown were detected by Western blotting. β-actin was set up as an internal control. Then, relative levels of p-AMPK/AMPK to β-actin in Huh7 cells from the same experiments as shown. *p < 0.05; **p < 0.01. Data represented the mean±SD. Statistical significance was analyzed by two-tailed unpaired Student’s t-test. Triplicate experiments independently with similar results. |
High Expression of LRP1B is Induced by HSF1 in HCC
In cancers, the regulation of LRP1B expression is often closely related to its promoter methylation status.26–28 To explore the high-expression mechanism of LRP1B in HCC, we used the upstream 5 kb sequence of LRP1B gene as a source to predict its promoter and then used MethPrimer software to scan the distribution of CpG islands of human LRP1B gene. The results showed that there were 4 potential CpG islands in the promoter region of human LRP1B gene, mainly located in exon 1 and upstream regions (0–437, +589-807, +1856-1982, +3103-3342) (sFigure 4). In order to further clarify the methylation status of the LRP1B gene promoter in HCC tissues, we used quantitative methylation-specific PCR (Q-MSP) to analyze the methylation level of the LRP1B gene promoter in 30 HCC samples and matched adjacent samples, but did not find the difference between HCC and matched adjacent samples (sFigure 4), suggesting that high-expression of LRP1B in HCC as compared to noncancerous tissues might result from other factors rather than its epigenetic regulation.
Our previous serial studies revealed that transcription factor-heat shock factor 1 (HSF1) was highly expressed in HCC and closely related to the malignant biological behavior of HCC cells as well as played a key role in the regulation of HCC metabolism.29–32 Therefore, to examine whether LRP1B expression can be regulated by HSF1 in HCC, we applied firstly TFBIND (http://tfbind.hgc.jp/) and ALGGEN-PROMO (http://alggen.lsi.upc.es) online software to analyze the transcription factor binding sites in the LRP1B promoter region and found that several potential HSF1-binding sites were simultaneously predicted (Figure 4A). Afterwards, we used the dual luciferase reporter assay to verify the actual binding. The result showed that, compared to the control group, the relative luciferase activity in cells co-transfected with HSF1 vector and wild-type LRP1B promoter was significantly increased; while the luciferase activity when co-transfected HSF1 vector with mutant LRP1B promoter (MT1 and MT2) was significantly decreased (Figure 4B). To explore the effect of HSF1 on LRP1B expression, we effectively down-regulated HSF1 expression in HCC cells by using a lentivirus-induced RNA interference system and found that LRP1B expression was significantly reduced after HSF1 knockdown in HCC cells (Figure 4C; sFigure 5). Collectively, the results above indicated that LRP1B expression can be directly regulated by HSF1 in HCC.
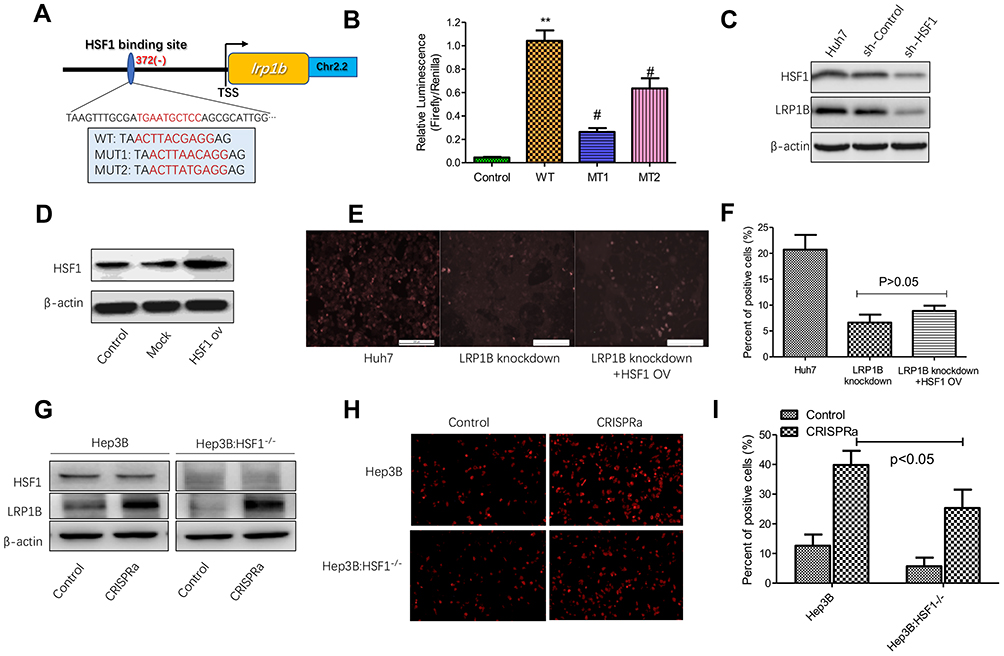 |
Figure 4 HSF1 regulates the expression of LRP1B and lipid metabolism in HCC cells. (A) Schematic representation of the sites of HSF1-binding in the LRP1B promoter region. The in-frame splicing indicates the sequences of LRP1B with an amino acid changed or not, where HSF1 bound at the transcription site. TSS: transcription site. ch2.2: chromosome 2.2. WT: wildtype; MUT1: mutation1; MUT2: mutation2. (B) Dual luciferase reporter assay was used to examine the effect of HSF1 on LRP1B promoter activity, **p<0.01 vs control, #p<0.05 vs WT. Control: HSF1+pGL3-basic+pRL-TK; WT: HSF1+LRP1B pro (wildtype) +pRL-TK; MT1: HSF1+LRP1B pro (MUT1) +pRL-TK; MT2: HSF1+LRP1B pro (MUT2) +pRL-TK. (C) Western blotting confirmed the efficiency of HSF1 knockdown in Huh7 cells. β-actin was regarded as an internal control. (D) HSF1 overexpression in Huh7 cells was measured by Western blotting. β-actin was regarded as an internal control. (E) Representative images of DiD staining in LRP1B-knockdown Huh7 cells with HSF1 overexpression. The red indicated the positive lipophilic cells (×100). (F) The percent of positive lipophilic cells collected from the same experiments as shown. (G) Western blotting was used for detecting the LRP1B transcriptional activation in Hep3B with HSF1 knockdown. (H) Representative images of DiD staining in Huh3B cells with transcriptional activation (×100). (I) The percent of positive cells collected from the same experiments as shown. *P<0.05. Data represented the mean±SD. Statistical significance was analyzed by two-tailed unpaired Student’s t-test. Triplicate experiments independently with similar results. |
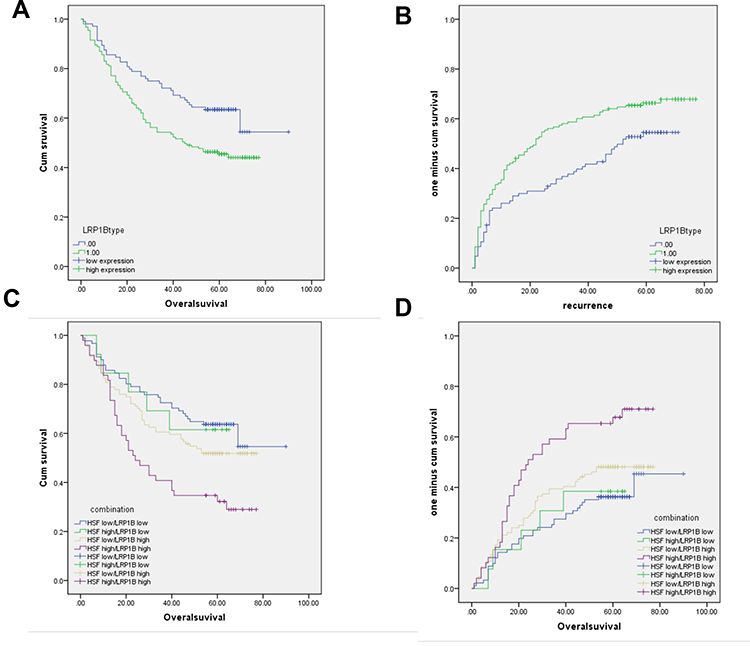 |
Figure 5 LRP1B and HSF1 is correlated with poor survival and prognosis in patients with HCC. (A–B), Kaplan-Meier analysis of cumulative overall survival time (OS) (A) and time to cumulative recurrence (TTR) (B) in HCC patients with LRP1B high expression compared to those with LRP1B low expression. (C–D), The cumulative OS (C) and TTR (D) analysis of HCC patients with simultaneous high expression of both LRP1B and HSF1. Statistical significance was analyzed by Log rank test. |
Involvement of HSF1 in LRP1B-Mediated Lipid Metabolism in HCC
Based on the findings above, we questioned whether HSF1 participates in lipid metabolism of HCC cells via regulating LRP1B. To address the issue, we upregulated HSF1 expression in HCC cells and found the upregulation of HSF1 promoted the intracellular lipid accumulation in HCC cells (sFigure 6). Subsequently, we upregulated HSF1 expression in LRP1B-knockdown HCC cells (Figure 4D) and observed by using DiD staining that the genetic upregulation of HSF1 in LRP1B deficiency condition slightly and non-significantly increased the number of staining positive cells as compared to cells with LRP1B knockdown, but maintained a significant decrease as compared to control cells without LRP1B knockdown (Figure 4E-F). This result suggested that HSF1 may be involved in lipid metabolism via affecting other bypasses except the LRP1B-mediated pathway.
To further support our hypothesis that HSF1 is involved in LRP1B-mediated lipid metabolism in HCC, we chose Hep3B cells, which harboring low LRP1B baseline expression, and down-regulated efficaciously HSF1 expression. Due to the fact that LRP1B mRNA sequence is too long (>15000 bp), which will lead to the dilemma of constructing LRP1B overexpression vector, we applied the CRISPRa transcription activation system to directly activate the transcription of LRP1B and upregulated its expression in Hep3B cells with HSF1 knockdown (Figure 4G); then measured the lipid content in these cells. The result showed that the activation of LRP1B transcription strikingly increased the number of staining positive cells in control Hep3B and HSF1-knockdown Hep3B; notably, when the direct activation of LRP1B transcription, staining positive number of Hep3B with HSF1 knockdown significantly reduced as compared to Hep3B without HSF1 knockdown (Figure 4H-I). This suggested that in HCC cells, LRP1B-mediated lipid metabolism requires mostly the existence of HSF1.
Prognostic Value of LRP1B Combined with HSF1 in HCC
Previous studies including our work have verified the clinical prognostic value of HSF1 in HCC.29,33 Herein, we further investigated the prognostic value of LRP1B combined with HSF1 in HCC through screening the expressions of LRP1B and HSF1 in 305 HCC samples with complete clinical information. The result from univariant analysis showed that LRP1B expression was correlated with patient age and serum AFP level (P = 0.038 and 0.008, respectively) (Table 1). Further results from Kaplan-Meier analysis showed that LRP1B expression represented a negative correlation with overall survival period of HCC patients (P = 0.006) (Figure 5A), while displayed a positive correlation with recurrence (P = 0.012) (Figure 5B); moreover, the OS of patients with simultaneous high expression of both LRP1B and HSF1 was significantly lower than that of patients with single high expression or simultaneous negative expression of LRP1B and HSF1 (P = 0.002) (Figure 5C), while the recurrence presented a significant increase in patients with simultaneous high expression of both LRP1B and HSF1 (P = 0.018) (Figure 5D). Furthermore, data from multivariate analysis showed that the simultaneous expression of HSF1 and LRP1B was an independent prognostic factor for OS (hazard ratio [HR]: 1.317; 95% CI: 1.121–1.549; P = 0.001) and recurrence (HR: 1.261; 95% CI: 1.093–1.456; P = 0.002) (Table 2).
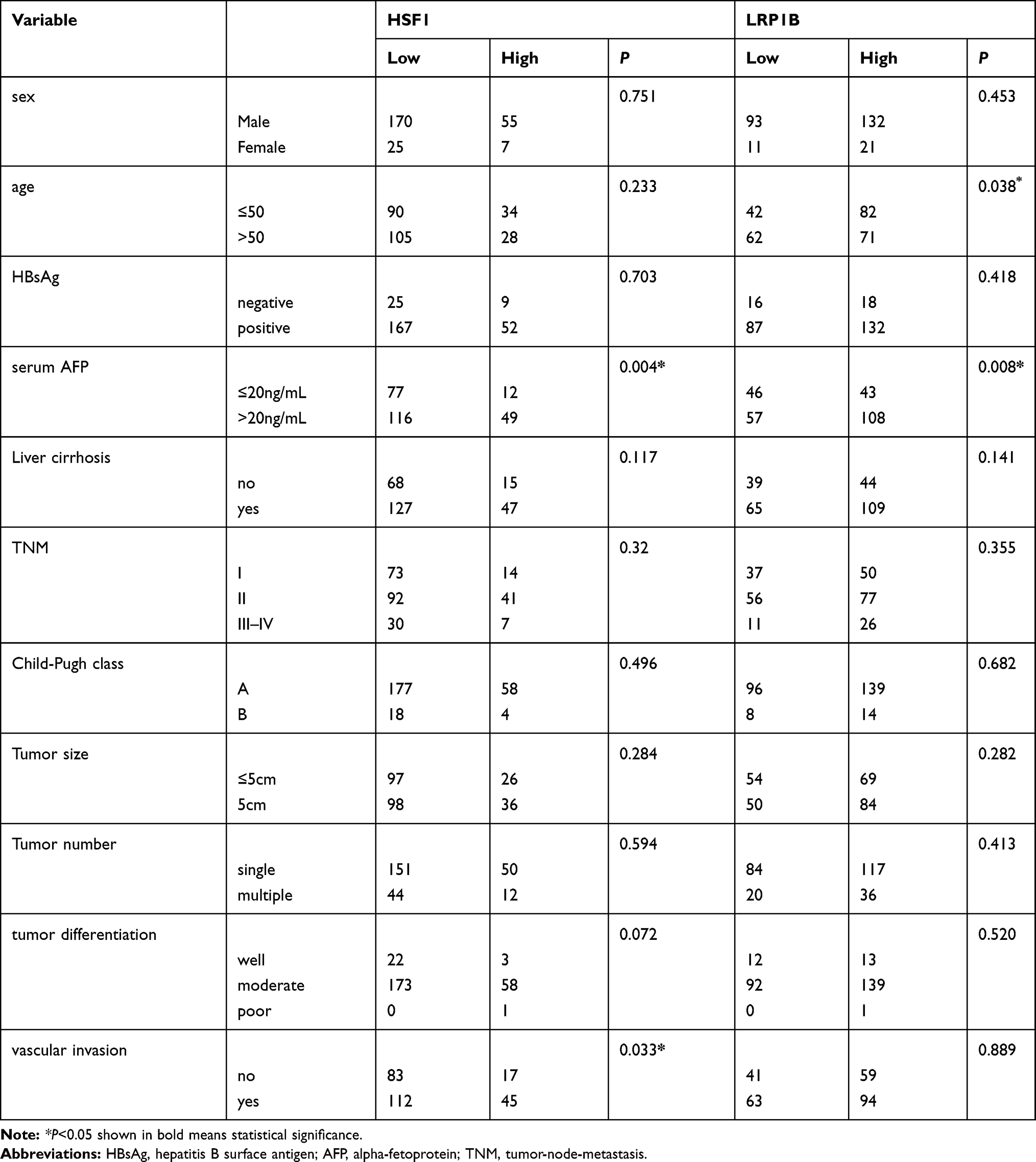 |
Table 1 Correlation of LRP1B and HSF1 Expression and Clinicopathologic Parameters in HCC Patients |
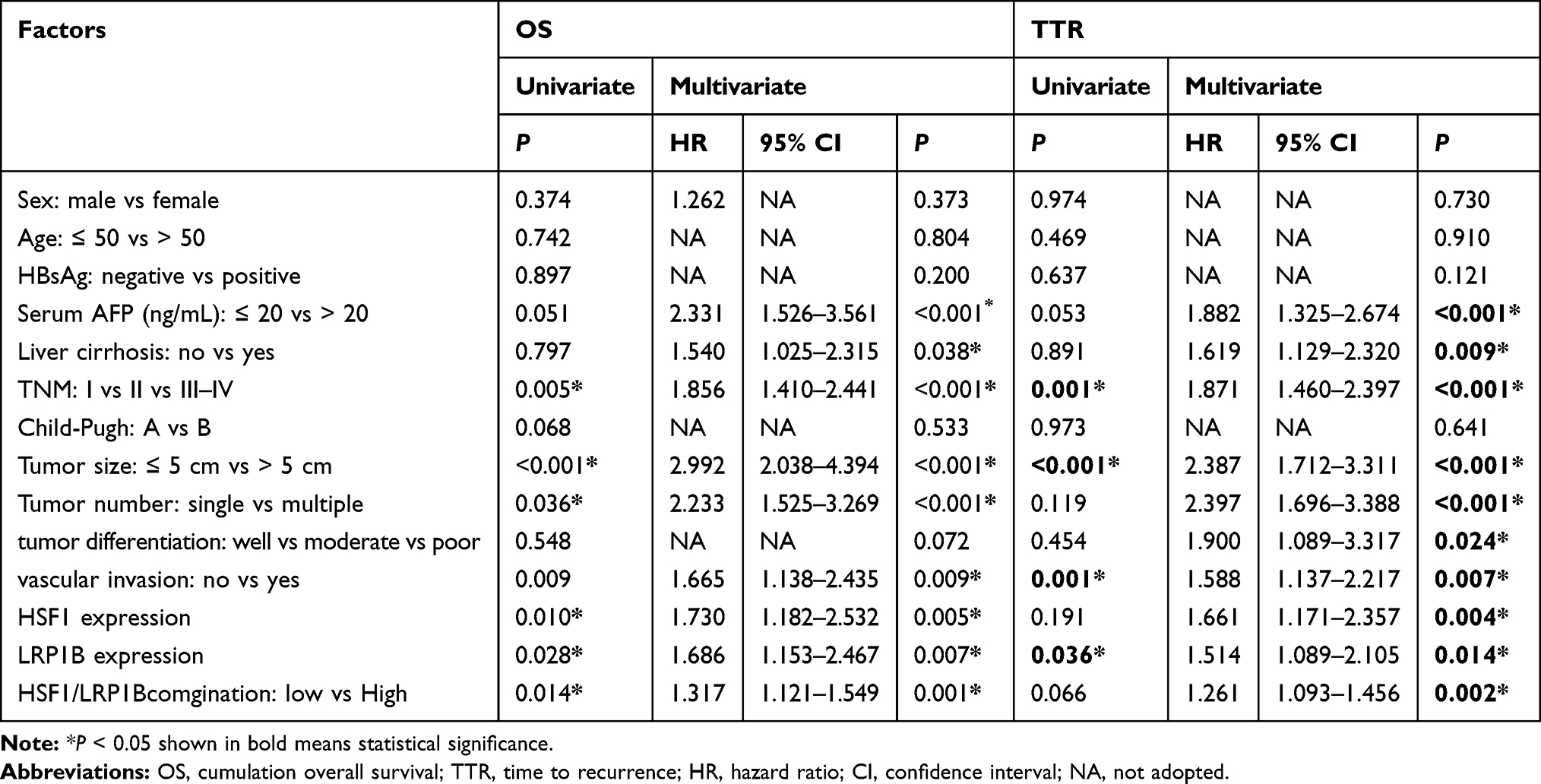 |
Table 2 Univariate and Multivariate Analysis of Different Prognostic Parameters in Patients with HCC Patients |
Synergistic Suppression of LRP1B Knockdown and HSF1 Inhibition in the Proliferation of HCC Cells
In recent years, high-fat diet has been demonstrated to promote the initiation and development of cancer34 and enhance the proliferation of cancer cells via a variety of oncogenic signaling.35,36 Therefore, we observed the effect of high-fat on the proliferation of liver cancer cells. The result from experiment in vitro showed that lipid-rich culture significantly upregulated the expressions of LRP1B and HSF1 in HCC cells (sFigure 7). Furthermore, we established the subcutaneous hepatoma in C57BL/6J mice with Hep1-6 cells and performed a high-fat diet feeding for 30 days. The result also showed that a high-fat diet enhanced the growth of allograft tumors and upregulated the expressions of LRP1B and HSF1 as well as FA synthesis-related enzymes while downregulated the expressions of β-oxidation-related enzymes (sFigure 7). In addition, based on the finding and observations above that simultaneous high expression of both LRP1B and HSF1 indicates the poor prognosis of patients with HCC, we sought to preliminarily explore the potential role of simultaneously targeted inhibition of LRP1B and HSF1 in treating HCC. Hence, we treated LRP1B-knockdown HCC cells with HSF1 specific inhibitor-KRIBB11 (10 μM) and observed the change of proliferation in these cells. The result showed that, compared to LRP1B knockdown or KRIBB11 treatment, LRP1B knockdown combined with KRIBB11 treatment significantly reduced the proliferation of HCC cells (Figure 6A). Moreover, we further found that LRP1B knockdown combined with KRIBB11 treatment in HCC cells significantly reduced the expression and activity of lipid synthesis-related enzymes, while enhancing the expression and activity of β-oxidation-related enzyme (Figure 6B-F).
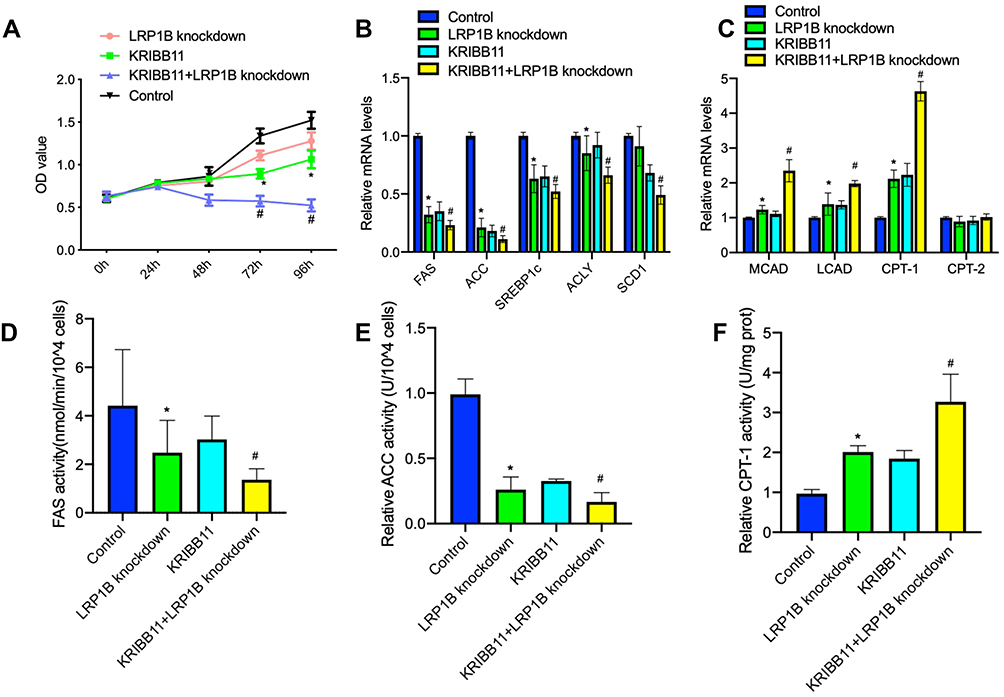 |
Figure 6 LRP1B knockdown combined with KRIBB11 treatment significantly reduced the proliferation of HCC cells. (A) Cell proliferation in Huh7 cells with single LRP1B knockdown or KRIBB11 treatment, LRP1B knockdown +KRIBB11 treatment was assessed by a CCK-8 assay. Untreated Huh7 was set up as a control. (B–C) The mRNA levels of several key enzymes for fatty acid synthesis-FAS, ACC, SREBP1c, ACLY, SCD1 (B) and important enzymes for β-oxidation of fatty acids-CPT-1, CPT-2, LCAD, MCAD (C) in Huh7 cells mentioned above were assessed by quantitative PCR. (D–F) Relative activity of FAS, ACC and CPT-1 in Huh7 cells mentioned above were assessed by associated ELISA assays. *P <0.05 vs control; # P <0.05 vs knockdown. Data represented the mean±SD. Statistical significance was analyzed by two-tailed unpaired Student’s t-test. Triplicate experiments independently with similar results. |
Discussion
In recent years, due to the development of multi-omics techniques including proteomics, genomics and metabolomics, the molecular feature of liver cancer has gradually been revealed.9,10,37 Notably, metabolic alteration is being regarded as a key hallmark of liver cancer but still remains unclear in HCC.10,38 Previous well-studied metabolic alterations in HCC are mainly glucose, glutamine and methionine metabolism,39–41 less attention has been paid to lipid metabolism features of HCC. As obesity- and NAFLD-related HCC cases increase in China, it is urgent to exploit the patterns and influences of lipid metabolism in HCC. Accumulating evidence shows that HCC is typically characterized by perturbations in fatty acid and lipid metabolism, mainly reflected in dysregulation of genes and serum metabolites involved in fatty acid synthesis and trafficking.42,43
As a large member of the low-density lipoprotein receptor family, LRP1B plays a dual role in lipid homeostasis, not only providing the essential lipids to maintain cell function, but also regulating the metabolism of cholesterol lipoproteins in the circulation.44 A majority of studies in the cancer field have demonstrated that LRP1B is negatively or lowly expressed in cancers including esophageal squamous carcinoma (ESC),28 non-small cell lung cancer (NSCLC),45 thyroid cancer,46 cervical cancer47 and melanoma,48 and identified LRP1B as a potential tumor suppressor in these cancers. Intriguingly, in contrast to the cancers mentioned above, HCC presents a predominantly strong expression of LRP1B. High expression of LRP1B in HCC was further identified in various cell lines, harboring different genetic backgrounds and representative characteristics, and clinical samples by us in the study. The result suggested a possibly unique role for LRP1B in HCC, different from tumor suppression in other majority cancers.
In view of the fundamental function of LRP1B as a member of the low-density lipoprotein receptor family, we focused mainly on the effect of LRP1B on lipid metabolism of HCC cells, although we also identified the inhibition of LRP1B in the proliferation, clone formation and pseudopodia formation of HCC cells in another study, suggesting a positive role of LRP1B in maintaining the malignant behaviors of HCC cells. In the study, we found that lipid accumulation, storage or lipid droplets were significantly reduced after LRP1B knockdown. The result indicated the important role of LRP1B in remaining intracellular lipid content in HCC cells. Meanwhile, based on the fact that lipids can provide sufficient energy and substances for cancer cells,49,50 it also suggested a potential therapeutic strategy for HCC treatment that targeting against LRP1B will suppress the energy production in HCC cells through limiting the supplement of lipid and lipoprotein.
It was reported that an upregulated lipogenic pathway exists in HCC.51 As the first rate-limiting enzyme for fatty acid synthesis, ACC can catalyze fatty acid synthesis and inhibit the oxidative decomposition of fatty acids while the other enzyme FAS is often in an over-activated state in cancers, leading to aberrated lipid metabolism.52 In contrast, as the rate-limiting enzyme in the process of fatty acid β-oxidation, CPT-1 controls fatty acids to enter the mitochondria.53 Through detecting the expressions and activities of these enzymes after LRP1B knockdown, we found the significant decrease of ACC and FAS and the increase of CPT-1 after LRP1B knockdown. It suggested that LRP1B-mediated lipid accumulation may result from regulating the balance between FA synthesis and β-oxidation. Furthermore, AMPK is a sensor of cell energy metabolism and its activation can inhibit the expression of SERBP-1c, an important upstream factor of ACC and FAS, finally leading to the suppression of de novo synthesis of fatty acid.54 More recently, it has been reported that upregulation of AMPK activity, a typical nutrient deficiency condition, promoted the expressions of CPT-1 and CPT-2 and finally initiated the β-oxidation in HCC.55 Hence, we observed the change of AMPK activation after LRP1B knockdown in HCC cells and found the upregulated activation of AMPK signaling. Herein, it was hypothesized that the involvement of the AMPK pathway may be in the regulation of LRP1B in lipid metabolism in HCC cells.
In the study, to further clarify the mechanism of LRP1B in lipid metabolism, we investigated which factors accounted for the high expression of LRP1B in HCC. Previous researchers have found that loss of LRP1B function in other cancers often results from homozygous deletion or CpG island hypermethylation-induced transcriptional silencing.28 Herein, we examined the methylation level of CpG island in the promotion of LRP1B gene in HCC tissues and matched adjacent tissues and found no significant difference. It suggested that high expression of LRP1B in HCC may not be directly related to epigenetic regulation. In addition to epigenetic regulation, the transcription of a gene is also closely related to upstream transcription factors. In our previous studies, HSF1 was identified as a key transcription factor, which played a pivotal role in regulating the metabolic feature of HCC cells.29–32 Hence, we investigated whether HSF1 can directly modulate the transcription of LRP1B and confirmed the hypothesis by using a dual luciferase report assay. Subsequently, we concluded that LRP1B-mediated lipid metabolism requires the existence of HSF1 in HCC cells. Nevertheless, the specific molecular mechanism needs to be further explored.
Finally, to evaluate the potential prognostic value of LRP1B combined with HSF1 in HCC, we analyzed the association of LRP1B and HSF1 expression with prognosis of 305 HCC patients and found that a high expression of LRP1B and HSF1 was closely related to shorter OS and a higher risk of recurrence; and the simultaneous expression of HSF1 and LRP1B was an independent prognostic factor for HCC patients. The result will provide evidence for the precise stratification of HCC patients in the future. At the same time, the finding encourages us to explore the possibility of HCC targeted therapy against LRP1B and HSF1. We applied, respectively, genetic and chemical inhibitors of LRP1B and HSF1 to combinedly treat HCC cells and demonstrated the synergistic suppression of LRP1B knockdown and HSF1 inhibition in the proliferation of HCC cells as well as activity of lipid metabolism-related enzymes. Pharmacologic inhibition of the HSF1 pathway is promising in the field of treating cancers.56 Our findings will provide a choice for making multidisciplinary therapeutic strategies for HCC in the future.
However, there are several limitations in our study. We had not planned to observe the direct effect of intracellular lipid accumulation through applying specific drugs on the growth of liver cancer cells, but an important study demonstrated recently that lipid synthesis promotes the tumorigenesis of hepatocytes;57 meanwhile we observed that a high-fat diet enhanced the growth of subcutaneous tumors in vivo. Altogether, it suggests a key relationship between lipogenesis and growth of HCC cells. In addition, the exhaustive molecular mechanism for explaining the association of LRP1B with HSF1 or AMPK, which consequently leads to lipid metabolism reprogramming in HCC, is still to be elucidated. Nevertheless, from the available data, we are able to reveal a novel unique oncogenic role of LRP1B in HCC by serving as a mediator in lipid metabolism, whereas the direct regulation of LRP1B transcription by HSF1 provides an insight for synergistically inhibiting the proliferation of HCC cells. Hence, targeting LRP1B-mediated lipid metabolism might be a potential therapeutic strategy for HCC treatment in the future.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China, China (81872492) and Shanghai Pujiang Program (No. 15PJD007).
Author Contributions
K.G. designed the experimental work, wrote the study documentation and the manuscript and was the chief investigator of the study. M.L. and J.H. participated in the experiments, analyzed data and wrote the manuscript. R.J. coordinated the acquisition, distribution and quality evaluation of clinical samples. H(x).C. performed the supplementary experiments in the revision process and wrote the revised manuscript. H(p).C. and L.L. contributed to statistical analysis and participated in the experimental works.
Disclosure
The authors declare no conflicts of interest.
References
1. Erratum. Erratum: global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2020;70(4):313. doi:10.3322/caac.21609
2. GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study. Lancet. 2018;392(10159):1736–1788. doi:10.1016/S0140-6736(18)32203-7
3. Kumari R, Sahu MK, Tripathy A, Uthansingh K, Behera M. Hepatocellular carcinoma treatment: hurdles, advances and prospects. Hepatic Oncology. 2018;5(2):HEP08. doi:10.2217/hep-2018-0002
4. Gao Q, Wang ZC, Duan M, et al. Cell Culture System for Analysis of Genetic Heterogeneity Within Hepatocellular Carcinomas and Response to Pharmacologic Agents. Gastroenterology. 2017;152(1):232–242. doi:10.1053/j.gastro.2016.09.008
5. De Mattia E, Cecchin E, Guardascione M, et al. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J Gastroenterol. 2019;25(29):3870–3896. doi:10.3748/wjg.v25.i29.3870
6. Lin Z, Lu D, Wei X, Wang J, Xu X. Heterogeneous responses in hepatocellular carcinoma: the achilles heel of immune checkpoint inhibitors. Am J Cancer Res. 2020;10(4):1085–1102.
7. Caruso S, O’Brien DR, Cleary SP, Roberts LR, Zucman-Rossi J. Genetics of HCC: novel approaches to explore molecular diversity published online ahead of print, 2020. Hepatology. 2020;2:457.
8. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019;120(12):1090–1098. doi:10.1038/s41416-019-0451-4
9. Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–261. doi:10.1038/s41586-019-0987-8
10. Gao Q, Zhu H, Dong L, et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell. 2019;179(2):561–577. doi:10.1016/j.cell.2019.08.052
11. Kalish BT, Fell GL, Nandivada P, Puder M. Clinically Relevant Mechanisms of Lipid Synthesis, Transport, and Storage. JPEN J Parenter Enteral Nutr. 2015;39(1_suppl):8S–17S. doi:10.1177/0148607115595974
12. Strickland DK, Gonias SL, Argraves WS. Diverse roles for the LDL receptor family. Trends Endocrinol Metab. 2002;13(2):66–74. doi:10.1016/s1043-2760(01)00526-4
13. Chen H, Chong W, Wu Q, et al. Association of LRP1B mutation with tumor mutation burden and outcomes in melanoma and non-small cell lung cancer patients treated with immune check-point blockades. Front Immunol. 2019;10:1113. doi:10.3389/fimmu.2019.01113
14. Zheng H, Bai L. Hypoxia induced microRNA-301b-3p overexpression promotes proliferation, migration and invasion of prostate cancer cells by targeting LRP1B. Exp Mol Pathol. 2019;111:104301. doi:10.1016/j.yexmp.2019.104301
15. Liu L, Ren M, Han S, Sun L, Zhu L. Expression level and clinical significance of low-density lipoprotein receptor-related protein 1B gene in cervical squamous cell carcinoma. Int J Clin Exp Pathol. 2018;11(3):1701–1706.
16. Ni S, Hu J, Duan Y, et al. Down expression of LRP1B promotes cell migration via RhoA/Cdc42 pathway and actin cytoskeleton remodeling in renal cell cancer. Cancer Science. 2013;104(7):817–825. doi:10.1111/cas.12157
17. Liu C-X, Ranganathan S, Robinson S, Strickland DK. γ-Secretase-mediated Release of the Low Density Lipoprotein Receptor-related Protein 1B Intracellular Domain Suppresses Anchorage-independent Growth of Neuroglioma Cells. J Biol Chem. 2007;282(10):7504–7511. doi:10.1074/jbc.M608088200
18. Elgendy M, Fusco JP, Segura V, et al. Identification of mutations associated with acquired resistance to sunitinib in renal cell cancer. Int J Cancer. 2019;145(7):1991–2001. doi:10.1002/ijc.32256
19. Zhu H, Yang B, Liu J, Wu W, Ling Y. Case Report of acute myeloid leukemia with “WT1, ATRX, CEBPA, CSMD1, IKZF1, and LRP1B mutation and translocation between chromosome 1 and 19” developing from Philadelphia-negative chronic myeloid leukemia after TKI therapy. Medicine. 2020;99(3):e18888. doi:10.1097/MD.0000000000018888
20. Wang L, Yan K, Zhou J, et al. Relationship of liver cancer with LRP1B or TP53 mutation and tumor mutation burden and survival. 2019 ASCO Abstract 2019 1573.
21. Yu H, Wang H, Xu H, et al. Overexpression of MTHFD1 in hepatocellular carcinoma predicts poorer survival and recurrence. Future Oncol. 2019;15(15):1771–1780. doi:10.2217/fon-2018-0606.
22. Camp RL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–7259. doi:10.1158/1078-0432.CCR-04-0713.
23. Marzolo M-P, Bu G. Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin Cell Dev Biol. 2009;20(2):191–200. doi:10.1016/j.semcdb.2008.10.005
24. Tian S, Li B, Lei P, et al. Sulforaphane Improves Abnormal Lipid Metabolism via Both ERS-Dependent XBP1/ACC &SCD1 and ERS-Independent SREBP/FAS Pathways. Mol Nutr Food Res. 2018;62(6):e1700737. doi:10.1002/mnfr.201700737
25. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
26. Lu YJ, Wu CS, Li HP, et al. Aberrant methylation impairs low density lipoprotein receptor-related protein 1B tumor suppressor function in gastric cancer. Genes Chromosomes Cancer. 2010;49(5):412–424. doi:10.1002/gcc.20752
27. Nakagawa T, Pimkhaokham A, Suzuki E, Omura K, Inazawa J, Imoto I. Genetic or epigenetic silencing of low density lipoprotein receptor-related protein 1B expression in oral squamous cell carcinoma. Cancer Science. 2006;97(10):1070–1074. doi:10.1111/j.1349-7006.2006.00283.x
28. Sonoda I, Imoto I, Inoue J, et al. Frequent Silencing of Low Density Lipoprotein Receptor-Related Protein 1B (LRP1B) Expression by Genetic and Epigenetic Mechanisms in Esophageal Squamous Cell Carcinoma. Cancer Res. 2004;64(11):3741–3747. doi:10.1158/0008-5472.CAN-04-0172
29. Zhang J-B, Guo K, Sun H-C, et al. Prognostic value of peritumoral heat-shock factor-1 in patients receiving resection of hepatocellular carcinoma. Br J Cancer. 2013;109(6):1648–1656. doi:10.1038/bjc.2013.488
30. Shen JH, Chen PH, Liu HD, Huang DA, Li MM, Guo K. HSF1/AMPKα2 mediated alteration of metabolic phenotypes confers increased oxaliplatin resistance in HCC cells. Am J Cancer Res. 2019;9(11):2349–2363.
31. Liu HT, Huang DA, Li MM, Liu HD, Guo K. HSF1: a mediator in metabolic alteration of hepatocellular carcinoma cells in cross-talking with tumor-associated macrophages. Am J Transl Res. 2019;11(8):5054–5064.
32. Liu D, Sun L, Qin X, et al. HSF1 promotes the inhibition of EMT-associated migration by low glucose via directly regulating Snail1 expression in HCC cells. Discov Med. 2016;22(120):87–96.
33. Kijima T, Prince T, Neckers L, Koga F, Fujii Y. Heat shock factor 1 (HSF1)-targeted anticancer therapeutics: overview of current preclinical progress. Expert Opin Ther Targets. 2019;10(5):369–377. doi:10.3389/fimmu.2019.01113
34. High-Fat Diet A. Promotes Intestinal Stemness and Tumorigenesis. Cancer Discov. 2016;6(5):OF12. doi:10.1158/2159-8290.CD-RW2016-048
35. Wang C, Li P, Xuan J, et al. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell Physiol Biochem. 2017;42(2):729–742. doi:10.1159/000477890
36. Huang M, Narita S, Numakura K, et al. A high-fat diet enhances proliferation of prostate cancer cells and activates MCP-1/CCR2 signaling. Prostate. 2012;72(16):1779–1788. doi:10.1002/pros.22531
37. Liu F, Qin L, Liao Z, et al. Microenvironment characterization and multi-omics signatures related to prognosis and immunotherapy response of hepatocellular carcinoma. Exp Hematol Oncol. 2020;9:10. doi:10.1186/s40164-020-00165-3
38. De Matteis S, Ragusa A, Marisi G, et al. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid Med Cell Longev. 2018;2018:7512159. doi:10.1155/2018/7512159
39. Mossenta M, Busato D, Bo M D, Glucose Metabolism TG. Oxidative Stress in Hepatocellular Carcinoma: role and Possible Implications in Novel Therapeutic Strategies. Cancers. 2020;12(6):1668. doi:10.3390/cancers12061668
40. Nwosu ZC, Piorońska W, Battello N, et al. Severe metabolic alterations in liver cancer lead to ERK pathway activation and drug resistance. EBioMedicine. 2020;54:102699.
41. Pascale RM, Peitta G, Simile MM, Feo F. Alterations of Methionine Metabolism as Potential Targets for the Prevention and Therapy of Hepatocellular Carcinoma. Medicina. 2019;55(6):296. doi:10.3390/medicina55060296
42. Nelson ME, Lahiri S, Chow JD, et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat Commun. 2017;8:14689. doi:10.1038/ncomms14689
43. Fages A, Duarte-Salles T, Stepien M, et al. Metabolomic profiles of hepatocellular carcinoma in a European prospective cohort. BMC Med. 2015;13:242. doi:10.1186/s12916-015-0462-9
44. Marschang P, Brich J, Weeber EJ, et al. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol Cell Biol. 2004;24(9):3782–3793. doi:10.1128/mcb.24.9.3782-3793.2004
45. Beer AG, Zenzmaier C, Schreinlechner M, et al. Expression of a recombinant full-length LRP1B receptor in human non-small cell lung cancer cells confirms the postulated growth-suppressing function of this large LDL receptor family member. Oncotarget. 2016;7(42):68721–68733. doi:10.18632/oncotarget.11897
46. Prazeres H, Torres J, Rodrigues F, et al. Chromosomal, epigenetic and microRNA-mediated inactivation of LRP1B, a modulator of the extracellular environment of thyroid cancer cells. Oncogene. 2011;30(11):1302–1317. doi:10.1038/onc.2010.512
47. Wang Y, Han S, You X, et al. The role of low density lipoprotein receptor-related protein 11 as a tumor promoter in cervical cancer. Cancer Manag Res. 2019;11:8081–8093. doi:10.2147/CMAR.S211912
48. Chen H, Chong W, Wu Q, Yao Y, Mao M. Corrigendum: association of LRP1BMutation With Tumor Mutation Burden and Outcomes in Melanoma and Non-small Cell Lung Cancer Patients Treated With Immune Check-Point Blockades. Front Immunol. 2019;10:1523. doi:10.3389/fimmu.2019.01523
49. Annabi B, Doumit J, Plouffe K, Laflamme C, Lord-Dufour S, Béliveau R. Members of the low-density lipoprotein receptor-related proteins provide a differential molecular signature between parental and CD133+ DAOY medulloblastoma cells. Mol Carcinog. 2010;49(7):710–717. doi:10.1002/mc.20645
50. Budhu A, Roessler S, Zhao X, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144(5):1066–1075. doi:10.1053/j.gastro.2013.01.054
51. Yamashita T, Honda M, Takatori H, et al. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J Hepatol. 2009;50(1):100–110. doi:10.1016/j.jhep.2008.07.036
52. Guebre-Egziabher F, Alix PM, Koppe L, et al. Ectopic lipid accumulation: A potential cause for metabolic disturbances and a contributor to the alteration of kidney function. Biochimie. 2013;95(11):1971–1979. doi:10.1016/j.biochi.2013.07.017
53. Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7(5):e2226. doi:10.1038/cddis.2016.132
54. Qu YY, Zhao R, Zhang HL, et al. Inactivation of the AMPK-GATA3-ECHS1 Pathway Induces Fatty Acid Synthesis That Promotes Clear Cell Renal Cell Carcinoma Growth. Cancer Res. 2020;80(2):319–333. doi:10.1158/0008-5472.CAN-19-1023
55. Sangineto M, Villani R, Cavallone F, et al. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers. 2020;12(6):1419. doi:10.3390/cancers12061419.
56. Fok JHL, Hedayat S, Zhang L, et al. HSF1 Is Essential for Myeloma Cell Survival and A Promising Therapeutic Target. Clin Cancer Res. 2018;24(10):2395–2407. doi:10.1158/1078-0432.CCR-17-1594
57. Guri Y, Colombi M, Dazert E, et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell. 2017;32(6):807–823. doi:10.1016/j.ccell.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.