")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Effects of intracerebral hemorrhage on 5-hydroxymethylcytosine modification in mouse brains
Authors Tang Y, Han S, Asakawa T , Luo Y, Han X, Xiao B, Dong Q, Wang L
Received 30 September 2015
Accepted for publication 7 January 2016
Published 15 March 2016 Volume 2016:12 Pages 617—624
DOI https://doi.org/10.2147/NDT.S97456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Yilin Tang,1,* Sha Han,1,* Tetsuya Asakawa,2,3 Yunhe Luo,1 Xiang Han,1 Baoguo Xiao,4 Qiang Dong,1,4 Liang Wang1,4
1Department of Neurology, Huashan Hospital of Fudan University, Shanghai, People’s Republic of China; 2Department of Neurosurgery, 3Department of Psychiatry, Hamamatsu University School of Medicine, Hamamatsu, Shizuoka, Japan; 4Institute of Neurology, Huashan Hospital of Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: The past decade has resulted in an increase in the knowledge of molecular mechanisms underlying brain injury induced by intracerebral hemorrhage (ICH). Recent advances have provided a link between epigenetic modification and the regulation of gene expression. 5-hydroxymethylcytosine (5hmC) converted from 5-methylcytosine by the ten-eleven translocation (TET) family of proteins has emerged as a new epigenetic modification. While the dynamics of 5hmC during cerebral ischemia have recently been reported, whether 5hmC is involved in ICH remains unexplored. In this study, we investigated the effects of ICH on DNA hydroxymethylation. We showed that the global level of 5hmC rapidly decreased as early as 24 hours after ICH and persisted until 72 hours. Furthermore, the level of 5hmC in the CpG-rich regions of Akt2, Pdpk1 and Vegf genes was significantly decreased with a minimum level observed at 48 hours or 72 hours. Decreased 5hmC was observed in parallel with an increase in 5-methylcytosine over this time course, and mRNA levels of Akt2, Pdpk1 and Vegf were downregulated upon ICH injury. Finally, Tet1, Tet2 and Tet3 mRNA levels were dramatically decreased in the ICH brain. Our study for the first time established the correlation between DNA hydroxymethylation and ICH injury. Further investigations should examine whether 5hmC modification could be a therapeutic target for the treatment of ICH injury.
Keywords: stroke, cerebral edema, DNA hydroxymethylation, TET protein
Introduction
Intracerebral hemorrhage (ICH) is a devastating form of stroke associated with high mortality rates.1,2 Many studies on animal models of ICH have been performed to elucidate the mechanism of the secondary damage induced by ICH, including altered gene expression as observed by several genomic profiling studies.3–5 However, how ICH causes the transcriptional changes is still unknown.
Recent studies have revealed a link between epigenetic modifications and the regulation of gene expression.6 DNA methylation is a covalent postreplicative modification of genomic DNA that suppresses gene expression and plays an important role in diverse biological processes, such as development and tumor suppression.7–9 This modification is mediated by DNA methyltransferases (DNMTs) and occurs predominantly on CpG dinucleotides.10 Increasing evidence has suggested that DNA methylation is important in ischemic brain damage. In the middle cerebral artery occlusion mice, DNA methylation levels were increased in the ischemic brain tissue. Administration of 5-aza-2′-deoxycytinine (a DNMT inhibitor) conferred neuroprotection in wild-type mice, and DnmtS/+ heterozygous mice exhibited reduced methylation and increased resistance to ischemic damage. However, mice without neuronal DNMT1 were not protected from cerebral ischemia.11 Decrease of Dnmt1 expression at day 4 postischemia might be related to ischemia-induced delayed neuronal death.12 These observations suggested that the dynamic modulation of Dnmt expression and the status of DNA methylation are important to prevent cell death in cerebral ischemia.
Recent studies have shown that 5-methylcytosine (5mC) can be converted to 5-hydroxymethylcytosine (5hmC) by the ten-eleven translocation (TET) family proteins through Fe(II) and alpha-ketoglutarate-dependent hydroxylation.13 5hmC is found in diverse cell types and mouse organs.14,15 The level of 5hmC is five to ten times higher in the mammalian brain than any other tissues.16,17 Mounting evidence has demonstrated that 5hmC is involved in the pathology of various neurological diseases, such as cerebral ischemia, Alzheimer’s disease, fragile X-associated tremor/ataxia syndrome, and Huntington’s disease.18–21 These observations suggested that 5hmC may function as a new epigenetic modification in addition to 5mC or as an intermediate state during DNA demethylation.14,22
Because DNA methylation and hydroxymethylation play key roles in cerebral ischemia, and as 5hmC is highly enriched in the brain, we proposed that DNA hydroxymethylation might also be involved in ICH injury. In this study, we investigated the global 5hmC levels and the effects on transcription in the brain of mice with ICH.
Materials and methods
Animals and ICH model
Male C57BL/6J mice (8–12 weeks old, body weight 25–35 g; Shanghai Institute of the Chinese Academy of Science, Shanghai, People’s Republic of China) were involved in this study. All animals were treated according to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Animal Care and Use Committee of Fudan University.
After anesthetization with 10% chloral hydrate (400 mg/kg; Sinopharm Chemical Reagent Co., Ltd., Shanghai, People’s Republic of China), the ICH model was established after a double injection of autologous blood (15 μL) into the brain parenchyma (basal ganglia) of the mice, as described previously.23 ICH mice were divided into three groups according to the duration of ICH procedure: ICH24h (n=12), ICH48h (n=12), and ICH72h (n=12). We also used sham mice (n=12) by inserting the injection needle into the basal ganglia without injection of the autologous blood. The blank control mice (n=12) were sacrificed without any surgery, deeply anesthetized using chloral hydrate (400 mg/kg).
Behavioral testing
To verify the success of the ICH model, a rating scale, based on a battery measuring the neurological function in mice described previously,24 was used to assess the neurological deficits of the animals. Briefly, two tasks, the postural reflex test (PRT) and the forelimb placing test (FPT), were included in the rating scale. The scores of PRT are 0–2 according to the level of deficits of the upper body posture while the animal was held by the tail. The scores of FPT included three subitems measuring the sensorimotor integration when forelimbs were placed responding to visual (dorsal placing 0–2; lateral placing 0–2), tactile (dorsal placing 0–2; lateral placing 0–2), and proprioceptive (0–2) stimuli. The total score was obtained by adding the scores of PRT and FPT, and the maximal score is 12, indicating the most severe deficits. After finishing the behavioral test, the animals were anesthetized, and mice were perfused with normal saline solution and 4% ice-cold paraformaldehyde through the left cardiac ventricle. The brains were removed and placed in formalin solution. The right hemisphere tissue was collected and stored at −80°C for DNA and RNA extraction.
Nissl staining
The brains were dehydrated in a 10% sucrose solution for 1 day and then 30% sucrose solution for 2–3 days, until the brain sank to the bottom of the bottle. Serial coronal sections (10 μm) were cut on a freezing microtome. Sections were stained with toluidine blue (Sigma-Aldrich Co., St Louis, MO, USA) for 20 minutes. After washing with distilled water, sections were treated with 95% ethanol for 30 seconds, covered with 50% glycerin and dried. Images were taken using a light microscope (Olympus Corporation, Tokyo, Japan) and a digital camera (Olympus Corporation).
DNA extraction
Genomic DNA was extracted from snap-frozen tissues (n=6 in each group) using the DNeasy TIANamp Genomic DNA Kit (Tiangen Biotech, Beijing, People’s Republic of China) following the manufacturer’s instructions. DNA concentrations were determined throughout by fluorometry using the HS dsDNA kit and QubitFluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Dot-blot assay
DNA dot-blot assay was performed as described previously with some modifications.25 Briefly, genomic DNA was spotted on a nitrocellulose membrane (Whatman plc, Maidstone, UK). The membrane was placed under an ultraviolet lamp for 20 minutes to crosslink the DNA and then blocked with 5% milk in Tris-buffered saline–Tween 20 for 1 hour, followed by incubation with the anti-5hmC (Active Motif, Carlsbad, CA, USA) antibody at 4°C overnight. After incubation with a horseradish peroxidase-conjugated secondary antibody (GeneScript Biotech Corporation, Piscataway Township, NJ, USA) for 1 hour at room temperature, the membrane was washed with Tris-buffered saline–Tween 20 three times and then scanned by a Typhoon scanner (GE Healthcare, Little Chalfont, UK). To control spotting, blots were stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2). Quantification of 5hmC was done by Image-Quanta software (GE Healthcare).
Quantitative real-time PCR analysis
Total RNA from mouse brains (n=6 in each group) was extracted by TRIzol reagent (Thermo Fisher Scientific), and total RNA from culture cells was extracted by the RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands). Complementary DNA was constructed using the TransScript First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, People’s Republic of China). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed using an Applied Biosystems 7500 Sequence Detection System with SYBR green labeling (Takara, Kusatsu, Japan) following the manufacturer’s instruction. β-Actin was used as an endogenous control. All primer sequences are listed in Table S1.
hMeDIP-qPCR and MeDIP-qPCR analyses
Genomic DNA from mouse brains was prepared using a phenol–chloroform method. The methylated DNA immunoprecipitation (MeDIP) and hydroxymethylated DNA immunoprecipitation (hMeDIP) assays were performed as previously described.13,26 Briefly, genomic DNA was denatured and then immunoprecipitated with an anti-5mC (Eurogentec, Liège, Belgium) or an anti-5hmC (Active Motif) antibody and protein G magnetic Dynabeads (Thermo Fisher Scientific). After washing three times, beads were treated with protein K for 4 hours. DNA was prepared with phenol–chloroform and precipitated using cold ethanol, and immunoprecipitated DNA was analyzed by qPCR. All primers are listed in Table S2.
Statistical analyses
Statistical analyses were performed with a two-tailed unpaired Student’s t-test. All data shown represent the results obtained from triplicate independent experiments with standard deviations (mean ± SD). The values of P<0.05 were considered statistically significant.
Results
Verification of ICH mouse model
The ICH model was evaluated by histopathological and behavioral analyses. Neuronal damage was examined by Nissl staining, which showed the apparent loss of neuron cells in the ICH groups (Figure 1A–C). In addition, we used the PRT and the FPT to evaluate motor functions. Compared with the control and sham groups, mice in ICH groups showed behavioral impairment in these tests (P<0.001) (Figure 1D). This histopathological and behavioral evidence confirmed the successful establishment of the ICH mouse model.
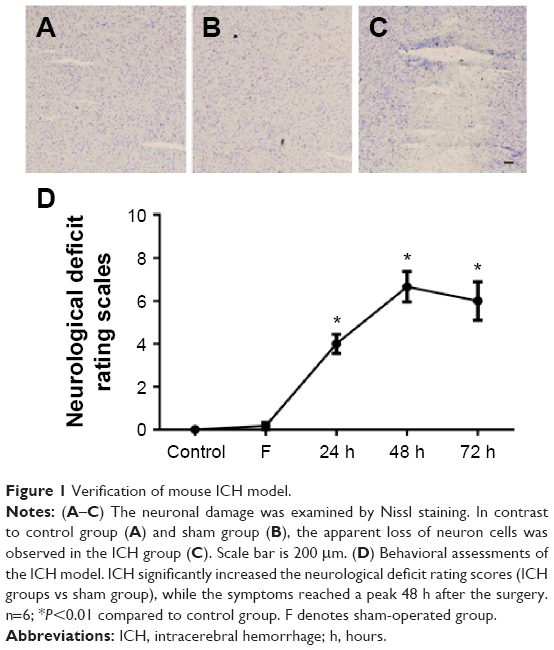 | Figure 1 Verification of mouse ICH model. |
Global 5hmC level was decreased in the brain of ICH mice
Recent studies showed that the level of 5hmC is dynamically regulated by metabolites.27 We next assessed whether the level of 5hmC in the mouse brain was altered after ICH using dot blotting. Global 5hmC levels were decreased by 51% within 24 hours after ICH injury (P<0.01) (Figure 2A and B). Moreover, the global 5hmC level was maintained at a low level until 72 hours after ICH injury. Thus, our data indicated that the global level of 5hmC in mouse brains is sensitive to ICH injury and is dynamically regulated.
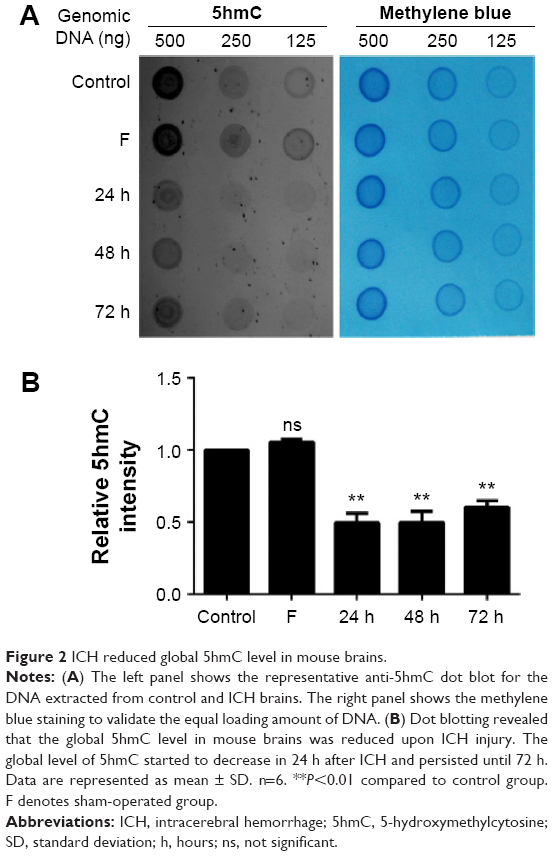 | Figure 2 ICH reduced global 5hmC level in mouse brains. |
ICH decreased 5hmC at three gene loci in mouse brains
Recent evidence showed that 5hmC can directly contribute to gene regulation in addition to being an intermediate in gene regulatory pathways.27,28 We therefore determined whether ICH injury could alter 5hmC levels at specific gene loci in mouse brains. A brain genomic profiling study revealed that growth factor-related genes and genes in the PI3K–Akt pathway show changes in expression after ICH.4 We thus selected genomic loci at CpG-rich regions in promoter regions of the genes Akt2, Pdpk1 and Vegf (Figure 3A–C). We then performed hMeDIP-qPCR and MeDIP-qPCR analyses to determine 5hmC or 5mC changes in the selected regions of these three genes, respectively. The hMeDIP-qPCR analysis demonstrated that 5hmC was significantly decreased on promoters of Akt2, Pdpk1 and Vegf genes in mouse brains 24 hours after ICH (P<0.05). Even at 72 hours after ICH, the 5hmC level on the promoters of the Akt2, Pdpk1 and Vegf genes decreased by 77%, 87%, and 75%, respectively (Figure 3D–F). The rapid decrease of 5hmC paralleled the increase of 5mC in these regions over the time course and reached its peak at 72 hours after ICH (Figure 3G–I). In addition, RT-qPCR analysis revealed that the mRNA levels of the three genes were downregulated upon ICH injury and reached their minimum at 24 hours or 48 hours (Figure 3J–L). Taken together, these results suggest that 5hmC can undergo dynamic changes, which are associated with the regulation of specific gene expression after ICH.
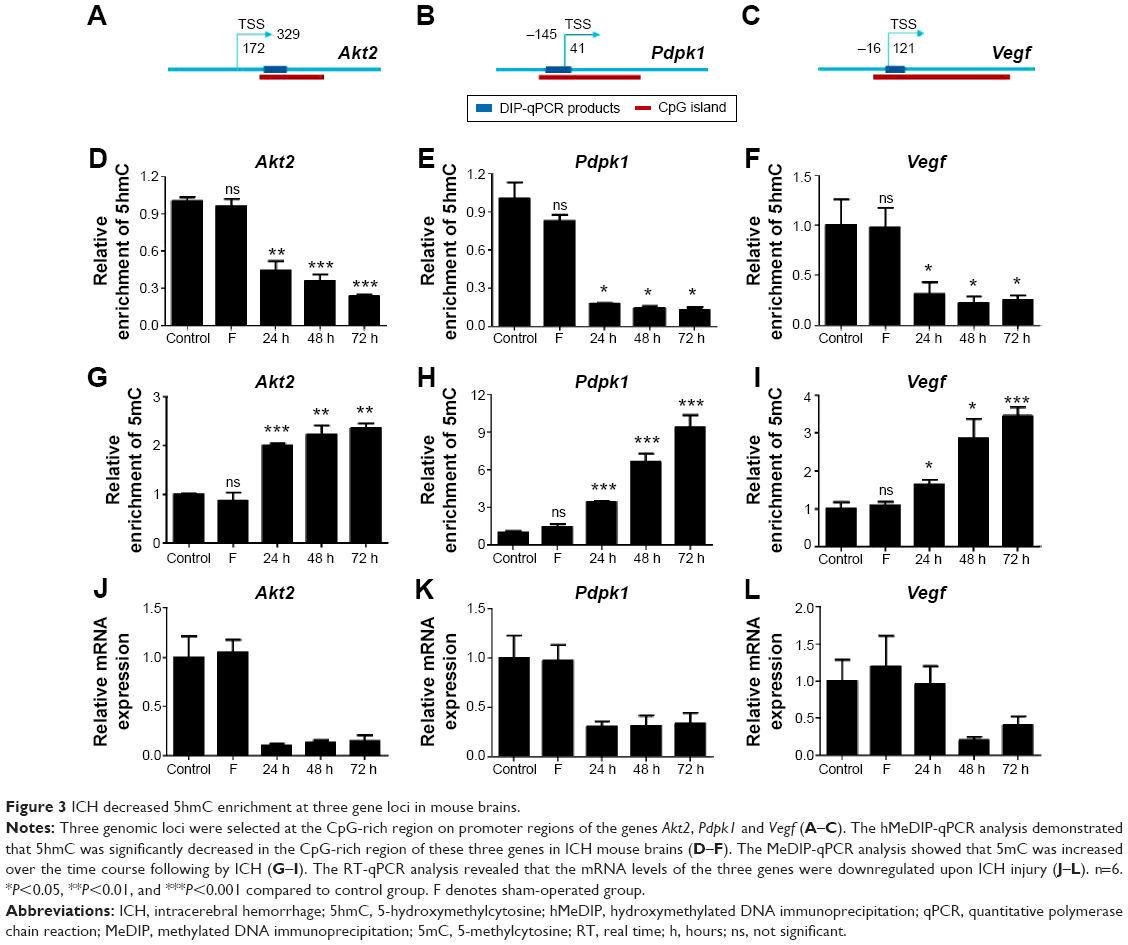 | Figure 3 ICH decreased 5hmC enrichment at three gene loci in mouse brains. |
TET expression is downregulated in ICH brain
As the generation of 5hmC is catalyzed by TET family proteins, we examined whether ICH has an impact on the expression of Tet family genes in the mouse brain by RT-qPCR. Our results showed that Tet1, Tet2, and Tet3 mRNA expression levels were significantly downregulated in the brain of the ICH mouse (Figure 4). These findings provide a potential molecular mechanism for the observed reduction of 5hmC in ICH.
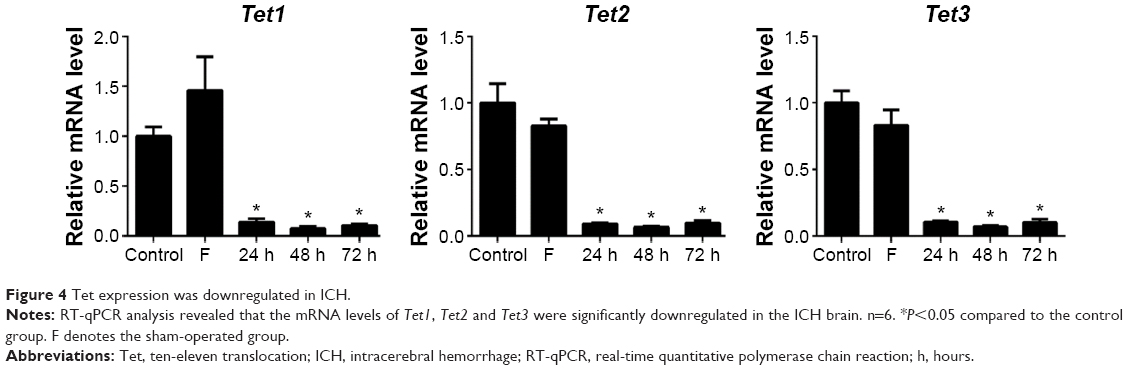 | Figure 4 Tet expression was downregulated in ICH. |
Discussion
This is the first report to implicate DNA hydroxymethylation in ICH. We observed the global 5hmC level reduction and 5hmC alteration on the three gene loci in ICH mouse brains. This reduction was in parallel with the increase of 5mC. These results indicate that the epigenetic regulatory system is involved in ICH injury.
Over the last decade, the mechanisms underlying ICH-induced brain injury have been demonstrated. The primary damage occurs within minutes to hours after the onset of bleeding and is mainly the result of mechanical damage associated with the mass effect.29 Secondary brain injury of ICH is mostly associated with hematoma toxicity,30 oxidative stress,31 and inflammation.32 Many genes and signaling pathways contribute to the unique pathophysiology of ICH.1 Growing knowledge in the field of epigenetics has dramatically changed our understanding of gene regulation. Multiple studies have shown that epigenetic modifications regulate a wide range of neuropathologic processes,33 and emerging evidence implicates a spectrum of epigenetic processes in the pathophysiology of stroke.34 Our results revealed the epigenetic mechanisms involved in ICH brain injury. The global level of 5hmC was significantly decreased 24 hours after ICH and persisted until 72 hours. Interestingly, an increasing trend was detected at 72 hours compared with the first 2 days, which could be partially explained by subsequent mechanisms after ICH-induced brain injury. Previous studies suggested that perihematomal edema progressed from ionic edema to vasogenic edema in the first 2 days, and it started to enter into the stage of resolution, which can stimulate angiogenesis at 3 days after ICH.35 Neutrophil infiltration was found in and around the hematoma, which peaks at 2–3 days and almost disappears at 3–7 days.32 The changes of the global 5hmC level in mouse brains might be partially consistent with the pathophysiology of ICH. However, in contrast to ICH brain injury, a recent study showed that overall 5hmC abundance was increased in the mouse brain after ischemia reperfusion.21 This discrepancy may be owing to the different pathophysiologies of hemorrhage and ischemic injury.
We selected the Akt2, Pdpk1 and Vegf genes to test whether ICH injury alters the 5hmC levels at specific gene loci in mouse brains. Previous reports showed that these genes are regulated after ICH, and their expression changes contribute to cell death after ICH.4 Our results showed that 5hmC was significantly decreased in the CpG-rich region of these three genes, and this was accompanied by an increase of 5mC. These data suggest that ICH injury alters the dynamics of cytosine methylation at specific gene loci. Future studies of the genome-wide changes of 5hmC and 5mC in the mouse brain upon ICH injury, including genes known to be upregulated and unchanged, may help further our understanding of the mechanisms underlying ICH injury response.
Methylation of DNA at the gene promoter region blocks the binding of transcriptional factors and thus represses gene expression. In contrast to 5mC, 5hmC increases the hydrophilicity and destabilizes the double helix and enhances binding of DNA-binding proteins.36,37 Whereas methylation has been linked to gene silencing, hydroxymethylation has been associated with actively transcribed genes.13,38,39 As expected, all three genes examined in this study were downregulated upon ICH injury accompanied by decreased 5hmC levels. This may be explained in part by the removal of the positive effect of 5hmC, but it is also possible that 5mC has a repressive effect on transcription.
The TET family contains three members, TET1, TET2, and TET3, which mediate the conversion of 5mC to 5hmC and its ultimate DNA demethylation.13 Our results showed that Tet expression was downregulated in the ICH brain and was associated with a decrease in 5hmC modification, which is consistent with previous research showing that a decrease in Tet resulted in reduced 5mC conversion to 5hmC and caused hypermethylation and transcription repression.40,41 However, several studies demonstrated that Tet1 downregulation leads to increased 5hmC and promotes the transcriptionally active state of genes.42 Because Tet1 is also involved in the repression of Polycomb-targeted developmental regulators, a dual function of Tet1 in both transactivation and gene silencing was revealed.42 This study could not determine whether Tet expression was responsible for the regulation of 5hmC. Future research with Tet knockout mice is needed to investigate the exact function of Tet during ICH injury.
Recent reports have indicated promising results of epigenetic therapies in human disease. Increased levels of 5mC have been shown to enhance cell death,43 and suppression of DNA methylation conferred resistance to ischemia.11,44 Other studies showed that 5hmC might be important in reversing the abnormal epigenetic regulation in cancer.45 Decrease of 5hmC modifications by inhibiting Tet2 activity can reduce infarct volume after ischemic injury.21 However, whether changes in DNA demethylation could be a therapeutic strategy for the treatment of ICH injury is still unknown. Future studies should examine whether regulated levels of 5hmC are associated with differences in outcome after ICH by targeting the 5hmC pathway.
Conclusion
In conclusion, this is the first study to investigate DNA hydroxymethylation in ICH injury. The mechanisms of 5hmC regulation and the function of 5hmC in ICH remain to be fully elucidated. Although the study of 5hmC is in its infancy, it has stimulated a lot of interest, in that 5hmC might have a potential role in mechanisms of injury and repair in ICH injury, which needs to be investigated further.
Acknowledgments
The authors are deeply indebted to all coworkers for this work. The work was especially supported by Hui Yang of the Institutes of Biomedical Sciences, Fudan University.
Disclosure
The authors report no conflicts of interest in this work.
References
Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. | ||
Woo D, Broderick JP. Spontaneous intracerebral hemorrhage: epidemiology and clinical presentation. Neurosurg Clin N Am. 2002;13:265–279. | ||
Carmichael ST, Vespa PM, Saver JL, et al. Genomic profiles of damage and protection in human intracerebral hemorrhage. J Cereb Blood Flow Metab. 2008;28:1860–1875. | ||
Lu A, Tang Y, Ran R, Ardizzone TL, Wagner KR, Sharp FR. Brain genomics of intracerebral hemorrhage. J Cereb Blood Flow Metab. 2005;26:230–252. | ||
Rosell A, Vilalta A, García-Berrocoso T, et al. Brain perihematoma genomic profile following spontaneous human intracerebral hemorrhage. PLoS One. 2011;6:e16750. | ||
Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. | ||
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. | ||
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. | ||
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. | ||
Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. | ||
Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport. 2001;12:3763–3766. | ||
Lee J, Park JH, Yan BC, et al. Effects of transient cerebral ischemia on the expression of DNA methyltransferase 1 in the gerbil hippocampal CA1 region. Neurochem Res. 2013;38:74–81. | ||
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. | ||
Münzel M, Globisch D, Carell T. 5-Hydroxymethylcytosine, the sixth base of the genome. Angew Chem Int Ed. 2011;50:6460–6468. | ||
Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. | ||
Globisch D, Münzel M, Müller M, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. | ||
Song C, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2010;29:68–72. | ||
Wang F, Yang Y, Lin X, et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum Mol Genet. 2013;22:3641–3653. | ||
Chouliaras L, Mastroeni D, Delvaux E, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging. 2013;34:2091–2099. | ||
Yao B, Lin L, Street RC, et al. Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2014;23:1095–1107. | ||
Miao Z, He Y, Xin N, et al. Altering 5-hydroxymethylcytosine modification impacts ischemic brain injury. Hum Mol Genet. 2015;24(20):5855–5866. | ||
Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. | ||
Rynkowski MA, Kim GH, Komotar RJ, et al. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–128. | ||
Belayev L, Saul I, Curbelo K, et al. Experimental intracerebral hemorrhage in the mouse: histological, behavioral, and hemodynamic characterization of a double-injection model. Stroke. 2003;34:2221–2227. | ||
Xu H, Ying Y, Wang ZT, Cheng KT. Identification of Dendrobium species by dot blot hybridization assay. Biol Pharm Bull. 2010;33:665–668. | ||
Mohn F, Weber M, Schubeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP). Methods Mol Biol. 2009;507:55–64. | ||
Yang H, Lin H, Xu H, et al. TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res. 2014;24:1017–1020. | ||
Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57:662–673. | ||
Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. | ||
Qureshi AI, Ali Z, Suri MFK, et al. Extracellular glutamate and other amino acids in experimental intracerebral hemorrhage: an in vivo microdialysis study. Crit Care Med. 2003;31:1482–1489. | ||
Tang J, Liu J, Zhou C, et al. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–1350. | ||
Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2006;27(5):894–908. | ||
Hwang J, Aromolaran KA, Zukin RS. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology. 2012;38:167–182. | ||
Qureshi IA, Mehler MF. Emerging role of epigenetics in stroke. Arch Neurol. 2010;67:1435–1441. | ||
Urday S, Kimberly WT, Beslow LA, et al. Targeting secondary injury in intracerebral haemorrhage. Nat Rev Neurol. 2015;11:111–122. | ||
Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(Hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. | ||
Wanunu M, Cohen-Karni D, Johnson RR, et al. Discrimination of methylcytosine from hydroxymethylcytosine in DNA molecules. J Am Chem Soc. 2011;133:486–492. | ||
Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. | ||
Thalhammer A, Hansen AS, El-Sagheer AH, Brown T, Schofield CJ. Hydroxylation of methylated CpG dinucleotides reverses stabilisation of DNA duplexes by cytosine 5-methylation. Chem Commun (Camb). 2011;47:5325–5327. | ||
Rudenko A, Dawlaty MM, Seo J, et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–1122. | ||
Zhang R, Cui Q, Murai K, et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13:237–245. | ||
Wu H, D’Alessio AC, Ito S, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. | ||
Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31:16619–16636. | ||
Endres M, Meisel A, Biniszkiewicz D, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. | ||
Rodger EJ, Chatterjee A, Morison IM. 5-hydroxymethylcytosine: a potential therapeutic target in cancer. Epigenomics. 2014;6:503–514. |
Supplementary materials
 | Table S1 Primer sequences for real-time qPCR analysis |
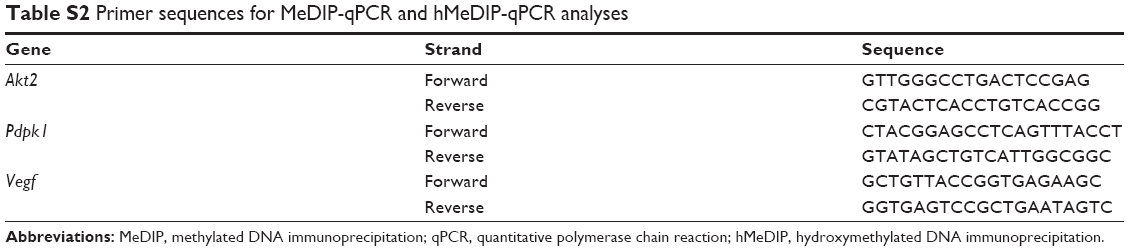 | Table S2 Primer sequences for MeDIP-qPCR and hMeDIP-qPCR analyses |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.