")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 7
Effects of food and alcohol on the pharmacokinetics of an oral, extended-release formulation of hydrocodone in healthy volunteers
Authors Farr S, Robinson C, Rubino CM
Received 9 July 2014
Accepted for publication 26 August 2014
Published 19 January 2015 Volume 2015:7 Pages 1—9
DOI https://doi.org/10.2147/CPAA.S70831
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Stephen J Farr,1 Cynthia Y Robinson,1 Christopher M Rubino2,3
1Zogenix, Inc., Emeryville, CA, 2Institute for Clinical Pharmacodynamics, Latham, NY, 3School of Pharmacy and Pharmaceutical Sciences, The State University of New York, University at Buffalo, Buffalo, NY, USA
Background: The purpose of this study was to evaluate the pharmacokinetics, bioavailability, and safety of oral extended-release hydrocodone (HC-ER) when administered with food or alcohol.
Methods: Two single-center, open-label, randomized, crossover studies were conducted in healthy volunteers. In a two-period food-interaction study, 12 subjects received HC-ER 20 mg after an overnight fast and a high-fat meal. In a three-period alcohol-interaction study, 30 naltrexone-blocked subjects received HC-ER 50 mg with a 0%, 20%, or 40% alcohol/orange juice solution after an overnight fast. Pharmacokinetic parameters were derived from plasma concentrations of hydrocodone and its metabolites.
Results: Exposure to hydrocodone after HC-ER 20 mg was similar in the fed and fasted states, as assessed by area under the plasma concentration versus time curve from time of dosing to time of last detectable concentration (AUC0–t; 316.14 versus 311.94 ng · h/mL); relative bioavailability (Frel) was 101.74%. Differences (fed versus fasted) in hydrocodone mean maximum plasma concentration (Cmax; 28.86 versus 22.74 ng/mL) and median time to Cmax (tmax; 6 versus 8 hours) were not clinically significant. Administration of 20% alcohol with HC-ER 50 mg did not increase systemic exposure relative to 0% alcohol (AUC0–t 878 versus 832 ng · h/mL; Frel 105%) or result in clinically meaningful changes in Cmax (51.8 versus 46.3 ng/mL) or tmax (5.44 versus 6.16 hours). Administration with 40% alcohol increased AUC0–t (1,008 ng · h/mL versus 832 ng · h/mL; Frel 120%) and Cmax (109 versus 46.3 ng/mL), and shortened tmax (2.43 versus 6.16 hours). Adverse events occurred in 10.0%, 24.1%, and 66.7% of subjects after 0%, 20%, and 40% alcohol, respectively.
Conclusion: HC-ER can be administered without regard to meals. While there was no evidence of "dose-dumping" (an unintended, rapid release in a short time period of all or most of the hydrocodone from HC-ER), even with 40% alcohol, as with all opioids, alcohol should not be ingested while using HC-ER.
Keywords: opioid, food interaction, alcohol interaction, bioavailability, norhydrocodone, hydromorphone
Introduction
Chronic pain is a public health problem in the USA, where it affects approximately 100 million adults.1 In terms of medical treatment and lost productivity, chronic pain is conservatively estimated to cost the nation between $560 and $635 billion annually (more than that for cardiovascular disease [$309 billion] and cancer [$243 billion] combined), a total representing approximately $2,000 per year for every living person in the USA.1 Despite shortcomings in data contributing to estimates of its prevalence, chronic pain1 appears to be rising as the USA population ages, grows more obese, incurs and endures injuries and diseases, and re-enters the health care system through health care reform or because of new advances in pain management.1
Hydrocodone (HC) has been used for decades in combination products, most commonly with acetaminophen (APAP). However, APAP-induced liver toxicity has become a growing concern, resulting in the US Food and Drug Administration recently mandating changes to both prescription and over-the-counter (OTC) medication labeling as well as reductions in the amount of APAP included in combination products.2 As recently reviewed by Blieden et al,3 6% of adults in the USA are prescribed APAP in doses that exceed the recommended 4 g/day limit and 30,000 patients are hospitalized for APAP toxicity each year. The majority of these cases involve unintentional overdose, largely related to opioid-APAP combinations in attempts at better pain relief.3 Furthermore, 69% of acute liver failure cases involving opioid-APAP combinations were in patients who used an immediate-release formulation of HC/APAP.3,4
Liver injury associated with APAP can limit dosing in combination formulations,5 but single-entity HC was not marketed in the USA until recently, when an extended-release (ER) formulation became available.6 The current commercial ER formulation of hydrocodone bitartrate (HC-ER; Zohydro ER®; Zogenix, Inc., Emeryville, CA, USA) is indicated for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate.6 The safety and effectiveness of HC-ER administered every 12 hours was demonstrated in patients with moderate-to-severe chronic low back pain.7
The pharmacokinetics of HC-ER were extensively studied in its clinical development program, and population pharmacokinetics indicate that irrespective of variability across and within subjects, the formulation provides consistent overall exposure and reliable sustained HC concentrations.8
Here we describe the effects of food on the pharmacokinetics of HC-ER. In addition, since some ER formulations can be subject to alcohol-induced “dose-dumping”, which has been defined as the “unintended, rapid drug release in a short period of time of the entire amount or a significant fraction of the drug contained in a modified-release dosage form”,9,10 we also evaluated the effects of alcohol on the pharmacokinetics of HC-ER.
Materials and methods
Two open-label, randomized studies were conducted in healthy volunteers after protocols were reviewed and approved by an independent ethics committee (research ethics committee, Queens University, Belfast, UK; food-interaction study) or an institutional review board (independent investigational review board, Plantation, FL, USA; alcohol-interaction study). Both studies were conducted at clinical research facilities in full accordance with the Good Clinical Practice: Consolidated Guideline approved by the International Conference on Harmonisation and applicable national and local laws and regulations.11 All subjects in both studies provided informed consent.
Study designs and treatments
Food-interaction study
The food-interaction study was a two-treatment, two-period crossover study to evaluate the rate and extent of HC absorption from HC-ER in both the fed and fasted states. During the first period, subjects received a single oral dose of HC-ER (20 mg capsule; supplied by Elan Corporation, plc, Athlone, Republic of Ireland) either after a 10-hour overnight fast (fasted state) or 30 minutes after receiving a high-fat breakfast (approximately 900–1,000 calories with 500–600 calories from fat; fed state). Subjects were treated in numerical order according to a randomization schedule provided before the start of the study. After a 7-day washout, subjects received a second 20 mg dose of HC-ER in the alternative (fed or fasted) state. Subjects were confined to the clinical research facility starting on the day before dosing and ending after the 36-hour pharmacokinetic sampling period following HC-ER administration.
Alcohol-interaction study
The alcohol-interaction study was a three-treatment, three-period crossover study to evaluate the effects of coingestion of alcohol on the safety, pharmacokinetics, and relative bioavailability of HC-ER. During the first period, subjects received a single 50 mg dose of HC-ER (supplied by Zogenix, Inc.) given with 240 mL of either a 40% alcohol/orange juice solution, a 20% alcohol/orange juice solution, or orange juice alone. Subjects received the alternative treatments during the second and third treatment periods, with subjects assigned to treatment sequence according to a randomization code. Treatment periods were separated by 4–5 days; all treatments were given after an overnight fast. To protect these opioid-naïve subjects from potential opioid-related adverse events, a 50 mg dose of naltrexone (purchased commercially) was administered approximately 12 hours (with a light snack) and 2 hours (fasted) prior to HC-ER administration and 10 hours (with a light snack) after HC-ER administration in each treatment period. Subjects were confined to the clinical research facility starting on the day before dosing and during the 48-hour pharmacokinetic sampling period after dosing in each period. Subjects were contacted by phone on day 18 of the final study period for follow-up and adverse event recording.
Subjects
Men and women in good health were eligible for both studies. Women had to be not pregnant and use contraception if not surgically sterile or postmenopausal.
Food-interaction study
The study planned for and enrolled 12 subjects. Subjects were eligible if they were 18–45 years of age and within 10% of ideal body weight (according to the Metropolitan Life Insurance Tables, 1983 edition). Subjects had to be free of significant disease as determined by medical history, physical examination, electrocardiogram (ECG), clinical chemistry, hematology, urinalysis, virology, and drug screen, and have no history of alcohol or drug abuse (at any time) or of smoking (within 6 months). They also had to have no history or signs of chronic obstructive pulmonary disease, no previous use of tricyclic antidepressants or monoamine oxidase inhibitors, no therapeutic use of narcotics within the previous year, no use of prescription medications with 2 weeks of study entry, no use of nonprescription or OTC medications within one week, and no hypersensitivity to APAP.
Alcohol-interaction study
Enrollment of 30 healthy subjects was planned in order to have 24 evaluable subjects. Subjects were eligible if they were 21–45 years of age, weighing at least 65 kg with body mass index ≥19 and ≤35 kg/m2. Subjects had to be nonsmokers for at least 3 months or light smokers (<10 pack-years) and have a history of moderate consumption of alcohol (admitted consumption of 7–21 drinks per week); subjects who were alcohol-naïve or had less than moderate alcohol intake were excluded, as were those who had excessive alcohol intake. Subjects with a corrected QT interval >450 msec or sitting blood pressure <110/45 mmHg at screening were also excluded. Subjects had to be in good health, as determined by laboratory profile, ECGs, and the investigator. They had to have no history or presence of significant disease, including diabetes, alcoholism, or drug abuse; no asthma or other chronic respiratory illness; no gastrointestinal dysmotility, irritable bowel syndrome, chronic constipation, or recent enteritis or surgery of the gastrointestinal tract; and no history of seizures or convulsions, head injury or other intracranial lesions, or a pre-existing increase in intracranial pressure. Subjects were not eligible if they had a history of hypersensitivity or idiosyncratic reaction to morphine, HC, or other opioids; hypersensitivity or idiosyncratic reaction to naltrexone, naloxone, or other opioid antagonists; or used hepatic enzyme-inducing drugs within the previous 3 months, prescription medications within the previous 14 days, or OTC medications within the previous 7 days.
Pharmacokinetic evaluation
Sampling and analytical methods
In the food-interaction study, blood samples were taken prior to administration of HC-ER and over 36 hours (0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 30, and 36 hours) following administration in each period. The venous blood samples were collected into 6 mL lithium heparin vacutainers from antecubital veins either through an indwelling catheter or by direct venipuncture. Plasma was separated by centrifugation at 2°C within one hour of collection, transferred into polypropylene tubes, and stored at −20°C to −80°C until analysis was performed. Concentrations in plasma of HC and its minor, though active O-demethylated metabolite, hydromorphone,12,13 were determined by a validated assay using liquid chromatography with tandem mass spectrometric detection (LC-MS/MS) (Bioanalytical Services, Athlone, Republic of Ireland). Following solid-phase extraction of HC, hydromorphone, and internal deuterated standards, samples were reconstituted in trifluoroacetic acid and acetonitrile, and loaded onto a liquid chromatography system equipped with a Sciex mass spectrometer. HC and hydromorphone were determined by multiple reaction monitoring transitions m/z 300→199 and m/z 286→185, respectively. Deuterated HC and hydromorphone were determined by multiple reaction monitoring transitions m/z 303→199 and m/z 289→185, respectively. The LC-MS/MS was performed using a PE Sciex API 3000 triple quadrupole mass spectrometer. The quantifiable range was 2.0–160 ng/mL for HC (precision 2.15%–4.45%) and 0.25–50 ng/mL for hydromorphone (precision 2.84%–13.11%).
In the alcohol-interaction study, blood samples were taken prior to administration of HC-ER and over 48 hours (0.25, 0.5, 0.75, 1.0, 1.25, 1.50, 2.0, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, 36, and 48 hours) following administration in each period. Blood sampling, processing, and storage was similar to that described above except that K2-EDTA was the anticoagulant. Concentrations in plasma of HC (quantifiable range 0.100–100.0 ng/mL; precision 3.4%–6.7%), its major N-demethylated metabolite, norhydrocodone12,13 (0.100–100 ng/mL; precision 2.3%–7.3%), as well as hydromorphone (0.100–100 ng/mL; precision 2.0%–8.0%) were determined by LC-MS/MS (Covance Global Clinical Pharmacology Inc., Madison, WI, USA). The method was similar to that described above. Norhydrocodone and deuterated norhydrocodone were determined by multiple reaction monitoring transitions m/z 286→199 and m/z 289→202, respectively.
In both studies, linear regression from concentration standards with a weighting factor of 1/concentration2 was used to construct the standard curves for determining sample concentrations of each analyte of interest.
Pharmacokinetic variables
Pharmacokinetic variables determined for HC and its metabolites included maximum observed concentration (Cmax), area under the concentration-time curve (AUC) from time 0 to infinity (AUC0–∞), AUC from time 0 to time of last measureable plasma concentration (AUC0–t), observed time of Cmax (tmax), terminal clearance half-life (t½), relative bioavailability (Frel), and terminal elimination rate constant (kel). Pharmacokinetic parameters were calculated, if appropriate, using commercial software (eg, WinNonlin, Pharsight Corporation, version 5.2, Mountain View, CA, USA).
Safety evaluation
Safety was assessed by adverse events, clinical laboratory test results (including serum alcohol in the alcohol-interaction study), vital signs measurements, ECG findings, and physical examination findings.
Statistical analyses
No formal sample size analysis was done; based on previous pharmacokinetic studies, a sample size of 12 subjects was determined to be sufficient to accurately characterize the pharmacokinetics for a treatment group. Because of the possibility of ethanol-associated emesis reducing the number of evaluable subjects, 30 subjects were enrolled in the ethanol interaction study. All enrolled subjects in the food-interaction study who completed the two treatment periods were included in the pharmacokinetic analyses, and all dosed subjects were included in the safety analysis. In the alcohol-interaction study, subjects who completed the 12-hour period immediately following HC-ER dosing, had no emesis within 4 hours of dosing, and had blood samples collected according to the protocol were included in pharmacokinetic analyses, and all subjects who met the inclusion criteria and received at least one dose of HC-ER were included in the safety analysis. Data were summarized using descriptive statistics (mean, standard deviation, median, minimum and maximum values, and coefficient variation percent); values are expressed as the mean ± standard deviation unless otherwise noted. Relative bioavailability was assessed by change in systemic exposure (AUC and Cmax) with and without food (food-interaction study) or with and without alcohol (alcohol-interaction study) for HC and its metabolites. No inferential statistical analyses were planned or conducted.
Results
Food-interaction study
A total of 12 healthy subjects (three male, nine female) were enrolled and completed the food-interaction study; all were included in the pharmacokinetics and safety analyses. All subjects were white, with a mean age of 22±4 (range 19–33) years and a mean weight of 64.8±7.2 (range 51.9–74.4) kg.
Plasma concentration-time profiles for HC in the fed and fasted state are shown in Figure 1 and pharmacokinetic parameters in Table 1. Overall, systemic absorption of single oral doses of HC-ER 20 mg was comparable in the fed and fasted states. AUC estimates for HC in the fed and fasted states were similar, and the Frel based on these estimates was close to 100%. The mean Cmax for HC was higher in the fed state compared with the fasted state (28.86 versus 22.74 ng/mL). The median tmax in the fed and fasted states was approximately 6 and 8 hours, respectively, and mean t½ was 4.9 and 6.5 hours, respectively (Table 1). For hydromorphone, Cmax and AUC values were approximately 6% of those for HC and were similar in both the fed and fasted states; median tmax was approximately 9 hours in the fed state and 7 hours in the fasted state (Table 1).
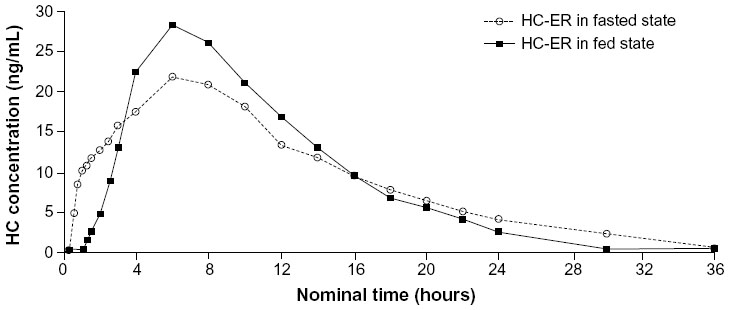 | Figure 1 Plasma concentration by time profiles for hydrocodone after oral administration of HC-ER 20 mg in the fasted (open symbols) and fed (closed symbols) states. |
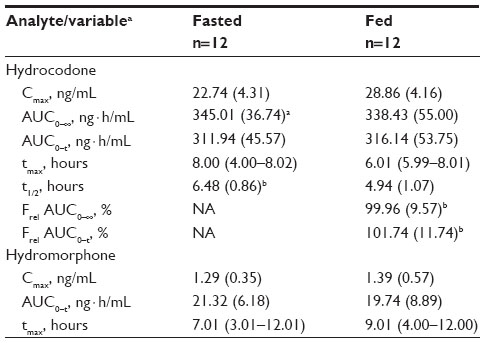 | Table 1 Pharmacokinetic parameters for hydrocodone and hydromorphone after administration of HC-ER 20 mg in fed and fasted states |
Alcohol-interaction study
A total of 30 healthy subjects were enrolled, and 28 completed all three treatment periods in the alcohol-interaction study (one subject withdrew consent and one was withdrawn due to noncompliance). The majority of subjects enrolled were male (28 of 30 [93.3%]) and white (22 of 30 [73.3%]). Their mean age was 32±7 (range 22–44) years, their mean weight was 87.2±9.9 (range 67.3–111.5) kg, and their mean body mass index was 28.5±3.1 (range 22.4–34.3) kg/m2. All 30 subjects were included in the safety and pharmacokinetic analyses. However, one subject had anomalously low HC and HC metabolite values after treatment with HC-ER and 40% alcohol. Therefore, pharmacokinetic analyses were conducted both including and excluding this subject, the latter to correct for any potential underestimation of the effect of 40% alcohol; data excluding this subject are presented here. Additionally, a total of ten subjects had emesis within 4 hours after dosing with 0% (one subject), 20% (one subject), and 40% alcohol (ten subjects) and were excluded from the respective analyses.
Plasma concentration-time profiles for HC after administration of HC-ER with and without alcohol are shown in Figure 2 and pharmacokinetic parameters for HC and its metabolites, norhydrocodone and hydromorphone, in Table 2. The mean Cmax for HC was similar after administration of HC-ER without alcohol and with 20% alcohol (46.3 and 51.8 ng/mL, respectively), and tmax was reached in a similar time frame (6.16 and 5.44 hours, respectively). Overall mean exposure (AUC0–∞) to HC was also similar after HC-ER without and with 20% alcohol (846 and 900 ng · h/mL; Frel 105%; Table 2). After administration of HC-ER with 40% alcohol, Cmax was approximately 2.3-fold higher than after HC-ER administered without alcohol (109 versus 46.3 ng/mL; Table 2). Further, tmax was reached in less than half the time (2.43 versus 6.16 hours; Table 2). Mean AUC0–∞ was approximately 20% higher after administration of HC-ER with 40% alcohol compared with HC-ER without alcohol (1,017 versus 846 ng · h/mL; Frel 119%; Table 2). Among individual subjects, the maximum increase in Cmax was 3.9-fold (from 43.6 ng/mL at 0% alcohol to 170 ng/mL at 40% alcohol).
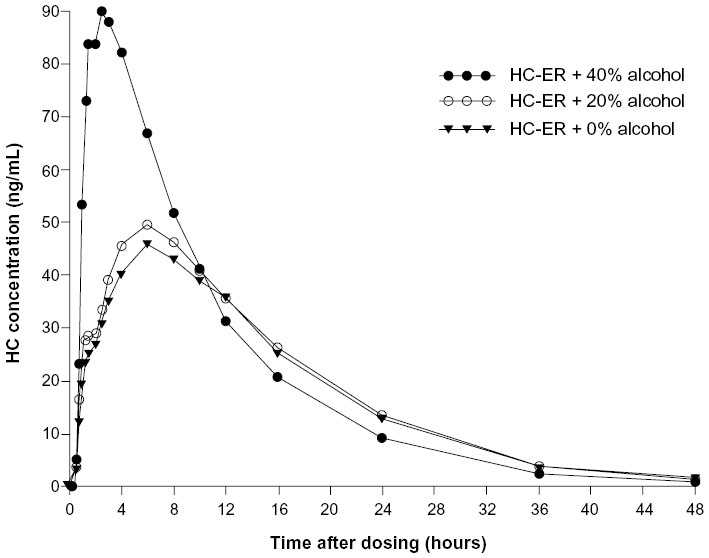 | Figure 2 Plasma concentration by time profiles for hydrocodone after oral administration of 50 mg HC-ER with or without alcohol. |
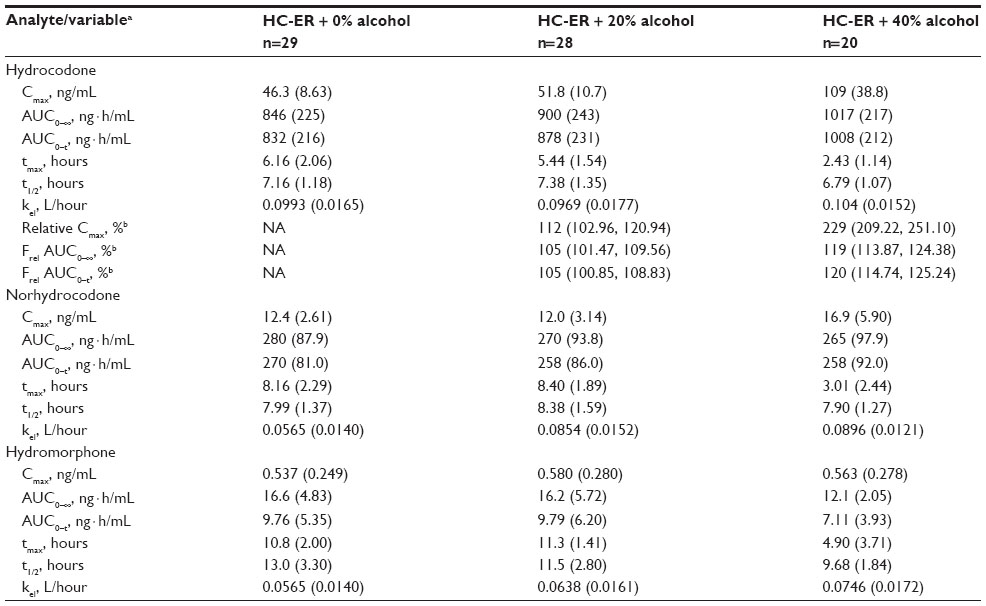 | Table 2 Pharmacokinetic parameters for hydrocodone, hydromorphone, and norhydrocodone after administration of HC-ER 50 mg with and without alcohol |
For the major metabolite norhydrocodone, Cmax values were approximately 16%–27% of those for HC, and AUC values were approximately 26%–33% of those for HC. For the minor metabolite hydromorphone, Cmax and AUC values were approximately 0.5%–1% and 1%–2% of those for HC, respectively (Table 2).
Safety
All 12 subjects (100%) in the food-interaction study experienced adverse events after HC-ER 20 mg given in the fasted or fed state. The most frequent adverse events were (fasted versus fed) nausea (50% versus 25%), pruritus (50% versus 67%), asthenia (42% versus 75%), dizziness (42% versus 33%), somnolence (17% versus 17%), headache (17% versus 0%), and pharyngitis (17% versus 0%). In the alcohol-interaction study, subjects had received naltrexone to block potential opioid-related adverse events after administration of HC-ER 50 mg with and without alcohol. Overall, three of 30 subjects (10.0%), seven of 29 (24.1%), and 20 of 30 (66.7%) experienced adverse events after receiving HC-ER with 0%, 20%, and 40% alcohol, respectively. The most frequent adverse events were headache (3.3% versus 13.8% versus 23.3%), vomiting (3.3% versus 3.4% versus 33.3%), dizziness (0% versus 3.4% versus 20.0%), nausea (0% versus 0% versus 16.7%), and abdominal pain (3.3% versus 3.4% versus 10.0%).
There were no clinically significant changes in clinical laboratory tests, vital signs, or ECGs in any subject in either study, and no deaths, serious adverse events, or other significant adverse events.
Discussion
HC-ER is the first single-entity HC product available and offers an alternative to chronic use of immediate-release HC/APAP products, thereby reducing the risks of APAP-induced liver injury. It is effective when administered twice daily in patients with moderate-to-severe chronic low back pain.7 Like other ER formulations of drugs, HC-ER is designed to deliver drug over longer periods of time compared with immediate-release formulations, and also like other ER drug formulations, the dose of HC contained in HC-ER (in the range of 10–50 mg per capsule) is higher than in corresponding immediate-release HC/APAP formulations (in the range of 5–10 mg).6,14 If the ER mechanism for any drug fails, releasing all or a substantial portion of the drug rapidly (ie, “dose-dumping”), serious or fatal adverse events could occur. The present studies indicated no evidence of dose-dumping of HC from the HC-ER formulation, even when the highest dose available (50 mg) was coingested with 40% alcohol. Nevertheless, plasma levels of HC were increased by coingestion of HC-ER with 40% alcohol, confirming that, as with all opioids, alcohol should not be ingested while using HC-ER due to the increased risk of central nervous system depression.
The present studies indicated that HC-ER can be administered without regard to meals. This can be of benefit to patients with chronic pain who, in order to maintain a consistent level of HC, need not take meals into consideration when taking HC-ER. In the food-interaction study, there was no increase in systemic exposure (as assessed by AUC) to HC after administration of 20 mg HC-ER in the presence or absence of food. Cmax was slightly higher under fed conditions. While the mechanism of this increase was not elucidated in the study, a high-fat meal can delay gastric emptying,15 and the higher Cmax value may be the result of increased absorption of the immediate-release component of ER formulations. Others have also reported that, for a different ER formulation of HC, AUC in the fed and fasted state met bioequivalence criteria but that Cmax was 40%–45% higher under fed conditions.16 Likewise, food has been shown to increase the Cmax of oxymorphone when administered in ER oxymorphone formulations,17,18 and it is recommended that oxymorphone ER be given either one hour before or 2 hours after eating.17
In our food-interaction study, the difference in HC Cmax in the fed state compared with the fasted state was modest, and there was no evidence of dose-dumping after administration of HC-ER in either condition. In addition, the type and frequency of adverse events were similar under both conditions. While the overall incidence of adverse events was high in the food-interaction study (100% under both conditions), the dose of HC-ER employed in the study (20 mg) was twice the recommended starting dose (10 mg) for HC-ER in opioid-naïve and opioid-nontolerant patients, in whom dose titration is recommended in order to minimize side effects.6 In addition, the subjects in the food-interaction study did not receive an opioid antagonist to block potential adverse events. Overall, given that individualized dosing is recommended for HC-ER,6 the slight increase in Cmax with food is not considered to be clinically important.
The dose of HC-ER used in the alcohol-interaction study was 50 mg, which was higher than that used in the food-interaction study (20 mg), and is the highest dose of HC-ER available.6 In the alcohol-interaction study, administration of 240 mL of 20% alcohol taken immediately with the HC-ER dose did not result in any increase in systemic exposure or evidence of dose-dumping relative to 0% alcohol. This amount of alcohol is approximately equivalent to three “shots” (ie, 1.5 fl oz or 44 mL per shot; 132 mL total) of 80-proof (ie, 40%) vodka, taken on an empty stomach. In contrast, ingestion of 240 mL of 40% alcohol (equivalent to 5–6 shots [220–264 mL] of 80-proof vodka on an empty stomach) increased the HC Cmax relative to 0% alcohol and to 20% alcohol. The rate of absorption of HC from HC-ER was also increased, as evidenced by the concomitant decrease in tmax. As with all opioids, alcohol should not be ingested while using HC-ER due to the potential for increased plasma levels of HC and the risk of central nervous system depression.
The incidence of adverse events was also highest in the alcohol-interaction study after subjects ingested HC-ER with 40% alcohol. Since subjects received naltrexone in the alcohol-interaction study, opioid-related side effects were not anticipated. Instead, vomiting was the most frequent adverse event observed with the combination of HC-ER and 40% alcohol. This may be due to the interaction of naltrexone with alcohol, and has been observed in other studies where the two have been coadministered.19
The mechanism behind the increased absorption of HC was not determined in this study but is likely to be due to partial early release of HC from the formulation’s sustained release microparticles in the stomach induced by the 40% alcohol. Increased absorption following coingestion with alcohol has been observed with other ER opioid formulations,17,19–24 and a comparison of alcohol interaction data for HC-ER and other marketed ER opioid products is shown in Table 3. For HC-ER, the overall mean Cmax ratio (2.3-fold increase) and maximum increase (3.9-fold increase) observed in any individual for 40% alcohol versus 0% alcohol treatments6 were within the range of increases observed for other currently marketed ER opioid products.17,21–24 While others have reported no substantial differences in mean Cmax with concomitant administration of 40% alcohol and a different ER formulation of HC,25 the data to date have been presented only in preliminary form.
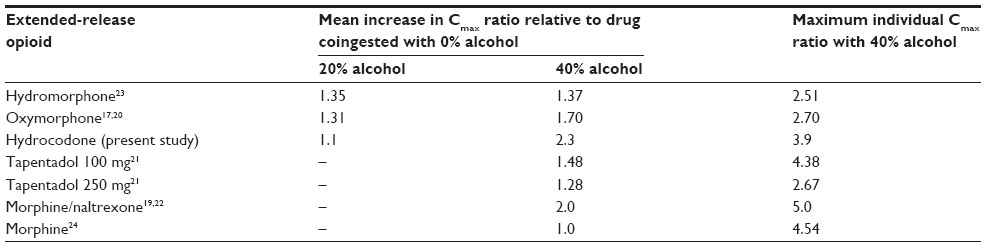 | Table 3 Cmax ratio of extended-release opioids administered with coingestion of alcohol |
Hydromorphone formed by the metabolism of hydrocodone via cytochrome P450 (CYP)2D6 has been implicated as a contributor to the analgesic effect of hydrocodone.26 This speculation is based in part on the 7-fold to 43-fold higher affinity for the opioid μ receptor exhibited by hydromorphone compared with the parent compound.13,27 However, Kaplan et al showed that inhibition of CYP2D6 had no effect on objective and subjective responses to hydrocodone in volunteers who were either poor or extensive CYP2D6 metabolizers, leading them to conclude that hepatic metabolism of hydrocodone had no influence on its effect.28 Based on the low relative exposure to hydromorphone compared with hydrocodone observed in the present studies, it appears unlikely that hydromorphone would contribute to the analgesic efficacy of HC-ER. Our studies were single-dose studies conducted in populations largely consisting of white subjects and thus extrapolation to other races may be limited.
Conclusion
Given the lack of differences in AUC0–t and the relatively small differences in Cmax, HC-ER can be administered without regard to meals. There was no evidence of “dose-dumping” (ie, an unintended, rapid release in a short period of time of the entire amount or a significant fraction of HC from HC-ER), even with 40% alcohol. Nevertheless, as with all opioids, alcohol should not be ingested while using HC-ER due to the potential for increased plasma levels of HC and risk of central nervous system depression.
Acknowledgments
The authors wish to acknowledge Elan Corporation for sponsoring the food-interaction study. The alcohol-interaction study was sponsored by Zogenix, Inc. The authors also wish to acknowledge that professional medical writing and editing was provided by Joyce Willetts of PharmaWrite, LLC (Princeton, NJ, USA), and was funded by Zogenix, Inc. This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP2 Guidelines”.
Disclosure
SJF and CYR are employees of and own stock in Zogenix, Inc. CMR is a consultant to Zogenix, Inc., and a Principal of the Institute for Clinical Pharmacodynamics, which has received grant funding from Zogenix for data analysis on several related studies (not including current study).
References
Institute of Medicine Committee on Advancing Pain Research. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC, USA: The National Academies Press; 2011. | |
US Food and Drug Administration. FDA recommends health care professionals discontinue prescribing and dispensing prescription combination drug products with more than 325 mg of acetaminophen to protect consumers. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm381644. Accessed May 9, 2014. | |
Blieden M, Paramore LC, Shah D, Ben-Joseph R. A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert Rev Clin Pharmacol. 2014;7(3):341–348. | |
Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364–1372. | |
Manchikanti L, Abdi S, Atluri S, et al. American Society of Interventional Pain Physicians (ASIPP) guidelines for responsible opioid prescribing in chronic non-cancer pain: part 2 – guidance. Pain Phys. 2012;15(Suppl 3):S67–S116. | |
Zohydro® ER (hydrocodone bitartrate) extended-release tablets [prescribing information]. San Diego, CA, USA: Zogenix, Inc.; 2013. | |
Rauck RL, Nalamachu S, Wild JE, et al. Single-entity hydrocodone extended-release capsules in opioid-tolerant subjects with moderate-to-severe chronic low back pain: a randomized double-blind, placebo-controlled study. Pain Med. 2014;15(6):975–985. | |
Melhem MR, Rubino CM, Farr SJ, Robinson CY. Population pharmacokinetic analysis for hydrocodone following the administration of hydrocodone bitartrate extended-release capsules. Clin Pharmacokinet. 2013;52(10):907–917. | |
Jedinger N, Khinast J, Roblegg E. The design of controlled-release formulations resistant to alcohol-induced dose dumping: a review. Eur J Pharm Biopharm. 2014;87(2):217–226. | |
Meyer RJ, Hussain AS. Awareness topic: mitigating the risks of ethanol induced dose dumping from oral sustained/controlled release dosage forms. Available from: http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4187B1_01_08-Alcohol-Induced.pdf. Accessed May 7, 2014. | |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6(R1). June 10, 1996. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed November 14, 2014. | |
Cone EJ, Heltsley R, Black DL, Mitchell JM, Lodico CP, Flegel RR. Prescription opioids. II. Metabolism and excretion patterns of hydrocodone in urine following controlled single-dose administration. J Anal Toxicol. 2013;37(8):486–494. | |
Navani DM, Yoburn BC. In vivo activity of norhydrocodone: an active metabolite of hydrocodone. J Pharmacol Exp Ther. 2013;347(2):497–505. | |
Vicodin® Vicodin ES® Vicodin HP® (hydrocodone bitartrate and acetaminophen) tablets, USP [prescribing information]. North Chicago, IL, USA: AbbVie Inc.; 2013. | |
Stacher G, Granser GV, Bergmann H, Kugi A, Stacher-Janotta G, Hobart J. Slow gastric emptying induced by high fat content of meal accelerated by cisapride administered rectally. Dig Dis Sci. 1991;36(9):1259–1265. | |
Darwish M, Bond M, Shu C, Robertson P, Tracewell W. Effect of food on the pharmacokinetics of the hydrocodone extended-release tablet in healthy volunteers. J Pain. 2012;13:S77. | |
Opana® ER (oxymorphone hydrochloride) extended-release tablets for oral use [prescribing information]. Malvern, PA, USA: Endo Pharmaceuticals, Inc.; 2014. | |
Benedek IH, Jobes J, Xiang Q, Fiske WD. Bioequivalence of oxymorphone extended release and crush-resistant oxymorphone extended release. Drug Des Devel Ther. 2011;5:455–463. | |
Johnson FK, Ciric S, Boudriau S, Kisicki J, Stauffer J. Effects of alcohol on the pharmacokinetics of morphine sulfate and naltrexone hydrochloride extended release capsules. J Clin Pharmacol. 2012;52(5):747–756. | |
Fiske WD, Jobes J, Xiang Q, Chang SC, Benedek IH. The effects of ethanol on the bioavailability of oxymorphone extended-release tablets and oxymorphone crush-resistant extended-release tablets. J Pain. 2012;13(1):90–99. | |
Nucynta® ER (tapentadol) extended-release oral tablets [prescribing information]. Titusville, NJ, USA: Janssen Pharmaceuticals, Inc.; 2013. | |
Embeda® (morphine sulfate and naltrexone hydrochloride) extended-release capsules for oral use [prescribing information]. New York, NY, USA: Pfizer Inc.; 2013. | |
Exalgo® (hydromorphone HCl) extended-release tablets for oral use [prescribing information]. Hazelwood, MO, USA: Mallinckrodt Brand Pharmaceuticals, Inc.; 2013. | |
Johnson F, Wagner G, Sun S, Stauffer J. Effect of concomitant ingestion of alcohol on the in vivo pharmacokinetics of Kadian (morphine sulfate extended-release) capsules. J Pain. 2008;9(4):330–336. | |
Darwish M, Bond M, Tracewell W, Robertson P. Assessing risk of alcohol-induced dose dumping with the use of a new extended-release hydrocodone formulation. J Pain. 2012;13:S76. | |
Otton SV, Schadel M, Cheung SW, Kaplan HL, Busto UE, Sellers EM. CYP2D6 phenotype determines the metabolic conversion of hydrocodone to hydromorphone. Clin Pharmacol Ther. 1993;54(5):463–472. | |
Hennies HH, Friderichs E, Schneider J. Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung. 1988;38(7):877–880. | |
Kaplan HL, Busto UE, Baylon GJ, et al. Inhibition of cytochrome P450 2D6 metabolism of hydrocodone to hydromorphone does not importantly affect abuse liability. J Pharmacol Exp Ther. 1997;281(1):103–108. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.