")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Effects of Avitinib on CYP450 Enzyme Activity in vitro and in vivo in Rats
Authors Shi Y, Meng D, Wang S , Geng P, Xu T, Zhou Q, Zhou Y, Li W, Chen X
Received 13 June 2021
Accepted for publication 5 August 2021
Published 21 August 2021 Volume 2021:15 Pages 3661—3673
DOI https://doi.org/10.2147/DDDT.S323186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Yong Shi,1,* Deru Meng,1,* Shuanghu Wang,1 Peiwu Geng,1 Tao Xu,2 Quan Zhou,1 Yunfang Zhou,1 Wanshu Li,3 Xugao Chen4
1Comprehensive Breast Health Center, Department of Thyroid and Breast Surgery, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, 323000, People’s Republic of China; 2Department of Pharmacy, Ningbo First Hospital, Ningbo, 315010, Zhejiang, People’s Republic of China; 3Department of Pharmacy, Ningbo Municipal Hospital of Traditional Chinese Medicine, Ningbo, 315010, Zhejiang, People’s Republic of China; 4Department of Radiology, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, 323000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xugao Chen
Department of Radiology, The Sixth Affiliated Hospital of Wenzhou Medical University, The People’s Hospital of Lishui, Lishui, 323000, People’s Republic of China
Tel/Fax +86578-2780081
Email [email protected]
Wanshu Li
Department of Pharmacy, Ningbo Municipal Hospital of Traditional Chinese Medicine, Ningbo, 315010, Zhejiang, People’s Republic of China
Tel/Fax +86574-87089100
Email [email protected]
Purpose: Avitinib is the first third-generation epithelial growth factor receptor (EGFR) inhibitor independently developed in China and is mainly used for treating non-small cell lung cancer. However, pharmacokinetic details are limited. This study explored the in vivo and in vitro effects of avitinib on cytochrome CYP450 enzymes metabolic activity.
Methods: A rapid and sensitive ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method was developed and validated for determining six probe substrates and their metabolites. Avitinib influence on activity levels of CYP isozymes was examined in vitro using human and rat liver microsomes (HLMs/RLMs). For in vivo studies, rats were pretreated with 30 mg/kg avitinib once daily for 7 days (avitinib multiple-doses group), 30 mg/kg avitinib on day 7 (avitinib single-dose group), or an equivalent amount of CMC-Na once daily for 7 days (control group), followed by intragastrical administration of the probe substrates (1 mg/kg tolbutamide and 10 mg/kg phenacetin, bupropion, chlorzoxazone, dextromethorphan, and midazolam). Plasma pharmacokinetics and IC50 values of the probe substrates were then compared. Pharmacokinetic parameters were determined using non-compartmental analysis implemented in a pharmacokinetic program.
Results: In vitro experiments revealed different inhibitory effects of avitinib on the six probe substrates with various IC50 values (bupropion, 6.39/22.64 μM; phenacetin, 15.79/48.36 μM; chlorzoxazone, 23.15/57.09 μM; midazolam, 27.64/59.6 μM; tolbutamide, 42.18/6.91 μM; dextromethorphan, 44.39/56.57 μM, in RLMs and HLMs respectively). In vivo analysis revealed significant differences (P < 0.05) in distinct pharmacokinetic parameters (AUC(0-t), AUC (0-∞), Cmax, MRT(0-t), MRT (0-∞), and CLz/F) for the six probe substrates after avitinib pretreatment.
Conclusion: A sensitive and reliable UPLC-MS/MS method was established to determine the concentration of six probe substrates in rat plasma. Avitinib had inhibitory effects on CYP450 enzymes, especially cyp2b1, cyp1a2 in RLMs, CYP2C9 in HLMs, and cyp1a2, cyp2b1, cyp2d1, and cyp2e1 in vivo. Our data recommend caution when avitinib was taken simultaneously with drugs metabolized by CYP450 enzymes.
Keywords: cytochrome, EGFR inhibitor, UPLC-MS/MS, rat liver microsome, drug-drug interaction
Introduction
Non-small cell lung cancer (NSCLC) is one of the most prevalent malignant tumors worldwide with mutations in epithelial growth factor receptor (EGFR) being the most common cause.1 NSCLC mortality rates are ranked first among malignant tumors, exhibiting severely low 5-year survival rates.2,3 Tyrosine kinase inhibitor (TKI) is one type of targeting drug for the treatment of NSCLCs carrying EGFR mutations. TKI treatment is effective for approximately 70% of NSCLC patients having EGFR mutations. However, acquired resistance is very common among patients taking first- or second-generation TKIs, especially for patients carrying the T790M mutation, which leads to EGFR-TKI drug failure. To overcome this problem third-generation EGFR-targeted drugs, such as avitinib (AC0010), have been recently developed.4–6
Avitinib is the first self-developed third-generation TKI in China and currently has entered the clinical trial stage. Clinical research has shown that patients taking avitinib for more than one month can significantly reduce their tumor volumes with high efficacy and safety.7–9 Avitinib is metabolized by the liver after oral administration through cytochrome (CYP) enzymes CYP2C19, CYP3A4, and CYP1A2, as well as other channels, and is mainly metabolized into seven metabolites, which are excreted in urine and feces.10–13 Unfortunately, there have been only a few reports published on the pharmacokinetic characteristics and drug-drug interactions of avitinib with other drugs since its entry into clinical trials.14
The CYP enzyme CYP450 is responsible for the metabolism of many drugs and plays an important role in drug metabolism. Accordingly, it is the subject of important preclinical drug metabolism research, including many of its subtypes, such as CYP1A2, CYP3A4, and CYP2C9, among others.15,16 Based on previously reports, many drugs can either inhibit or induce CYP450 activity, leading to changes in plasma drug concentrations. These drug-drug interactions can potentially produce undesirable interactions and affect treatment efficacy and safety.17 For instance, the combination of many drugs can induce CYP3A4 mRNA expression and increase drug metabolic toxicity in the body.18 Experiments using rat models have also provided evidence that the active ingredients of Andrographis paniculata can induce CYP1A1/2 and CYP2B activity and alter the metabolic activity of CYP1A1 and CYP1A2. Moreover, some drugs, such as tanshinone and glycyrrhetinic acid, can inhibit the metabolic activity of specific CYP450 enzymes and may increase drug exposure time.19,20 The use of cocktails is a well-known method for drug-drug interaction studies and it is widely employed in the evaluation of drug efficacy, both in vitro and in vivo.21 Cocktails allow for rapid and routine simultaneous measurement of enzyme activity of multiple isozymes, thereby significantly increasing sample throughput.22
In the current study, we established an ultra high performance liquid chromatography- tandem quadrupole mass spectrometry (UPLC-MS/MS) method for the detection of six probe substrates (phenacetin for cyp1a2 and CYP1A2, bupropion for cyp2b1 and CYP2B6, tolbutamide for cyp2c11 and CYP2C9, dextromethorphan for cyp2d1 and CYP2D6, chlorzoxazone for cyp2e1 and CYP2E1, and midazolam for cyp3as and CYP3As, Figure 1) in rat plasma and investigated the effect of avitinib on CYP450 enzyme activity in rats, RLMs and HLMs. Our findings should provide a theoretical basis for the rational clinical usage of avitinib, therapeutic drug monitoring, and clinical reduction of drug interactions.
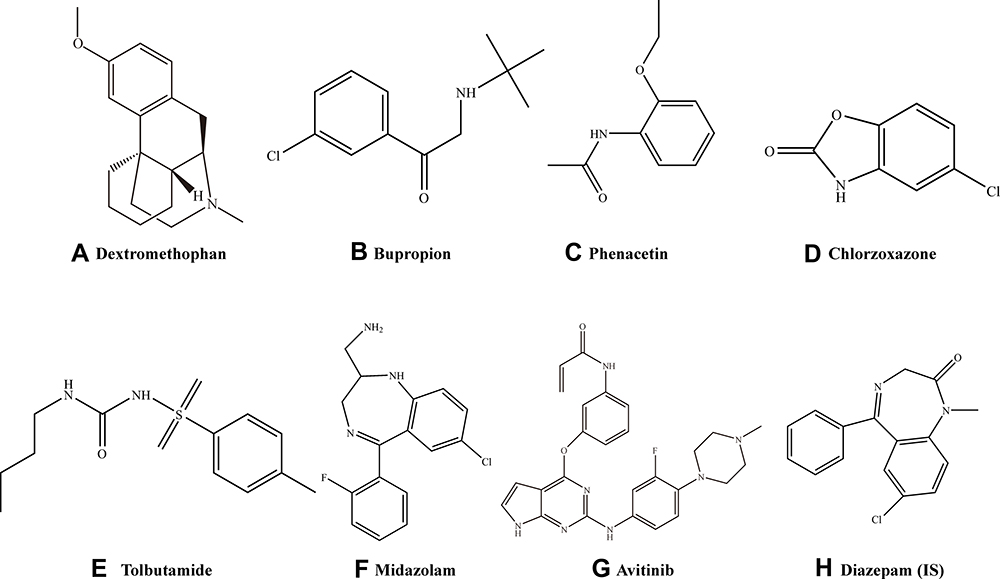 |
Figure 1 Probe substrate structures. (A) Dextromethorphan, (B) bupropion, (C) phenacetin (D) chlorzoxazone, (E) tolbutamide, (F) midazolam, (G) avitinib, and (H) diazepam, internal standard (IS). |
Methods
Chemicals and Reagents
Avitinib (purity >98%) was purchased from Send Pharmaceutical Technology Co., Ltd (Shanghai, China). Diazepam (purity >98%) was purchased from King York Pharmaceutical Co., Ltd. (Tianjin, China) and used as the internal standard. Hydroxybupropion, α-hydroxymidazolam, acetaminophen, and formic acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phenacetin, tolbutamide, dextromethorphan, and chlorzoxazone were purchased from Powered by J&K Chemical Ltd. (Beijing, China). Bupropion hydrochloride, and nordextromethorphan were purchased from Tokyo Chemical Industry (Tokyo, Japan). Midazolam was purchased from Nhwa Pharmaceutical Co., Ltd (Jiangsu, China). Hydroxy tolbutamide and 6-hydroxychlorzoxazone were purchased from Toronto Research Chemicals (Toronto, Canada). Nicotinamide adenine dinucleotide phosphate (NADPH) was purchased from Roche (Shanghai, China). BCA Protein Assay Kit was purchased from Thermo Scientific (USA). HLM was purchased from PrimeTox (Wuhan, China). Rat liver microsomes (RLMs) were prepared in our laboratory. Ultra-pure water was prepared using a Millipore Milli-Q purification system (Bedford, MA, USA). UPLC-grade acetonitrile and methanol were purchased from Fisher Scientific Co., (Waltham, MA, USA). Rat blank plasma samples from drug-free rats were supplied by the Laboratory Animal Center of Wenzhou Medical University. All other chemicals for this study were reagent grade and used without further purification.
Instrumentation and Analytical Conditions
A UPLC-MS/MS system consisting of an ACQUITY I-Class UPLC and XEVO TQD triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA) equipped with an electrospray ionization (ESI) interface was used to analyze the compounds. The ACQUITY UPLC system had a Binary Solvent Manager (BSM) and Sample Manager-Flow Through Needle (SM-FTN). Masslynx 4.1 software (Waters Corp.) was used for data acquisition and instrument control.
Probe substrates and their metabolites, as well as the internal standard diazepam, were separated using a UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm; Waters Corp.) and maintained at 40°C. The initial mobile phase consisted of acetonitrile and formic acid water (0.1% formic acid) with a linear gradient elution flow rate of 0.4 mL/min and an injection volume of 2 μL. The linear elution gradient consisted of acetonitrile content increasing from 10% to 50% within 0.6 min, rapidly increasing from 50% to 80% between 0.6 and 1.0 min, and increasing from 80% to 95% from 1.0 to 2.0 min. The acetonitrile content was then maintained at 95% for 0.5 min and finally dropped to 10% within 0.1 min. The total run time of the analytes was 3 min. After each injection, the sample manager underwent a needle wash process with methanol-water.
Mass spectrometric detection was performed on a triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) interface in positive mode. Nitrogen was used as the desolvation gas (1000 L/h) and cone gas (50 L/h). The ion monitoring voltage conditions were as follows: capillary voltage, 2.5 kV; source temperature, 150°C; and desolvation temperature, 500°C. Multiple reaction monitoring (MRM) methods were used for quantitative analysis.
Preparation of RLMs and Protein Concentration Quantification
Male Sprague-Dawley (SD) rats, body weight 220–250 g, were weighed and anesthetized with 10% chloral hydrate (0.35 mL/100 g) followed by hepatic perfusion with phosphate buffered saline (PBS). The liver was excised, rinsed with ice-cold saline (0.9% NaCl), weighed, and homogenized. The homogenized liquid was subsequently centrifuged at 9000 × g for 30 min at 4°C. The supernatant was collected and centrifuged at 105,000 × g for 60 min at 4°C. The precipitate was resuspended in a buffer solution of 0.15 M KCl-PBS at pH 7.4. After repeating the above procedure, the final sample was stored in KCl-PBS containing 0.25 mol/L sucrose at −80°C until use. The protein concentrations of the RLMs were determined using the BCA Protein Assay Kit (Pierce Thermo Scientific).
Effect of Avitinib on Probe Substrate Metabolism in vitro
To determine the half-maximal inhibitory concentration (IC50), based on previous studies,26–28 the incubation mixtures were prepared in a total volume of 200 μL as follows: 0.44 mg/mL RLMs or 0.5 mg/mL HLMs, 100 mM potassium phosphate buffer, cocktail probes (40μM phenacetin, 20 μM bupropion, 100 μM tolbutamide, 10 μM dextromethorphan, 40 μM chlorzoxazone, and 5 μM midazolam), a series concentration of avitinib (0, 1, 2.5, 5, 10, 25, 50 and 100 μM), after pre-incubation in a 37°C water bath for 5 min, 1 mM NADPH was added to the mixture to start the reaction. Thirty minutes later, the mixture was transferred to ice and 200 μL of acetonitrile added to stop the reaction and precipitate the protein. Then, 20 μL of diazepam (500 ng/mL) was added and the mixture then vortexed and centrifuged 13,800 × g for 5 min. Finally, 2 μL of the supernatant was injected into the ACQUITY I-Class UPLC and XEVO TQD triple quadrupole mass spectrometer for separation and analysis.
Animal Experiments
For the evaluation of drug interactions, male Sprague-Dawley (SD) rats, body weight 220–250 g, were obtained from the Wenzhou Medical University Experimental Animal Center. The animals were housed in a specific pathogen-free (SPF) facility under 12 h light-dark cycles with access to rodent cubes and sterile water. All experimental procedures and protocols were reviewed and approved by the Animal Ethics Committee of Wenzhou Medical University in accordance with the “Guidelines for the Care and Use of Laboratory Animals” (ID Number: wydw2019-650). Water was freely available but the diet was prohibited for 12 h prior to the experiment.
Effects of Avitinib on Probe Drugs Pharmacokinetics in vivo
Eighteen SPF-grade healthy male SD rats were randomly selected, divided into multiple-dose avitinib group (Group A), single-dose avitinib group (Group B), and control group (Group C), with six rats each. Group A and Group B rats were orally administered 30 mg/kg avitinib daily for seven days or on day seven only, respectively, while Group C rats were orally administered carboxymethyl cellulose sodium (CMC-Na) daily for seven days. In the seventh day, 30 min later after oral administration of avitinib, the rats were intragastrically administered the six probe drugs simultaneously, including 1 mg/kg tolbutamide and 10 mg/kg each of phenacetin, bupropion, chlorzoxazone, dextromethorphan and midazolam. All rats were fasted for 12 h before sampling. Blood was collected from the tail veins and collected directly into clean centrifuge tubes containing heparin. The samples were immediately centrifuged at 3000 × g for 10 min, the supernatant separated and transferred to another centrifuge tube, and the samples stored at −20°C until use.
Sample Preparation
Before analysis, the plasma samples were thawed to room temperature. A 20 μL aliquot of the diazepam internal standard working solution (0.5 μg/mL) was added to 50 μL of the collected plasma sample in a 1.5 mL centrifuge tube, followed by the addition of 200 μL of acetonitrile. The tubes were thoroughly mixed for 1.0 min and centrifuged at 13,000 × g for 10 min. Finally, 2 μL of the supernatant was injected into the UPLC-MS/MS system for separation and analysis.
Statistical Analysis
Non-compartmental analysis was performed using Drug and Statistics (DAS) version 3.2.8 software (The People’s Hospital of Lishui, China) to calculate the pharmacokinetic parameters.23–25 Average plasma concentration-time curves were drawn according to the mean drug concentrations at each time point. The IC50 value was calculated using GraphPad version 8 software (GraphPad Software Inc., San Diego, CA, USA). Statistical analyses were performed using one-way analysis of variance (SPSS 19.0, Chicago, IL, USA). A value of P <0.05 was regarded as statistically significant.
Results
Method Validation
After optimization of the incubation condition, UPLC-MS/MS analysis was performed to determine the metabolites present in one separation. This method was then validated by evaluating the linearity, accuracy, and inter-day and intra-day precision. As shown in Table 1, the correlation coefficients within the selected concentrations were all greater than 0.99. The accuracies were in the range of 85–115% with less than 15% inter-day or intra-day deviation. Typical UPLC-MS/MS chromatograms of blank matrix spiked with the probe substrates, their metabolites, and the diazepam internal standard are shown in Figures 2 and 3.
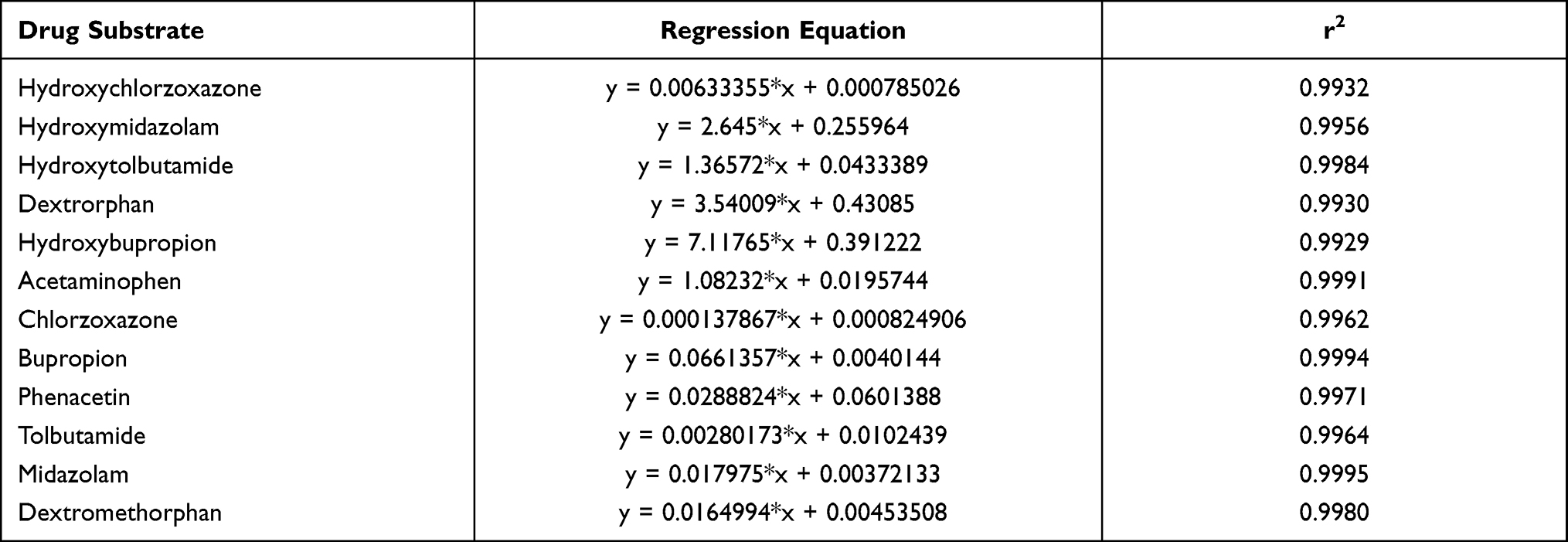 |
Table 1 Standard Curve Analysis of Six Probe Substrates and Their Metabolites |
 |
Figure 2 Typical UPLC-MS/MS chromatograms of blank matrix spiked with hydroxychlorzoxazone, hydroxymidazolam, hydroxytolbutamide, internal standard diazepam, dextrorphan, hydroxybupropion, and acetaminophen. |
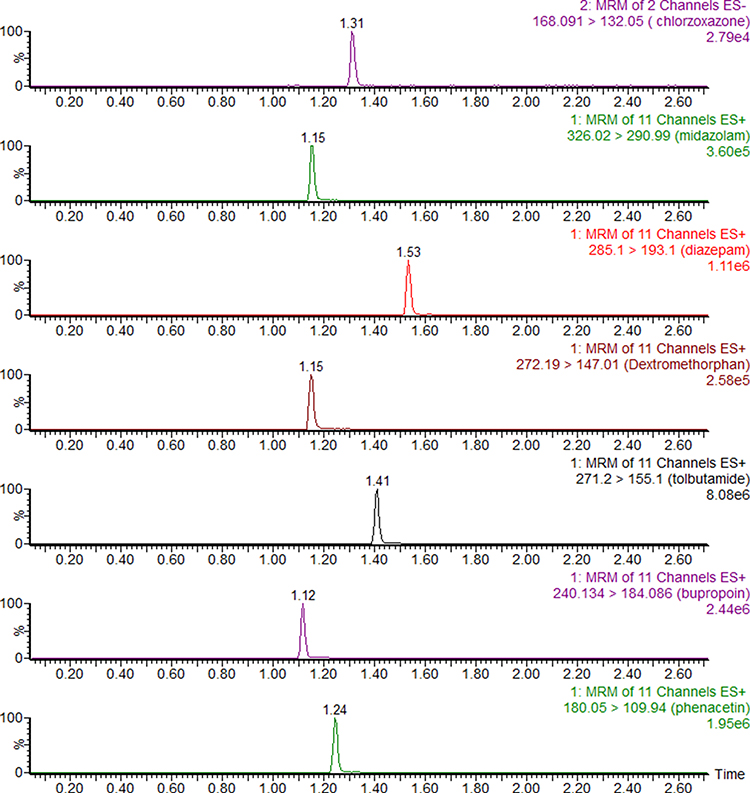 |
Figure 3 Typical UPLC-MS/MS chromatograms of blank plasma spiked with chlorzoxazone, midazolam, internal standard diazepam, dextromethorphan, tolbutamide, bupropion, and phenacetin. |
In vitro Inhibitory Effect of Avitinib on CYP450 in RLMs and HLMs
An in vitro incubation system was used to calculate the IC50 values of inhibitors, the equation is Y=100/(1+10^(X-LogIC50)). Serial concentrations of avitinib ranging from 1–100 μM were co-incubated with the probe substrates under the optimized incubation conditions to determine the IC50 values (Figures 4 and 5). The IC50 values of avitinib against the probe drugs were 6.39/22.64 μM for bupropion; 15.79/48.36 μM for phenacetin, 23.15/57.09 μM for chlorzoxazone; 27.64/ 59.6 μM for midazolam; 42.18/6.91 μM for tolbutamide; 44.39/56.57 μM for dextromethorphan, in RLMs and HLMs respectively. These findings indicated that avitinib exhibited a weak inhibitory effect on the majority of CYP450 isoforms, including cyp1a2, cyp2c11, cyp2d1, cyp2e1, and cyp3as in RLMs; CYP1A2, CYP2B6, CYP2D6, CYP2E1, and CYP3As in HLMs. For the cyp2b1 and CYP2C9, inhibition was moderate.
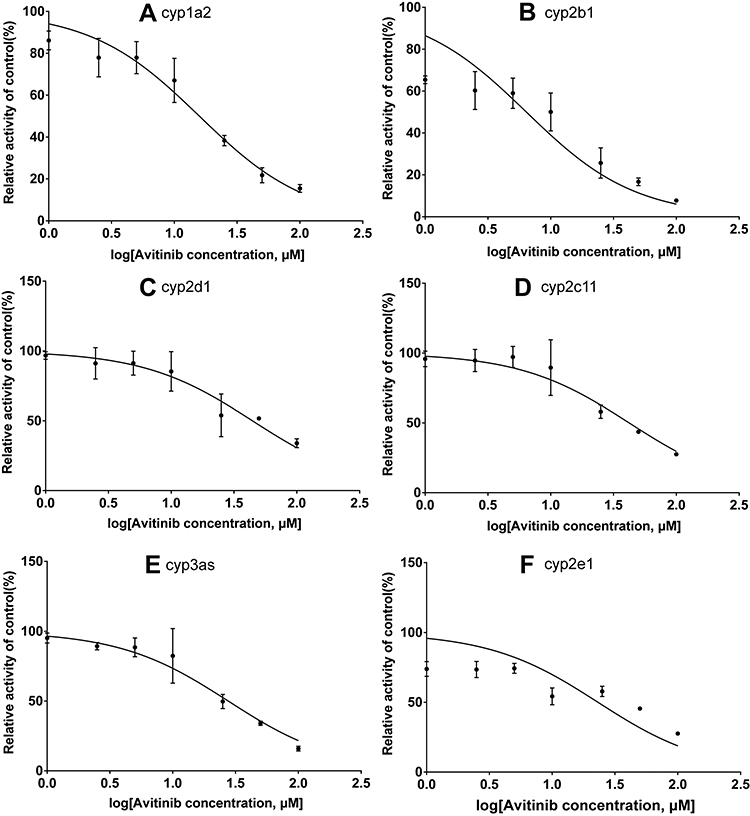 |
Figure 4 IC50 values for avitinib against cytochrome enzymes in rat liver microsomes. (A) cyp1a2, (B) cyp2b1, (C) cyp2d1, (D) cyp2c11, (E) cyp3as, and (F) cyp2e1. Mean ± SD, n = 3. |
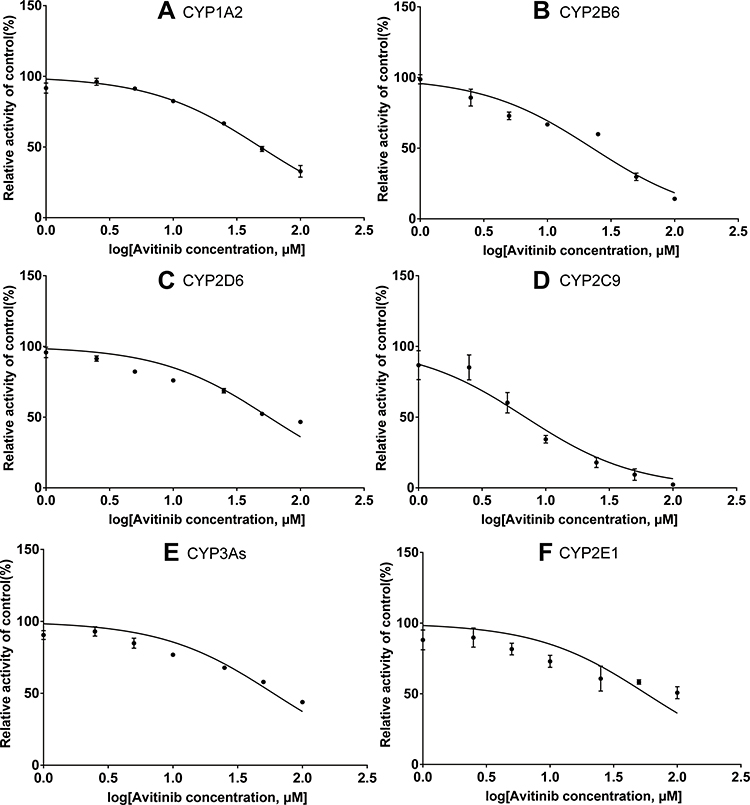 |
Figure 5 IC50 values for avitinib against cytochrome enzymes in human liver microsomes. (A) CYP1A2, (B) CYP2B6, (C) CYP2D6, (D) CYP2C9, (E) CYP3As, and (F) CYP2E1. Mean ± SD, n = 3. |
In vivo Inhibitory Effect of Avitinib on CYP450 in Rats
The metabolic activities of phenacetin, bupropion, tolbutamide, dextromethorphan, chlorzoxazone, and midazolam were evaluated in order to examine the inductive potency of avitinib on CYP450 enzymes in rats. Plasma concentration-time curves of the probe substrates after oral administration, both with and without avitinib pretreatment, are shown in Figure 6. Comparison of the main pharmacokinetic parameters of the three experimental groups of rats are shown in Tables 2Tables 3–4.
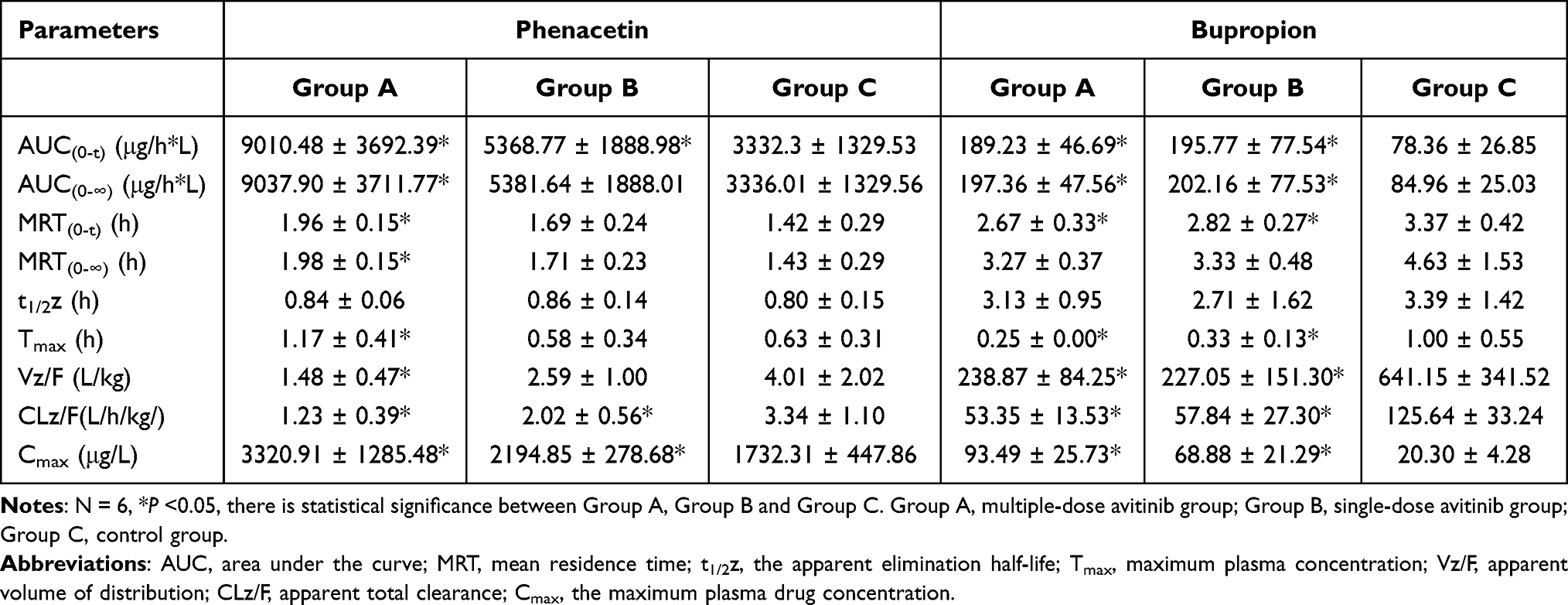 |
Table 2 Pharmacokinetic Parameters of Cyp1a2 and Cyp2b1 Enzymes on Probe Substrates Phenacetin and Bupropion in Group a, Group B and Group C Rats |
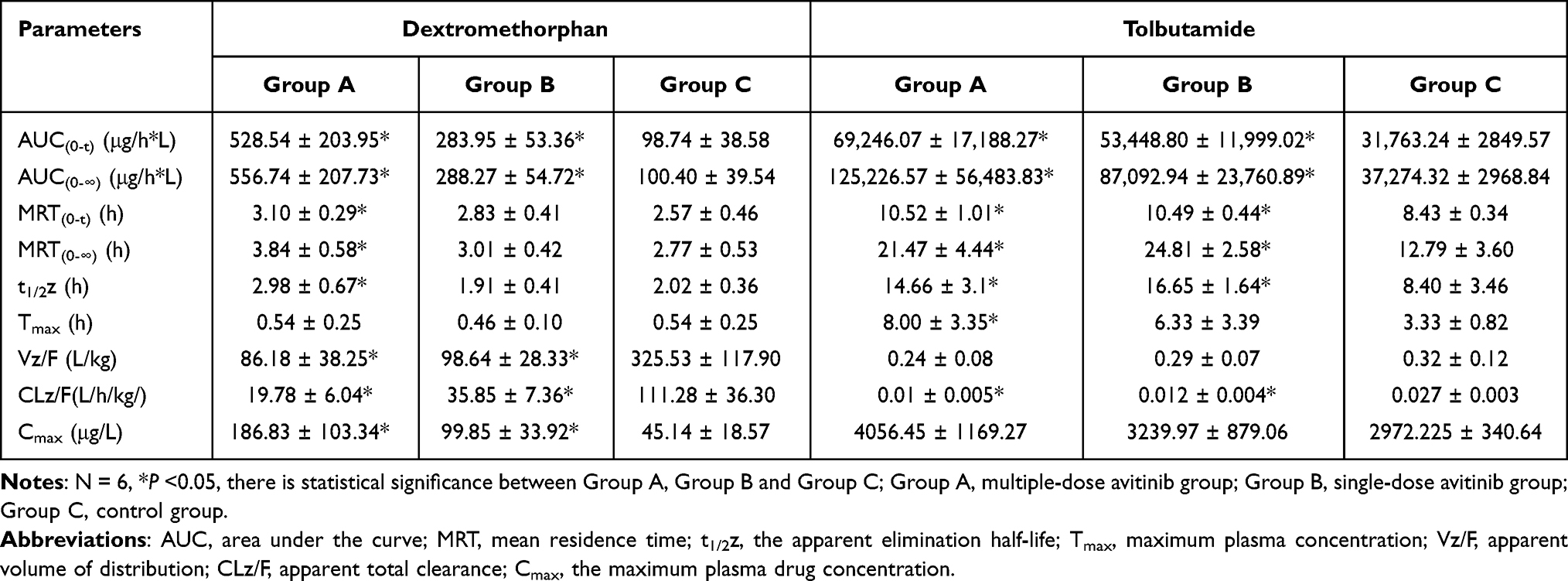 |
Table 3 Pharmacokinetic Parameters of Cyp2d1 and Cyp2c11 Enzymes on Probe Substrates Dextromethorphan and Tolbutamide in Group A, Group B and Group C Rats |
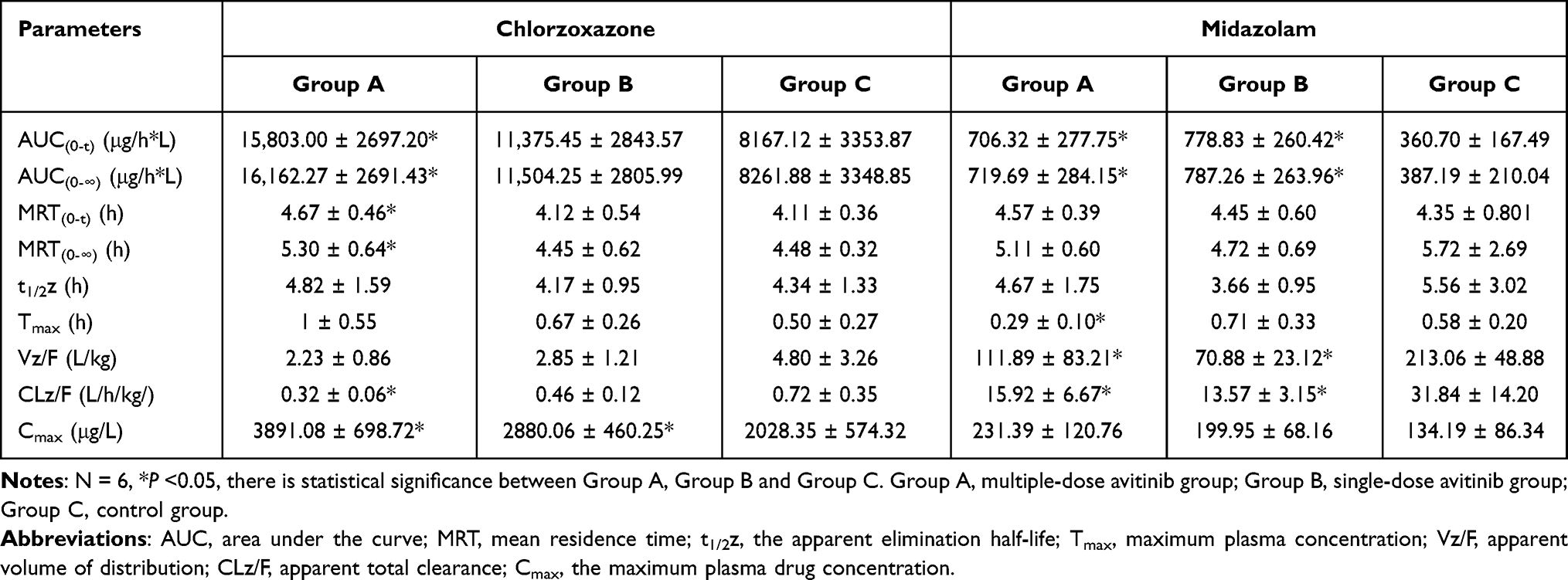 |
Table 4 Pharmacokinetic Parameters of Cyp2e1 and Cyp3as Enzymes on Probe Drug Chlorzoxazone and Midazolam in Group A, Group B, and Group C Rats |
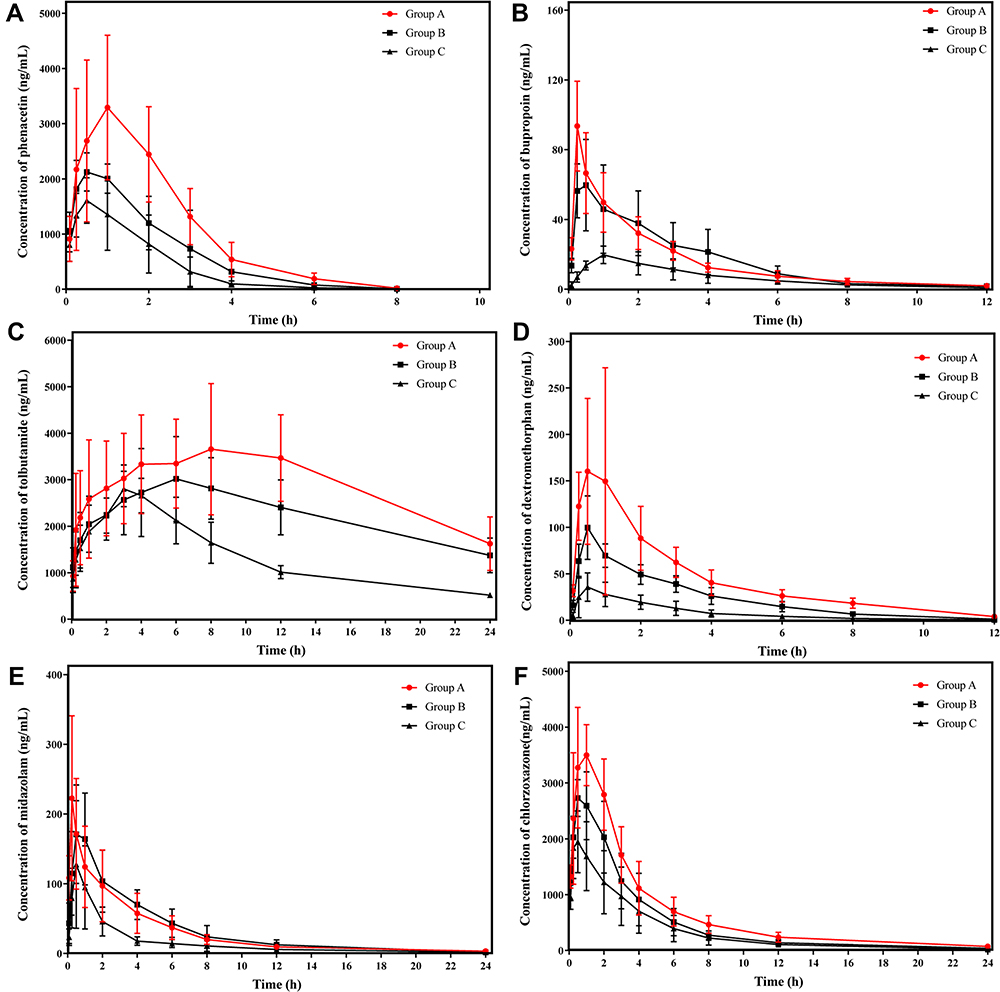 |
Figure 6 Concentration-time curves of probe substrates in experimental groups of rats. (A) phenacetin, (B) bupropion, (C) tolbutamide, (D) dextromethorphan, (E) midazolam, and (F) chlorzoxazone. Group A (●), Group B (■), and Group C (▲) rats. Mean ± SD, n = 6. Group A, multiple-dose avitinib group; Group B, single-dose avitinib group; Group C, control group. |
For phenacetin, when multiple-doses or a single-dose of 30 mg/kg avitinib was co-administered with the probe substrates, the area under the curve (AUC) (0-∞) values significantly increased 1.71-fold or 0.61-fold, respectively, the maximum plasma drug concentration (Cmax) values significantly increased 0.92-fold or 0.27-fold, respectively, and the mean residence time (MRT)(0-∞) values increased 0.38-fold or 0.20-fold, respectively, compared with that of the control group. In addition, avitinib significantly decreased the apparent volume of distribution during the terminal phase (Vz/F) and apparent total clearance of the drug from plasma after oral administration (CLz/F) of phenacetin. Specifically, the Vz/F of phenacetin was reduced 63.09% and 49.62% for the multiple-doses and single-dose treatments, respectively, compared with that of the control group while the CLz/F was reduced 63.17% and 39.52%, respectively. However, no significant change in the half-time elimination (t1/2) values were observed.
For bupropion, when multiple-doses or a single-dose of 30 mg/kg avitinib was co-administrated with the probe substrates, the AUC (0-∞) values significantly increased by 1.41-fold and 1.5-fold, respectively, the Cmax values significantly increased 3.61-fold and 2.39-fold, respectively, compared with the control group. Again, avitinib significantly decreased the Vz/F and CLz/F of bupropion. Specifically, the Vz/F of bupropion was reduced 62.74% and 64.59% for the multiple-doses and single-dose treatments, respectively, compared with that of the control group while the CLz/F was reduced 57.54% and 53.96%, respectively. No significant changes were observed in the t1/2 values or the time to reach maximum plasma concentration following administration (Tmax) values.
For dextromethorphan, when multiple-doses or a single-dose of 30 mg/kg avitinib was co-administrated with the probe substrates, the AUC (0-∞) values significantly increased 4.55-fold and 1.87-fold, respectively, the Cmax values significantly increased 3.13-fold and 1.21-fold, respectively, and the MRT (0-∞) values increased 0.39-fold and 0.09-fold, respectively, compared with that of the control group. Avitinib again significantly decreased the Vz/F and CLz/F values of dextromethorphan. Specifically, the Vz/F of dextromethorphan was reduced 73.53% and 69.7% for the multiple-doses and single-dose treatments, respectively, compared with that of the control group while the CLz/F was reduced 82.22% and 67.78%, respectively. No significant changes in the Tmax values were observed.
In reference to tolbutamide, chlorzoxazone, and midazolam, the AUC (0-∞) values for the multiple-doses and single-dose treatments were significantly increased by 2.36-fold and 1.34-fold (tolbutamide), 0.96-fold and 0.39-fold (chlorzoxazone), and 0.86-fold and 1.03-fold (midazolam), respectively. No significant changes were observed in the MRT(0-∞), t1/2, or Tmax values for tolbutamide. The Cmax values for chlorzoxazone were significantly increased by 3.14-fold and 1.21-fold for the multiple-doses and single-dose treatments, respectively, compared with that of the control group. No significant difference in Cmax was observed for tolbutamide or midazolam. These results indicated that avitinib was an inhibitor of CYP450-related metabolism of the probe substrates.
Discussion
Clinically, patients with cancer are commonly administered several drugs to treat their cancer and related diseases. It has been reported that 20–30% of all adverse reactions observed in clinical trials are caused by drug-drug interactions. CYP450 enzymes are responsible for the metabolism of a variety of drugs in which drug-drug interactions often occurred.29 Inhibition or induction of CYP450 enzyme activity can lead to changes in plasma drug concentrations in patients, which in turn can affect the therapeutic efficacy and safety of the drugs.30 Cocktail-based methods are the most commonly used approach for evaluating CYP450 enzyme activity levels and can reflect the increases or decreases in enzymatic activity by measuring the content of the remaining substrates or their metabolites.31 To better understand the potential effect of avitinib on the enzyme activity of CYP450, six different probe substrates were included in the cocktail used in the current study.
We investigated the potential drug-drug interactions in vitro using RLMs/HLMs and in vivo by performing UPLC-MS/MS analysis to detect the probe substrates and their metabolites in the plasma of rats. We then calculated the IC50 values of avitinib for the six probe substrates. The in vitro results indicated that avitinib had a direct inhibitory effect on all the probe substrates in this study by using the RLMs/HLMs incubation system. Our experimental in vitro approach was relatively simple to perform and the results to a certain extent reflected the inhibitory effects among drugs and avoided the interference of various internal factors. To further clarify the in vitro experiment results, an in vivo experiment in rats was also conducted using a combination of various avitinib treatments and the probe substrates. We determined that avitinib in rats also exhibited inhibitory effects on the pharmacokinetics of some P450 enzymes. The main pharmacokinetic parameters (AUC(0-t), and AUC(0-∞)) of phenacetin, bupropion, dextromethorphan, tolbutamide, and chlorzoxazone all increased significantly in the Group A animals compared with those in Group C. Meanwhile, the CLz/F and Vz/F of phenacetin, bupropion, dextromethorphan, chlorzoxazone, and midazolam decreased, indicating that avitinib may have affected them by prolonging exposure time and reducing their clearance rates. All these results indicated that avitinib was able to inhibit metabolism of the probe substrates in vivo.
Our findings provided a fundamental basis for the rational decision to combine the use of avitinib and other drugs in clinic. The in vivo experiment in rats indicated that avitinib exhibited various degrees of inhibitory effects on the six probe substrates, indicating avitinib would especially impact drugs that are metabolized by liver drug enzymes CYP2B6, CYP2D6, and CYP3As. Consistent with previous reports, we speculate that the inhibitory effects of avitinib on these P450 enzymes may have been caused by other proteins that were able to increase the expression of CYP450 enzymes.32,33 In addition, avitinib also exhibited inhibitory effects on CYP2B6 and CYP1A2. Therefore, mutual inhibition of metabolism may occur in vitro when avitinib is incubated with many drugs that are substrates or inhibitors of CYP2B6 and CYP1A2.34 Inhibitory effects of drug interaction often improve drug efficacy and can likely cause clinical adverse reactions and may even be life-threatening for drugs with a narrow therapeutic window. Based on our current findings, we propose that attentions should be paid in clinical practice when administering avitinib with other drugs metabolized by P450 enzymes. However, this study still has some limitations, multiple-dose or administration route of avitinib are still needed in the future. Moreover, we only used RLMs/HLMs and animal models to simulate drug-drug interaction, it is not sufficient information to guide the clinical medication due to interspecific differences. Further human studies including clinical patients are needed to confirm our results and help avoid cute adverse reactions caused by combination therapy including avitinib.
Conclusion
In this study, an in vitro incubation system of RLMs and an UPLC-MS/MS method for the detection of six CYP450 enzyme probe substrates (phenacetin, bupropion, dextromethorphan, tolbutamide, midazolam and chlorzoxazone) and their metabolites were developed and evaluated. Through these methods, avitinib was found to exhibit inhibitory effects on CYP450 enzymes in vitro. Results from the rat model showed that avitinib exhibited the same inhibitory effects on the six CYP450 probe drugs used in our study. Our results provide a theoretical basis for the combined application of avitinib and other CYP450-mediated drugs in the clinic.
Acknowledgments
This work was supported by grants funded by Public Welfare Technology Research Funding Project of Zhejiang (LGF21H310002), Key Research and Development Project of Lishui (2020ZDYF12, 2021ZDYF13 & 2021ZDYF15), Public Welfare Technology Research Funding Project of Lishui (2020GYX18 & 2020GYX23), Ningbo Science & Technology Bureau (2016A610153), Taizhou Science and Technology Project (1802ky29), Medical and Health Science and Technology Plan Project of Zhejiang Province (2021KY411 & 2019KY804).
Disclosure
The authors report no conflicts of interest and no disclosures to declare.
References
1. Roskoski R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395–411. doi:10.1016/j.phrs.2018.11.014
2. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–1953. doi:10.1002/ijc.31937
3. Qiang H, Chang Q, Xu J, et al. New advances in antiangiogenic combination therapeutic strategies for advanced non-small cell lung cancer. J Cancer Res Clin Oncol. 2020;146(3):631–645. doi:10.1007/s00432-020-03129-6
4. Xu X, Mao L, Xu W, et al. AC0010, an irreversible EGFR inhibitor selectively targeting mutated EGFR and overcoming T790M-induced resistance in animal models and lung cancer patients. Mol Cancer Ther. 2016;15:2586–2597. doi:10.1158/1535-7163.MCT-16-0281
5. Zhang YC, Chen ZH, Zhang XC, et al. Analysis of resistance mechanisms to abivertinib, a third-generation EGFR tyrosine kinase inhibitor, in patients with EGFR T790M-positive non-small cell lung cancer from a Phase I trial. EBioMedicine. 2019;43:180–187. doi:10.1016/j.ebiom.2019.04.030
6. Wang H, Pan R, Zhang X, Si X, Wang M, Zhang L. Abivertinib in patients with T790M-positive advanced NSCLC and its subsequent treatment with osimertinib. Thorac Cancer. 2020;11:594–602. doi:10.1111/1759-7714.13302
7. Huang S, Pan J, Jin J, et al. Abivertinib, a novel BTK inhibitor: anti-Leukemia effects and synergistic efficacy with homoharringtonine in acute myeloid leukemia. Cancer Lett. 2019;461:132–143. doi:10.1016/j.canlet.2019.07.008
8. Xu X. Parallel Phase 1 clinical trials in the US and in China: accelerating the test of avitinib in lung cancer as a novel inhibitor selectively targeting mutated EGFR and overcoming T790M-induced resistance. Chin J Cancer. 2015;34:285–287. doi:10.1186/s40880-015-0029-3
9. Ma Y, Zheng X, Zhao H, et al. First-in-human phase I study of AC0010, a mutant-selective EGFR inhibitor in non-small cell lung cancer: safety, efficacy, and potential mechanism of resistance. J Thorac Oncol. 2018;13:968–977. doi:10.1016/j.jtho.2018.03.025
10. Zheng X, Wang W, Zhang Y, et al. Development of an LC-MS/MS method for quantifying two main metabolites of abivertinib in human plasma. Biomed Chromatogr. 2020;34:e4704. doi:10.1002/bmc.4704
11. Attwa MW, Kadi AA, Abdelhameed AS. Reactive intermediates and bioactivation pathways characterization of avitinib by LC-MS/MS: in vitro metabolic investigation. J Pharm Biomed Anal. 2019;164:659–667. doi:10.1016/j.jpba.2018.11.033
12. Wang W, Zheng X, Wang H, Wang L, Jiang J, Hu P. Development of an UPLC-MS/MS method for quantification of Avitinib (AC0010) and its five metabolites in human cerebrospinal fluid: application to a study of the blood-brain barrier penetration rate of non-small cell lung cancer patients. J Pharm Biomed Anal. 2017;139:205–214. doi:10.1016/j.jpba.2017.02.057
13. Wang L, Zheng X, Wang W, Hu P, Jiang J. An LC-MS/MS method for quantification of AC0010, a novel mutant-selective epidermal growth factor receptor (EGFR) inhibitor, and its metabolites in human plasma and the application to a pharmacokinetic study. J Pharm Biomed Anal. 2017;141:9–18. doi:10.1016/j.jpba.2017.03.051
14. Wu Q, Jiang H, Wang S, et al. Effects of avitinib on the pharmacokinetics of osimertinib in vitro and in vivo in rats. Thorac Cancer. 2020;11:2775–2781. doi:10.1111/1759-7714.13587
15. Chu LL, Pandey RP, Jung N, Jung HJ, Kim EH, Sohng JK. Hydroxylation of diverse flavonoids by CYP450 BM3 variants: biosynthesis of eriodictyol from naringenin in whole cells and its biological activities. Microb Cell Fact. 2016;15:135. doi:10.1186/s12934-016-0533-4
16. Huang Y, Zheng SL, Zhu HY, Xu ZS, Xu RA. Effects of aescin on cytochrome P450 enzymes in rats. J Ethnopharmacol. 2014;151:583–590. doi:10.1016/j.jep.2013.11.016
17. Albaugh DR, Fullenwider CL, Fisher MB, Hutzler JM. Time-dependent inhibition and estimation of CYP3A clinical pharmacokinetic drug-drug interactions using plated human cell systems. Drug Metab Dispos. 2012;40:1336–1344. doi:10.1124/dmd.112.044644
18. Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet. 2011;36:1–16. doi:10.1007/s13318-011-0024-2
19. Zhou S, Gao Y, Jiang W, Huang M, Xu A, Paxton JW. Interactions of herbs with cytochrome P450. Drug Metab Rev. 2003;35:35–98. doi:10.1081/DMR-120018248
20. Huang LH, Zhong YM, Xiong XH, et al. The disposition of oxymatrine in the vascularly perfused rat intestine-liver preparation and its metabolism in rat liver microsomes. J Pharm Sci. 2016;105:897–903. doi:10.1016/j.xphs.2015.11.012
21. de Andres F, Llerena A. Simultaneous determination of cytochrome P450 oxidation capacity in humans: a review on the phenotyping cocktail approach. Curr Pharm Biotechnol. 2016;17:1159–1180. doi:10.2174/1389201017666160926150117
22. Ebner T, Ishiguro N, Taub ME. The use of transporter probe drug cocktails for the assessment of transporter-based drug-drug interactions in a clinical setting-proposal of a four component transporter cocktail. J Pharm Sci. 2015;104:3220–3228. doi:10.1002/jps.24489
23. Wang SH, Lin ZX, Su K, et al. Effect of curcumin and pirfenidone on toxicokinetics of paraquat in rat by UPLC-MS/MS. Acta Chromatographica. 2018;30:26–30. doi:10.1556/1326.2017.00175
24. Wang SH, Zhang ZG, Yu Z, Han C, Wang XQ. Pharmacokinetic study of delavinone in mice after intravenous and oral administration by UPLC-MS/MS. Biomed Res Int. 2019;2019:3163218. doi:10.1155/2019/3163218
25. Wang SH, Dong YW, Su K, et al. Effect of codeine on CYP450 isoform activity of rats. Pharm Biol. 2017;55:1223–1227. doi:10.1080/13880209.2017.1297466
26. Wang Y, Wang C, Wang S, et al. Cytochrome P450-based drug-drug interactions of vonoprazan in vitro and in vivo. Front Pharmacol. 2020;11:53. doi:10.3389/fphar.2020.00053
27. Zhou Y, Hua A, Zhou Q, et al. Inhibitory effect of lygodium root on the cytochrome P450 3A enzyme in vitro and in vivo. Drug Des Devel Ther. 2020;14:1909–1919. doi:10.2147/DDDT.S249308
28. Zhou Y, Tu Y, Zhou Q, et al. Evaluation of acacetin inhibition potential against cytochrome P450 in vitro and in vivo. Chem Biol Interact. 2020;329:109147. doi:10.1016/j.cbi.2020.109147
29. Orr ST, Ripp SL, Ballard TE, et al. Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J Med Chem. 2012;55:4896–48933. doi:10.1021/jm300065h
30. Pilgrim JL, Gerostamoulos D, Drummer OH. Review: pharmacogenetic aspects of the effect of cytochrome P450 polymorphisms on serotonergic drug metabolism, response, interactions, and adverse effects. Forensic Sci Med Pathol. 2011;7:162–184. doi:10.1007/s12024-010-9188-3
31. Spaggiari D, Geiser L, Daali Y, Rudaz S. A cocktail approach for assessing the in vitro activity of human cytochrome P450s: an overview of current methodologies. J Pharm Biomed Anal. 2014;101:221–237. doi:10.1016/j.jpba.2014.03.018
32. Yan ZY, Caldwell GW. Metabolism profiling, and cytochrome P450 inhibition & induction in drug discovery. Curr Top Med Chem. 2001;1:403–425. doi:10.2174/1568026013395001
33. Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535–567. doi:10.1146/annurev.pharmtox.41.1.535
34. Zhou SF, Xue CC, Yu XQ, et al. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29:687–710. doi:10.1097/FTD.0b013e31815c16f5
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.