")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Effects of Anterior Pituitary Adenomas’ Hormones on Glucose Metabolism and Its Clinical Implications
Authors Li M , Zhang J, Yang G, Zhang J, Han M , Zhang Y, Liu Y
Received 18 November 2022
Accepted for publication 2 February 2023
Published 13 February 2023 Volume 2023:16 Pages 409—424
DOI https://doi.org/10.2147/DMSO.S397445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Mengnan Li,1,2 Jian Zhang,1,2 Guimei Yang,1,2 Jiaxin Zhang,1,2 Minmin Han,1,2 Yi Zhang,3 Yunfeng Liu1
1Department of Endocrinology, First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2First Clinical Medical College, Shanxi Medical University, Taiyuan, People’s Republic of China; 3Department of Pharmacology, Shanxi Medical University, Taiyuan, People’s Republic of China
Correspondence: Yi Zhang, Department of Pharmacology, Shanxi Medical University, Taiyuan, People’s Republic of China, Email [email protected] Yunfeng Liu, Department of Endocrinology, First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China, Tel +86 18703416196, Email [email protected]
Abstract: Pituitary adenomas have recently become more common and their incidence is increasing yearly. Functional pituitary tumors commonly secrete prolactin, growth hormones, and adrenocorticotropic hormones, which cause diseases such as prolactinoma, acromegaly, and Cushing’s disease, but rarely secrete luteinizing, follicle-stimulating, thyroid-stimulating, and melanocyte-stimulating hormones. In addition to the typical clinical manifestations of functional pituitary tumors caused by excessive hormone levels, some pituitary tumors are also accompanied by abnormal glucose metabolism. The effects of these seven hormones on glucose metabolism are important for the treatment of diabetes secondary to pituitary tumors. This review focuses on the effects of hormones on glucose metabolism, providing important clues for the diagnosis and treatment of related diseases.
Keywords: pituitary adenoma, glucose metabolism, prolactin, growth hormones, glucocorticoids
Introduction
The incidence of pituitary adenomas, which are common benign tumors of the anterior pituitary gland, is increasing annually, accounting for approximately 14% of all intracranial tumors.1 Approximately a third to a half (36–54%) of pituitary tumors are nonfunctional.2–5 Nonfunctioning adenomas do not secrete hormones and may cause mass effects such as visual deficits, hypopituitarism, or headaches. Some nonfunctional adenomas may not cause symptoms and are incidentally discovered during neuroimaging.6 The other half of pituitary adenomas (46–64%) are functional tumors that secrete hormones.7–9 The common secreted hormones are prolactinoma (PRL; 32–51%), growth hormones (GH; 9–11%), and adrenocorticotropic hormones (ACTH; 3–6%). The less commonly secreted hormones are thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and melanocyte-stimulating hormone (MSH). Functional adenomas cause clinical symptoms mediated by the excessive secretion of hormones, leading to acromegaly, Cushing’s disease (CD), galactorrhea, hypogonadism, and other symptoms.
Pituitary tumors are often accompanied by impaired glucose tolerance or diabetes mellitus, which is often an early manifestation of these tumors. The presence of glucose metabolism disorders further increases the risk of cardiovascular disease-associated morbidity and mortality in patients with pituitary tumors.10 In addition, diabetes mellitus may also affect the treatment plan for patients with pituitary tumors because the therapeutic agents for pituitary tumors may affect glucose metabolism.11 In this review, we highlight the roles of seven hormones in glucose metabolism respectively and provide basic theoretical support for the clinical diagnosis and treatment of related glucose metabolism disorders.
PRL and Glucose Metabolism
The main manifestations of prolactinoma are hypogonadism and galactorrhea. In women, hypogonadism can lead to irregular menstruation, amenorrhea, sexual dysfunction, infertility, and reduced intersex bone mineral production.12 Patients who suffer from hyperprolactinemia often have fractures due to decreased bone mineral density.13 Moreover, large PRL-producing tumors can cause compressive symptoms, including headaches, vision changes, and hydrocephalus.14,15
PRL is a polypeptide hormone that is released by lactating cells from the anterior pituitary. Hypothalamus controls PRL secretion differently from other pituitary hormones because it is primarily suppressed by extracting dopamine from the tuberoinfundibular dopamine neurons (TIDA).16 Moreover, as prolactin receptors (PRLRs) are present in almost all organs, PRL affects more physiological processes than other pituitary hormones.17 Therefore, PRL plays several roles in endocrine metabolism and growth and development, water and electrolyte balance, and brain and behavior.16,18,19
PRL is not conducive to glucose metabolism in patients with prolactinoma who often have obesity and metabolic disorders.20,21 Compared to healthy individuals, individuals with hyperprolactinemia often have glucose intolerance and decreased insulin sensitivity.22–24 Data from female mouse studies showed that long-term high PRL levels are associated with increased appetite, weight gain, obesity, insulin resistance, and glucose intolerance, supporting the notion that high PRL levels are detrimental to metabolic homeostasis.25,26 Moreover, hyperprolactinemia has been associated with impaired metabolism in a study using the euglycemic hyperinsulinemic clamp technique,27 as well as an increase in the homeostatic model assessment (HOMA) index28 and a reduction of insulin sensitivity index (ISI)29 in both obese and lean patients.
Hyperprolactinemia increases blood glucose levels via two mechanisms. Hyperprolactinemia leads to hypoadiponectin. Adiponectin can enhance insulin-mediated inhibition of hepatic gluconeogenesis30 and improve insulin sensitivity by stimulating glucose uptake and fatty acid oxidation of fat and muscle cells,31,32 supporting the notion that patients with hyperprolactinemia may exhibit biochemical signs of both metabolic syndromes and insulin resistance.33 Hyperprolactinemia can also induce gonadotropin suppression, which may indirectly induce insulin resistance in both men and women.34,35
In conclusion, prolactinomas and hyperglycemia may occur in the same patient, requiring a comprehensive hypoglycemic treatment in accordance with the patient’s blood glucose levels.
GH and Glucose Metabolism
Acromegaly is a clinical syndrome characterized by the excessive secretion of GH, which usually occurs in pituitary adenomas.36 Typical facial features of acromegaly include thickened lips, enlarged nose and ears, nasolabial folds, and lordosis.37 The incidence of diabetes in patients with acromegaly is higher than that in the general population. More than 50% of patients diagnosed with acromegaly develop impaired glucose metabolism.38 Moreover, the presence or absence of diabetes is an important predictor of increased mortality in acromegaly.39
GH plays a direct role in glucose metabolism. It induces insulin resistance, leading to impaired glucose metabolism.40 Patients with acromegaly are characterized by reduced insulin sensitivity and impaired hepatic and extrahepatic insulin activity.41,42 GH mediates the action of insulin mainly by regulating p85α43 in fat tissues (Figure 1). Excessive GH levels promote the expression of p85α, which reduces the activity of insulin-mediated phosphoinositide 3-kinase (PI3K), leading to insulin resistance.44 In patients with obesity, the inhibition of GH activity reverses insulin resistance.45 In addition, GH acts on the liver,46 skeletal muscles,47 brain,48 kidneys,49 and other organs, leading to insulin resistance and increased glucose output.
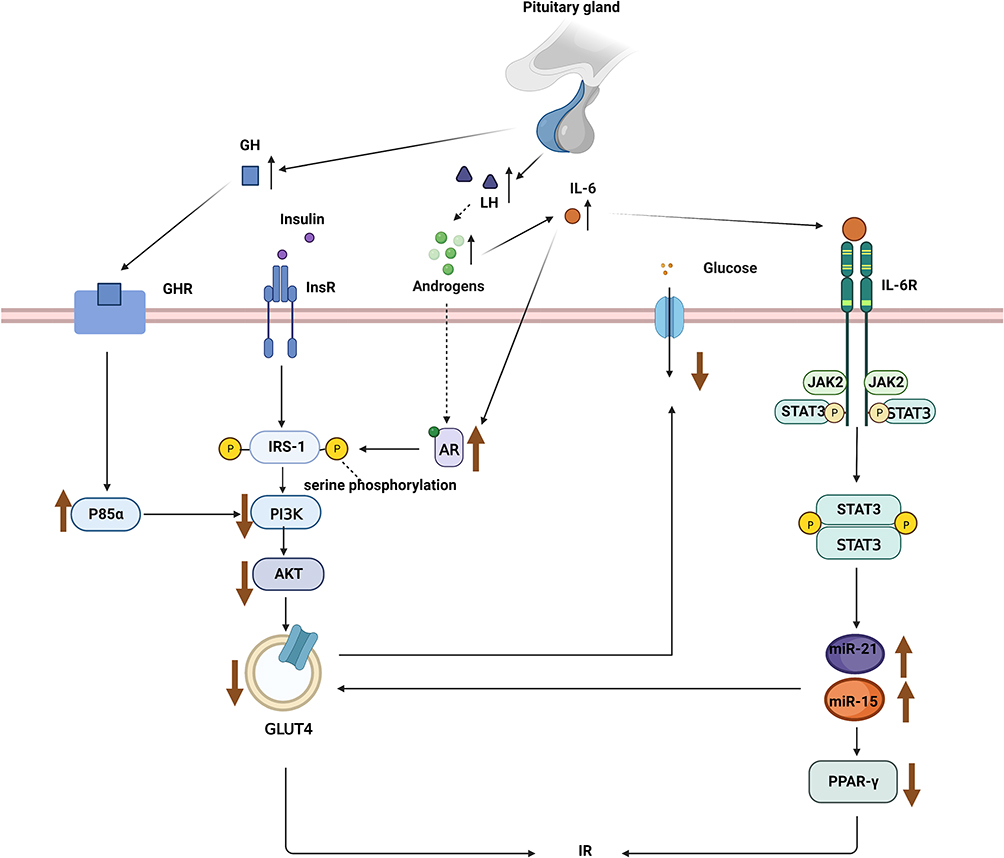 |
Figure 1 The regulation mechanism of luteinizing hormone (LH) and growth hormone (GH) on glucose homeostasis. LH indirectly affects glucose homeostasis by interfering with related signaling pathways through androgens and inflammatory mediators, while GH interferes with insulin signaling through p85α. Abbreviations: IRS-1, insulin receptor substrate-1; PI3K, phosphoinositide 3-kinase; GLUT4, glucose transporter 4; AR, androgen receptor; JAK2, Janus tyrosine kinase 2; STAT3, signal transducers and activators of transcription 3; PPAR-γ, peroxisome proliferator activated receptor γ; IL-6, interleukin 6. |
The indirect effect of GH on glucose metabolism is mediated by insulin-like growth factor 1 (IGF-1), a peptide hormone with a 50% homology to proinsulin. Similar to insulin, IGF-1 enhances glucose uptake and oxidative/non-oxidative glucose metabolism, which is positively correlated with the dose of IGF-1. GH binds to GH receptors, which are primarily located in hepatic cells, leading to the synthesis and secretion of IGF-1 in the liver, which improves insulin sensitivity.50,51 Moreover, IGF-1 can inhibit endogenous (mainly liver) glucose production, but is not as effective as insulin,52–54 and acts on the IGF-1 receptors in the brain55 to increase insulin sensitivity56 and decrease food intake.57
Although the direct and indirect roles of GH in glucose regulation are opposites, the overall effect is still the result of direct action. On the one hand, chronic excessive GH can cause insulin resistance, and these roles largely offset the favorable effect of IGF-1 on insulin sensitivity. On the other hand, IGF-1 is not as effective as insulin, and IGF-1 resistance is also observed in skeletal muscles.58
In conclusion, GH is implicated in the pathogenesis of diabetes and its extensive effects are important in the diagnosis, prevention, and treatment of diabetes. The hypoglycemic treatment for acromegaly is the same as that for diabetes. Generally, lowering GH levels increases insulin sensitivity, and glycemic control ensues.59
ACTH and Glucose Metabolism
Cushing syndrome (CS) is a general condition caused by excessive secretion of cortisol from the adrenal gland. The most common cause of CS is ACTH-secreting pituitary tumors called CD, accounting for about 80–85%.60 Typical symptoms of CS include full moon face, buffalo back, centripetal obesity, purple stripes, hypertension and hypokalemia, osteoporosis, and diabetes. Moreover, alterations in glucose metabolism are present in 50% of patients with CD.61
To the best of our knowledge, few studies have investigated the direct relationship between ACTH and glucose homeostasis, reporting that ACTH regulates glucose metabolism mainly through downstream glucocorticoids (GCs). Here, we discuss the effects of GCs on glucose metabolism from the following aspects (Figure 2).
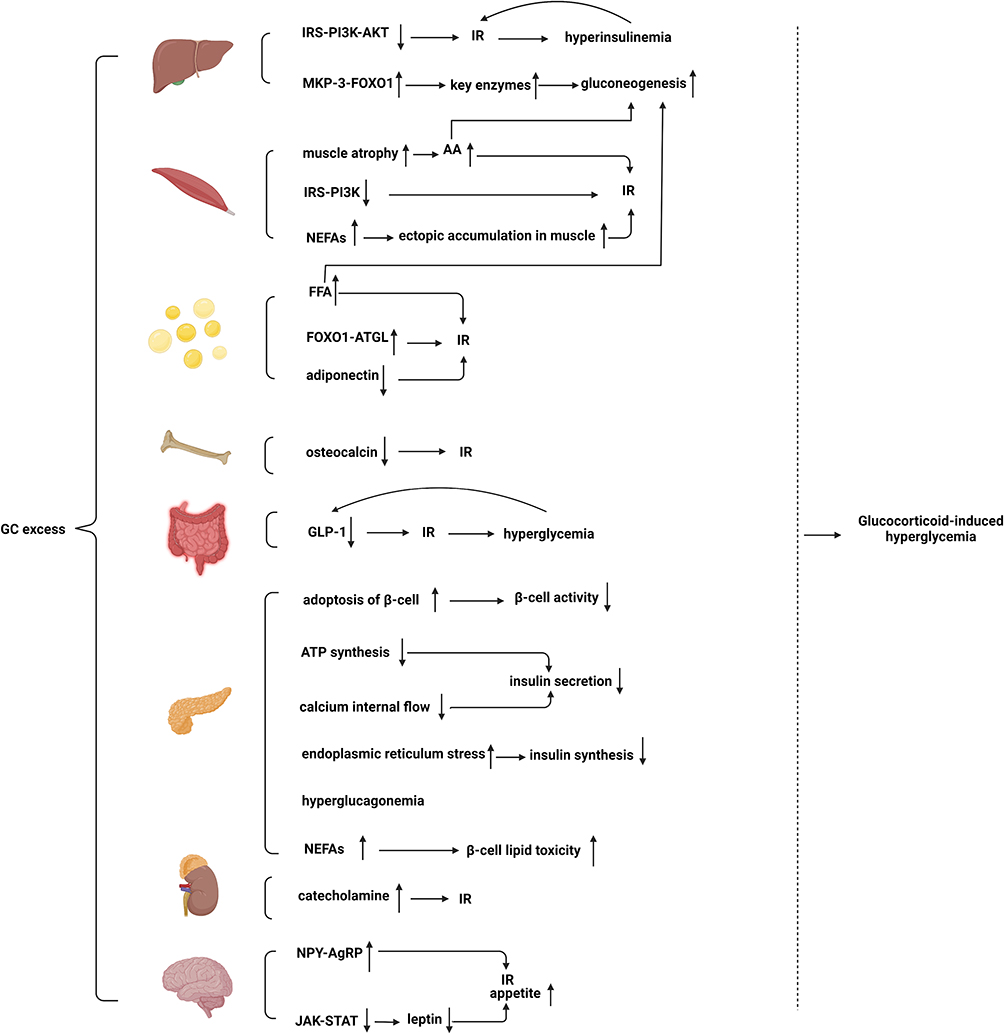 |
Figure 2 Excessive GCs act on the above-mentioned organs to induce glucose metabolism disorders and eventually lead to hyperglycemia. Abbreviations: IRS, insulin receptor substrate; PI3K, phosphoinositide 3-kinase; MKP-3, mitogen-activated protein kinase phosphatase-3; FOXO1, forkhead box protein O1; AA, amino acid; FFA, free fatty acids; NEFAs, non-esterified fatty acids; ATGL, adipose triglyceride lipase; NPY, nerve peptide Y; AgRP, agouti-related protein; JAK, Janus tyrosine kinase; STAT, signal transducers and activators of transcription. |
Hepatic tissue plays a major role in regulating glucose levels. The first key pathway is the regulation of key gluconeogenic enzymes. GCs act on target genes of gluconeogenic enzymes, including phosphoenolpyruvate carboxykinase and glucose-6-phosphatase.62 A study has suggested that GC-induced forkhead box protein O1 (FOXO1) upregulation stimulates hepatic glucose output.63 GCs upregulate FOXO1 by promoting mitogen-activated protein kinase phosphatase-3 (MKP-3) expression, a liver gluconeogenic activator.64 Moreover, Gluconeogenesis is further induced by lipolysis in adipose tissues and protein hydrolysis in the skeletal muscles.65 The second key pathway is insulin resistance mediated by the overexpression of GCs. Impaired phosphorylation of the downstream messenger of the insulin cascade (IRS-PI3K-AKT) in rats treated with GCs leads to compensatory hyperinsulinemia, which in turn increases insulin resistance in the liver through a vicious pre-cycle.66 GCs increase the blood glucose level by its effects on the liver as described above.
The skeletal muscle accounts for approximately 80% of insulin-mediated glucose uptake. GCs directly hinder insulin signaling in skeletal muscle cells to inhibit glucose uptake, thereby leading to GC-induced diabetes. Dexamethasone decreases the activity and expression of insulin substrate 1 (IRS1) and phosphoinositide-3-kinase regulatory subunit 1 (PI3Kr1) in rodent skeletal muscle cells,67 which are the primary target genes for skeletal muscle glucocorticoid receptor (GR), and further inhibits insulin-induced glucose transporter 4 (GLUT4) recruitment to skeletal muscle surfaces.68,69 In addition to the above mechanisms, patients with long-term exposure to corticosteroid drugs can develop extensive muscle atrophy, leading to the development of myopathy.70 In these patients, increased muscle proteolysis and passivated protein synthesis lead to elevated serum amino acid levels,71–73 which can indirectly impede glycogen synthesis and glucose transport.74 GCs also increase the circulation of non-esterified fatty acids (NEFAs) from adipose tissue and its ectopic accumulation75 in skeletal muscles, further exacerbating insulin resistance.76 In addition, elevated serum amino acids and NEFAs also provide substrates for liver gluconeogenesis.
GCs play various roles in adipose tissues77 such as the promotion of lipolysis and insulin-antagonistic mechanisms.78 Exposure to GCs causes systemic elevated fatty acid and triglycerides levels, leading to insulin resistance66 and increased hepatic gluconeogenesis.65 To date, accumulating evidence has indicated that 5 days of exposure to prednisolone results in increased levels of basal free fatty acids (FFA) and disruption of insulin-mediated inhibition of FFA, which induces increases in insulin and fasting glucose levels, suggesting that GCs induce insulin resistance.79 In vitro tests also have suggested that excessive exposure to GCs results in insulin resistance.77 Moreover, lipolysis can provide glycerol and fatty acids as gluconeogenic substrates, which in turn provide energy for gluconeogenesis and increase allosteric activation of gluconeogenesis by increasing hepatic acetyl Coenzyme A.77 GC-induced decrease in insulin signaling in fatty tissue is also associated with reduced glucose uptake. A previous study has reported that prednisolone may indirectly activate adipose triglyceride lipase (ATGL) transcription via FOXO1, suggesting that GCs block the insulin signaling pathway to prevent insulin-mediated inhibition.79 In addition, excessive levels of GCs can reduce adiponectin expression and secretion levels in fatty tissue.30 Because adiponectin enhances fatty acid oxidation in the muscles and insulin inhibition of hepatic gluconeogenesis,31,32 excessive GCs increase glucose and insulin resistance.
Osteocalcin is a bone-derived hormone whose non-carboxylated form (Glu-OC) is essential to glucose and energy metabolism.80 Osteocalcin is an insulin-sensitizing hormone released from osteoblasts.81 Interestingly, osteocalcin-deficient mice develop abnormal glucose tolerance and insulin resistance owing to reduced insulin expression and pancreatic β-cell proliferation.82 Osteocalcin mediates its functions both directly and indirectly. Glu-OC directly activates the putative receptor GPRC6A (G protein-coupled receptor, family C, group 6, member A) in the pancreas, leading to insulin production and β-cell proliferation.83 Glu-OC indirectly increases insulin secretion through glucagon-like peptide-1 (GLP-1)84 in a mechanism that may be referred to as bone-gut-metabolism flow. Moreover, Glu-OC negatively affects glucose metabolism independent of GLP-1, possibly by activating FOXO1 and the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α).85 Notably, long-term exposure to GCs induces a reduction in circulating osteocalcin, leading to insulin resistance.86 Therefore, these findings demonstrate the role of bones in regulating energy metabolism.
GLP-1 is synthesized from intestinal enteroendocrine L-cells and secreted after nutrient intake.87 GLP-1 mainly exhibits insulinotropic effects but has many other roles, such as stimulating β-cell proliferation and reducing insulin resistance, inhibiting gastric emptying, glucagon release, and food intake.88 Moreover, hypercortisolemia may affect the incretin system because excessive GCs cause a decrease in GLP-1 secretion. Accumulating evidence further indicates that dexamethasone reduces cyclic adenosine monophosphate (cAMP) production, which in turn impairs protein kinase A activity and ultimately GLP-1 secretion.89 GC-induced reduction of GLP-1 action ultimately leads to insulin resistance and impaired glucose tolerance, which accelerates the development of diabetes and further increases blood glucose levels,90 which in turn leads to decreased GLP-1 secretion, creating a vicious cycle.91
GCs impair β-cell activity and affect insulin secretion and synthesis. A study on human and mouse islets in vitro indicated that prednisolone decreases β-cell activity by inducing apoptosis of β-cells and that blocking GRs reverses this effect.92,93 Furthermore, chronic hypercortisolism results in pancreatic β-cell dysfunction. Excessive GCs lead to reduced ATP synthesis and calcium internal flow by decreasing the expression levels of type 2 glucose transporters and glucose kinases, leading to impaired metabolism in β-cells and glucose uptake, and ultimately to reduced insulin secretion.94 Treatment with prednisolone in INS1 (rat pancreatic β-cell line) induces endoplasmic reticulum stress and impairs the biosynthesis and secretion of insulin. In rats, chronic high GCs levels lead to hyperglucagonemia by affecting the function and mass of pancreatic cells,95,96 which also increases blood glucose levels. In addition to the aforementioned mechanism of GC-induced β-cell failure, GC-induced increase in NEFAs can lead to β-cell lipid toxicity, which further increases blood glucose levels.97
Pheochromocytoma is associated with preoperative diabetes mellitus.98 The literature suggests that catecholamine leads to pheochromocytoma-associated diabetes by impairing insulin sensitivity and secretion.99 These effects are mediated by complex mechanisms, including the direct action of catecholamine, the subsequent inhibition of glucose uptake in muscle cells, and high concentrations of FFA.100–102 Cortisol is important for the adrenal myelin, which synthesizes, stores, and secretes catecholamine in chromaffin cells. Excessive cortisol levels aggravate adrenaline-mediated glucose homeostasis.103
GCs also mediate the development of obesity and diabetes partly by inducing the hypothalamic arcuate nucleus to stimulate appetite.104 GCs can stimulate appetite and induce insulin resistance in animals by upregulating the mRNA of nerve peptide Y (NPY) and agouti-related protein (AgRP).105,106 However, physiologically, leptin inhibits appetite through leptin signaling in NPY-AgRP neurons.107 In rats, GCs impede the effects of leptin by impairing the leptin-dependent Janus tyrosine kinase (JAK)-signal transducers and activators of the transcription(STAT) signaling pathway in the hypothalamus.108
In conclusion, it is imperative to carefully assess the phenotype of patients with CS with impaired glucose tolerance and, if possible, conduct accurate insulin secretion and sensitivity tests before initiating treatment because CS further impairs glucose tolerance.109
Gonadotropin and Glucose Metabolism
In most cases, gonadotropin is characterized by the secretion of physiologically inactive hormones, therefore, its release does not cause any clinical symptoms. Functional gonadotroph-secreting tumors are scarce and account for only 0.2% of pituitary adenomas.110 Therefore, the diagnosis of gonadotropinomas is most usually based on the immunohistochemical analysis. However, in rare cases, gonadotropinomas secrete bioactive hormones, including LH and FSH.111,112 In vivo, functional gonadotropin adenomas are associated with changes in hormone levels. In patients with gonadotropinomas, estradiol levels are usually high whereas LH levels are low because of negative feedback from estrogen or damage to normal glands from pituitary tumors.113 Moreover, premenopausal women with functional gonadotropin adenomas have clinical manifestations of oligomenorrhea, amenorrhea, galactorrhea, or infertility whereas men usually have enlarged testicles or hypogonadism.112
We found little literature on changes in glucose metabolism caused by functional gonadotropin tumors, therefore, we will elucidate the independent role of FSH and LH in glucose metabolism respectively.
FSH and Glucose Metabolism
FSH adenomas can induce typical clinical manifestations of excessive hormone levels, which vary by age and sex, such as menstrual dysfunction and spontaneous ovarian hyperstimulation syndrome, enlarged male testicles, and precocious puberty in children of the same sex.
FSH receptor (FSHR) is a class A G-protein-coupled receptor rich in leucine repeats belonging to the glycoprotein hormone receptor (GPHRs) subfamily. GPHRs consist of FSHR and LH/choriogonadotropin receptor (LH/CGR). FSH binds to and activates FSHR on the surface of female ovarian granulosa cells and male testicular Sertoli cells, leading to folliculogenesis and spermatogenesis, respectively.114 Extragonadal tissues, including fat tissues,115 the biliary epithelium,116 liver tissues,117 and bone118 also express functional FSHR, which may be associated with glucose metabolism.
Animal experiments show that FSH induces gluconeogenesis to increase blood glucose levels. In mice, FSH enhances gluconeogenesis through G-protein-coupled receptor kinase 2 (GRK2) in the livers.119 Moreover, increasing evidence indicates that FSH levels are positively correlated with the homeostasis model of assessment-insulin resistance (HOMA-IR) and glucose.120 However, data from clinical studies also showed that FSH was negatively correlated with HOMA-IR during the menopausal transition. In contrast to previous animal studies, a cross-sectional study suggested that FSH is a protective factor.121,122 Women with premenopausal polycystic ovary syndrome (PCOS), who have lower FSH levels,123 are more likely to suffer from insulin resistance and diabetes,122 supporting the notion that FSH is a protective factor. Consistently, a case-control study has also reported the same conclusion.124
Although FSH is involved in various mechanisms of glucose metabolism, its effect on glucose metabolism is controversial in animal experiments and clinical trials, and its role requires further exploration.
LH and Glucose Metabolism
LH is produced and released by the anterior pituitary gland and plays an important role in female follicular growth stimulation, oocyte maturation, ovulation, and estrogen production.125 Androgen hypersecretion in PCOS is mainly due to LH hypersecretion and can result in insulin resistance and diabetes in women with PCOS.126,127 LH hypersecretion leads to increased synthesis and secretion of androgens, interfering with the insulin signaling pathway (IRS-1-PI3K-Akt-Glut) in peripheral tissues.128 It is mainly manifested as significantly increased serine phosphorylation of insulin receptor substrate-1 (IRS-1) stimulated by insulin (p-IRS-1 S636/639) and significantly decreased phosphorylation of insulin-stimulated Akt (p-AKT S473), as well as downregulated glucose transport genes including glucose transporter 2 (GLUT2) and GLUT-4. LH increases blood glucose levels via the insulin resistance mechanism described above.
In addition to mediating insulin resistance via the above pathway, some studies have shown that androgens are closely related to inflammatory factors.129,130 Chronic inflammation is also an important mechanism of insulin resistance. A recent study has reported elevated interleukin 6 (IL-6) levels in PCOS due to chronic inflammation.131 IL-6 can promote androgen receptors, leading to insulin resistance.132 In PCOS,133 IL-6 can also mediate the JAK2/STAT3 signaling pathway whose activation can stimulate the expression of miR-21 and miR-155 to downregulate the expression of peroxisome proliferator-activated receptor γ(PPAR-γ) and GLUT4, thereby indirectly causing insulin resistance.131 In addition to IL-6, the levels of tumor necrosis factor-α (TNF-α), IL-1α, and other inflammatory cytokines are significantly increased whereas IL-4, IL-10, and other anti-inflammatory cytokines are significantly decreased in women with PCOS,128 which can further lead to insulin resistance (Figure 1).
Collectivize, most studies have shown that LH causes insulin resistance through hyperandrogenemia and inflammation, which increase blood glucose levels. Although the exact mechanism by which excessive levels of androgens cause inflammatory states remains unclear, we speculate that LH has a glycemic effect.
TSH and Glucose Metabolism
Pituitary thyrotropin-secreting tumors (TSH tumors) are characterized by the spontaneous secretion of TSH, which is not affected by the negative feedback of thyroid hormones. Oversecretion of thyroxine (T4) and triiodothyronine (T3) due to continued overstimulation of TSH is classified as “central hyperthyroidism”.134 The clinical presentation of patients with TSH tumors is similar to the signs and symptoms of hyperthyroidism, often accompanied by compression of the optic chiasm and pituitary cells, resulting in visual defect or loss and impaired anterior pituitary. However, some patients with untreated TSH tumors have no symptoms.134,135
Some studies have shed new light on the relationship between excess TSH, hepatic gluconeogenesis, and insulin resistance. TSH inhibits cAMP-regulated transcriptional coactivators 2 (CRTC2) phosphorylation and upregulates p-CREB (cAMP-response element binding protein) by activating the TSHR/cAMP/ protein kinase A (PKA) pathway. CRTC2 then enters the nucleus to bind p-CREB, resulting in increased PEPCK and G6P expression, which can promote hepatic gluconeogenesis.136 A linear regression analysis has suggested that TSH is also independently and positively associated with HOMA-IR.137 Moreover, recent in vitro studies in mice have reported that TSH stimulates nuclear factor-kappa B (NF-κB) DNA-binding activity via the CAMP-PKA-dependent pathway to increase TNF-α transcriptional activity.138,139 The possible mechanisms by which TNF-α impairs insulin signal transduction include downregulating the expression of IRS-1, inhibiting tyrosine phosphorylation of IRS-1, and increasing serine/threonine phosphorylation of IRS-1. Reduced tyrosine phosphorylation and downregulation of IRS-1 expression inhibit PI3K activity, causing insulin resistance and downregulation of GLUT4 expression.140 In summary, the molecular mechanism of TSH-induced glucose metabolism disorders are mediated by gluconeogenesis and insulin resistance (Figure 3).
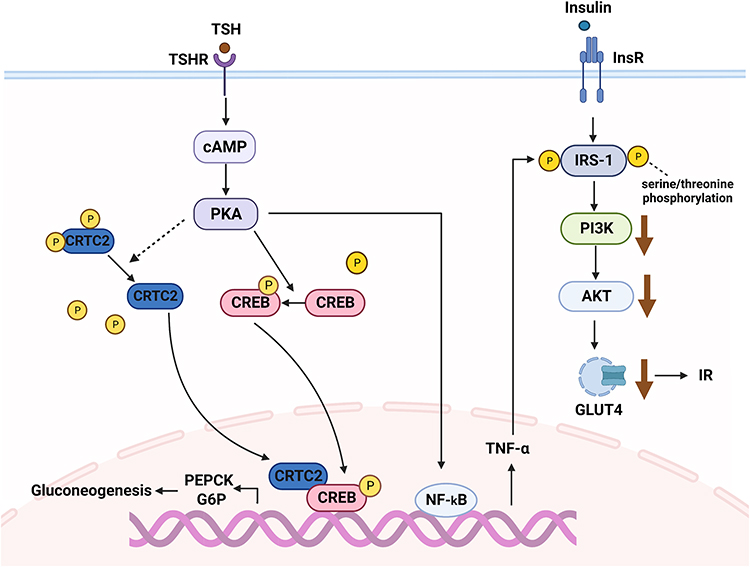 |
Figure 3 The regulatory mechanism of thyroid stimulating hormone (TSH) on glucose metabolism. TSH induces gluconeogenesis and insulin resistance through the TSHR/cAMP/PKA pathway. Abbreviations: CRTC2, cAMP-regulated transcriptional coactivators 2; cAMP, cyclic adenosine monophosphate; CREB, cAMP-response element binding protein; PEPCK, phosphoenolpyruvate carboxykinase; G6P, glucose-6-phosphatase; PKA, protein kinase A; TSHR, thyroid stimulating hormone receptor; IRS-1, insulin receptor substrate 1; PI3K, phosphoinositide 3-kinase. |
In addition to TSH, peripheral excessive thyroid hormone levels also influence blood glucose. Under a physiological state, thyroid hormones affect glucose metabolism by hindering insulin activity and promoting hepatic gluconeogenesis and glycogenolysis. In contrast, thyroid hormones can also upregulate the gene expression of phosphoglycerate kinase and GLUT-4 to promote glycolysis. They can enhance glucose processing and use in peripheral tissues via a synergistic action with insulin.141 Therefore, under a physiological state, thyroid hormones have a dual effect on glucose metabolism. However, patients with hyperthyroidism exhibit elevated blood glucose levels. A large clinical trial suggested that impaired glucose tolerance is mainly the result of insulin resistance in the liver of patients with hyperthyroidism.142 Mechanistically, increased blood glucose levels may be caused by altered transcription and translation of genes related to gluconeogenesis and glycogen metabolism143 and increased GLUT2 expression on the liver cell membrane.141 In adipose tissues, thyroid hormones accelerate insulin degradation by promoting lipolysis and increasing the concentration of FFA.144 Moreover, severe thyrotoxicosis can irreversibly destroy pancreatic tissues, thereby disrupting glucose homeostasis.145
In conclusion, we summarized various pathophysiological mechanisms of TSH tumors in regulating glucose metabolism, mainly from the perspective of TSH and thyroid hormones, which may provide a therapeutic strategy for treating hyperglycemic TSH tumors.
MSH and Glucose Metabolism
Three types of MSH (α-, β-, and γ-, MSH) are secreted by the adenohypophysis. These peptide hormones are derived from proopiomelanocortin (POMC). MSH stimulates the conversion of tyrosine into melanin within melanocytes, leading to hyperpigmentation. In addition, owing to the wide distribution of its receptors, MSH also has a variety of biological activities, including energy homeostasis, food intake, and inflammatory cytokine release.146
Among the receptors of MSH, melanocortin 3 receptor (MC3-R)147 and melanocortin 4 receptor (MC4-R) are involved in energy metabolism, and MC4-R is highly involved in food intake and energy expenditure.148 Moreover, α-MSH can bind to MC4R to reduce insulin release and increase insulin sensitivity.149 The absence of or mutations in the POMC system can lead to the development of obesity and diabetes in animals and humans. In mice, α-MSH deficiency leads to the development of type 2 diabetes.150 In humans, mutations in β-MSH can also lead to overeating and obesity,151 which can lead to the accumulation of adipose tissue in the body. Adipose tissue promotes insulin resistance and type 2 diabetes by releasing FFA, inflammatory cytokines, and other factors.152 Inflammatory factors, such as TNF-α and IL-6, play an important role in insulin resistance. A previous study has shown that α-MSH inhibits the transcription of pro-inflammatory cytokines, such as IL-6 and TNF-α, by blocking the nuclear translocation of NF-κB.153 Therefore, α-MSH can increase insulin sensitivity by downregulating inflammatory factors in the adipose tissues of obese mice, alleviating the effects of obesity (Figure 4).
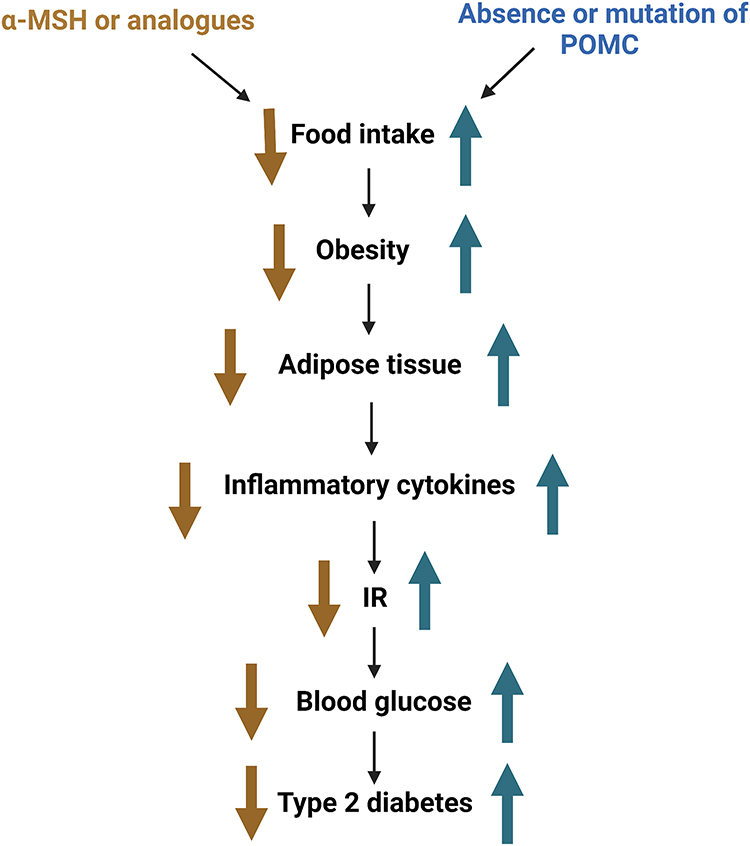 |
Figure 4 Relationship between the melanocortin system and diabetes. Abbreviation: POMC, proopiomelanocortin. |
In conclusion, the melanocortin system can suppress appetite and reduce body weight, which in turn can reduce blood glucose and lipid levels. These changes can impede the progression of type 2 diabetes and may act as therapeutic targets. These previous studies suggest that α-MSH or its analogs may be useful for the treatment of obesity and type 2 diabetes. However, inevitably, there are certain side effects, such as effects on the cardiovascular system.154 Therefore, follow-up studies are needed to comprehensively determine the components and effects of this system and their potential as therapeutic targets.
Prospects of Treatment
Excessive secretion of seven hormones in pituitary tumors plays an important role in the regulation of glucose metabolism. In patients with pituitary tumors, diabetes increases the mortality rate and affects the choice of drugs for the treatment of these tumors. Several drugs have been found to affect glucose metabolism. In the first-line treatment of acromegaly, first-generation long-acting somatostatin analogs, such as octreotide, inhibit insulin secretion, which may potentially affect glucose homeostasis in patients with acromegaly.155 However, a meta-analysis has shown that the change in glycated hemoglobin level during treatment with octreotide class is ≤0.5%,156 indicating that the effect of octreotide on glucose metabolism is modest. Long-acting pasireotide, a second-generation somatostatin analog, is associated with frequent complications, such as hyperglycemia and diabetes, compared to first-generation somatostatin analogs.157 The incidence of hyperglycemic events in the pasireotide group (28.7%) was significantly higher than that in the octreotide group (8.3%).158 Second-line treatment for acromegaly involves GH receptor antagonists such as pegvisomant. Compared with octreotide, pegvisomant has a more favorable effect on glucose homeostasis, especially in reducing fasting blood glucose levels, improving glucose tolerance, and increasing insulin sensitivity.159,160 In patients with prolactinoma, dopamine agonists, including bromocriptine (BRC) and cabergoline, are the standard treatment of choice.161,162 The use of both agents results in significant improvements in blood glucose levels, regardless of the presence or absence of hyperprolactinemia.163 In particular, rapidly released BRC can induce improvements in glucose tolerance and weight loss,164 and BRC-quick release has been approved as a complementary therapy for type 2 diabetes mellitus in the United States.165 In patients with CD, the first choice of treatment is surgical resection of pituitary adenoma, however, some patients often require drug treatment for various reasons. Drugs targeting pituitary corticotropin adenomas should inhibit tumor growth and reduce ACTH secretion. However, only pasireotide is currently approved for patients with failed or inoperable surgery.166 Pasireotide can not only treat acromegaly but also inhibit ACTH synthesis and release. However, pasireotide also inhibits insulin secretion, thereby affecting glucose homeostasis. Moreover, six percent of patients discontinue treatment because of hyperglycemia.167 Therefore, new drugs are needed to treat CD.
In this review, we summarized the pharmacological treatment of several common pituitary adenomas and their effects on glucose metabolism. Therefore, accurate metabolic assessment and monitoring of blood glucose levels before and after treatment should be performed when necessary. Interestingly, most pituitary adenomas show improvement in glucose metabolism after treatment and do not require additional hypoglycemic therapy. However, hypoglycemic therapy is needed if the blood glucose level of some patients is poorly controlled or if it is still not corrected after treatment for pituitary tumors. In general, hypoglycemic treatment is similar to that observed in patients with diabetes. However, attention should be paid to the effects of some hypoglycemic drugs on primary diseases and the interactions between drugs. In conclusion, we need to balance benefits and risks to determine the best treatment for patients.
Conclusions and Prospects
In summary, this study reviews the effects of seven complex pituitary hormones on glucose metabolism via different mechanisms. Our review indicates that excessive hormone levels would lead to an imbalance of blood glucose homeostasis, and abnormal blood glucose levels might be the early manifestation of some diseases, which provides clues for the early diagnosis and treatment of related diseases. New drugs based on the above mechanism need to be explored further. Compared to other reviews, the present review is more extensive and comprehensive. However, the roles of some hormones remain controversial and require further study.
Abbreviations
PRL, prolactin; GH, growth hormone; ACTH, adrenocorticotropic hormone; TSH, thyroid stimulating hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; MSH, melanocyte-stimulating hormone; TIDA, tuberoinfundibular dopamine neurons; PRLRs, prolactin receptors; HOMA, homeostatic model assessment; ISI, insulin sensitivity index; BMI, body mass index; IRS-1, insulin receptor substrate-1; PI3K, phosphoinositide 3-kinase; IGF-1, insulin-like growth factor 1; CS, Cushing’s syndrome; CD, Cushing’s disease; ACT, cortisol-secreting adrenocortical tumor; GC, glucocorticoid; FOXO1, forkhead box protein O1; IRS1, insulin substrate 1; PI3Kr1, phosphoinositide-3-kinase regulatory subunit 1; AKT, protein kinase B; GLUT4, glucose transporter 4; GLUT2, glucose transporter 2; NEFAs, non-esterified fatty acids; FFA, free fatty acids; ATGL, adipose triglyceride lipase; GPRC6A, G protein-coupled receptor, family C, group 6, member A; GLP-1, glucagon-like peptide-1; CAMP, cyclic adenosine monophosphate; IGT, impaired glucose tolerance; GR, glucocorticoid receptor; UPR, unfolded protein response; NPY, nerve peptide Y; AgRP, agouti-related protein; JAK, Janus tyrosine kinase; STAT, signal transducers and activators of transcription; FSHR, follicle-stimulating hormone receptor; GPHRs, glycoprotein hormone receptors; CGR, choriogonadotropin receptor 2; GRK2, G-protein-coupled receptor kinase 2; PCOS, polycystic ovary syndrome; IL-6, interleukin 6; PPAR-γ, peroxisome proliferator activated receptor γ; TNF-α, tumor necrosis factor-α; IL-18, interleukin 18; T4, thyroxine; T3, triiodothyronine; CRTC2, cAMP-regulated transcriptional coactivators 2; CREB, cAMP-response element binding protein; PEPCK, phosphoenolpyruvate carboxykinase; G6P, glucose-6-phosphatase; PKA, protein kinase A; NF-κB, nuclear factor-kappa B; TyG, triglyceride glucose; POMC, proopiomelanocortin; MC3-R, melanocortin 3 receptor; MC4-R, melanocortin 4 receptor; MKP-3, mitogen-activated protein kinase phosphatase-3; PGC1α, peroxisome proliferator-activated receptor-γ coactivator-1α; BRC, bromocryptotine.
Acknowledgments
The coauthors thank the National Natural Science Foundation of China (81973378, 82073909), Research Project Supported by Shanxi Scholarship Council of China (2020-0172) for their support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14(Suppl5):v1–v49. doi:10.1093/neuonc/nos218
2. Pekic S, Stojanovic M, Popovic V. Contemporary issues in the evaluation and management of pituitary adenomas. Minerva Endocrinol. 2015;40(4):307–319.
3. Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol. 2010;72(3):377–382. doi:10.1111/j.1365-2265.2009.03667.x
4. Fontana E, Gaillard R. Epidémiologie des adénomes hypophysaires : étude dans une agglomération urbaine de suisse[Epidemiology of pituitary adenoma: results of the first Swiss study]. Rev Med Suisse. 2009;5(223):2172–2174. French.
5. Gruppetta M, Mercieca C, Vassallo J. Prevalence and incidence of pituitary adenomas: a population based study in Malta. Pituitary. 2013;16(4):545–553. doi:10.1007/s11102-012-0454-0
6. Lake MG, Krook LS, Cruz SV. Pituitary adenomas: an overview. Am Fam Physician. 2013;88(5):319–327.
7. Tjörnstrand A, Gunnarsson K, Evert M, et al. The incidence rate of pituitary adenomas in western Sweden for the period 2001–2011. Eur J Endocrinol. 2014;171(4):519–526. doi:10.1530/eje-14-0144
8. Raappana A, Koivukangas J, Ebeling T, Pirilä T. Incidence of pituitary adenomas in Northern Finland in 1992–2007. J Clin Endocrinol Metab. 2010;95(9):4268–4275. doi:10.1210/jc.2010-0537
9. Agustsson TT, Baldvinsdottir T, Jonasson JG, et al. The epidemiology of pituitary adenomas in Iceland, 1955–2012: a nationwide population-based study. Eur J Endocrinol. 2015;173(5):655–664. doi:10.1530/eje-15-0189
10. Osorio RC, Oh JY, Choudhary N, Lad M, Savastano L, Aghi MK. Pituitary adenomas and cerebrovascular disease: a review on pathophysiology, prevalence, and treatment. Front Endocrinol. 2022;13. doi:10.3389/fendo.2022.1064216
11. Frara S, Maffezzoni F, Mazziotti G, Giustina A. Current and emerging aspects of diabetes mellitus in acromegaly. Trends Endocrinol Metab. 2016;27(7):470–483. doi:10.1016/j.tem.2016.04.014
12. Kleinberg DL, Noel GL, Frantz AG. Galactorrhea: a study of 235 cases, including 48 with pituitary tumors. N Engl J Med. 1977;296(11):589–600. doi:10.1056/nejm197703172961103
13. Koppelman MC, Kurtz DW, Morrish KA, et al. Vertebral body bone mineral content in hyperprolactinemic women. J Clin Endocrinol Metab. 1984;59(6):1050–1053. doi:10.1210/jcem-59-6-1050
14. Mah PM, Webster J. Hyperprolactinemia: etiology, diagnosis, and management. Semin Reprod Med. 2002;20(4):365–374. doi:10.1055/s-2002-36709
15. Poon A, McNeill P, Harper A, O’Day J. Patterns of visual loss associated with pituitary macroadenomas. Aust N Z J Ophthalmol. 1995;23(2):107–115. doi:10.1111/j.1442-9071.1995.tb00138.x
16. Grattan DR. 60 years Of neuroendocrinology: the hypothalamo-prolactin axis. J Endocrinol. 2015;226(2):T101–T122. doi:10.1530/joe-15-0213
17. Auriemma RS, De Alcubierre D, Pirchio R, Pivonello R, Colao A. Glucose abnormalities associated to prolactin secreting pituitary adenomas. Front Endocrinol. 2019;10:327. doi:10.3389/fendo.2019.00327
18. Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19(3):225–268. doi:10.1210/edrv.19.3.0334
19. Bernard V, Young J, Binart N. Prolactin - a pleiotropic factor in health and disease. Nat Rev Endocrinol. 2019;15(6):356–365. doi:10.1038/s41574-019-0194-6
20. Perić B, Kruljac I, Šundalić S, et al. Obesity and hypercholesterolemia in patients with prolactinomas: could DHEA-S and growth hormone be the missing link? Endocr Res. 2016;41(3):200–206. doi:10.3109/07435800.2015.1135444
21. Greenman Y, Tordjman K, Stern N. Increased body weight associated with prolactin secreting pituitary adenomas: weight loss with normalization of prolactin levels. Clin Endocrinol. 1998;48(5):547–553. doi:10.1046/j.1365-2265.1998.00403.x
22. Berinder K, Nyström T, Höybye C, Hall K, Hulting AL. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary. 2011;14(3):199–207. doi:10.1007/s11102-010-0277-9
23. Landgraf R, Landraf-Leurs MM, Weissmann A, Hörl R, von Werder K, Scriba PC. Prolactin: a diabetogenic hormone. Diabetologia. 1977;13(2):99–104. doi:10.1007/bf00745135
24. Serri O, Beauregard H, Rasio E, Hardy J. 21.Decreased sensitivity to insulin in women with microprolactinomas. Fertil Steril. 1986;45(4):572–574.
25. Perez Millan MI, Luque GM, Ramirez MC, et al. 22.Selective disruption of dopamine D2 receptors in pituitary lactotropes increases body weight and adiposity in female mice. Endocrinology. 2014;155(3):829–839. doi:10.1210/en.2013-1707
26. Ratner LD, Stevens G, Bonaventura MM, et al. 23.Hyperprolactinemia induced by hCG leads to metabolic disturbances in female mice. J Endocrinol. 2016;230(1):157–169. doi:10.1530/joe-15-0528
27. Tuzcu A, Yalaki S, Arikan S, Gokalp D, Bahcec M, Tuzcu S. 24.Evaluation of insulin sensitivity in hyperprolactinemic subjects by euglycemic hyperinsulinemic clamp technique. Pituitary. 2009;12(4):330–334. doi:10.1007/s11102-009-0183-1
28. Tuzcu A, Bahceci M, Dursun M, Turgut C, Bahceci S. 25.Insulin sensitivity and hyperprolactinemia. J Endocrinol Invest. 2003;26(4):341–346. doi:10.1007/bf03345182
29. Yavuz D, Deyneli O, Akpinar I, et al. 26. Endothelial function, insulin sensitivity and inflammatory markers in hyperprolactinemic pre-menopausal women. Eur J Endocrinol. 2003;149(3):187–193. doi:10.1530/eje.0.1490187
30. de Oliveira C, de Mattos AB, Biz C, Oyama LM, Ribeiro EB, Do Nascimento CM. 27.High-fat diet and glucocorticoid treatment cause hyperglycemia associated with adiponectin receptor alterations. Lipids Health Dis. 2011;10:11. doi:10.1186/1476-511x-10-11
31. Fruebis J, Tsao TS, Javorschi S, et al. 28.Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98(4):2005–2010. doi:10.1073/pnas.98.4.2005
32. Yamauchi T, Kamon J, Minokoshi Y, et al. 29.Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295. doi:10.1038/nm788
33. de Assunção Alves Rodrigues LF, Campos SM, Miranda PA, et al. Prolactinoma: a condition associated with hypoadiponectinemia. Horm Metab Res. 2012;44(11):832–838. doi:10.1055/s-0032-1321832
34. Carr MC. The emergence of the metabolic syndrome with menopause. J Clin Endocrinol Metab. 2003;88(6):2404–2411. doi:10.1210/jc.2003-030242
35. Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47(7):1175–1187. doi:10.1007/s00125-004-1448-x
36. Hannon AM, Thompson CJ, Sherlock M. Diabetes in patients with acromegaly. Curr Diab Rep. 2017;17(2):8. doi:10.1007/s11892-017-0838-7
37. Vilar L, Vilar CF, Lyra R, Lyra R, Naves LA. Acromegaly: clinical features at diagnosis. Pituitary. 2017;20(1):22–32. doi:10.1007/s11102-016-0772-8
38. Alexopoulou O, Bex M, Kamenicky P, Mvoula AB, Chanson P, Maiter D. Prevalence and risk factors of impaired glucose tolerance and diabetes mellitus at diagnosis of acromegaly: a study in 148 patients. Pituitary. 2014;17(1):81–89. doi:10.1007/s11102-013-0471-7
39. Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev. 2010;31(3):301–342. doi:10.1210/er.2009-0033
40. Elkeles RS, Wright AD, Lowy C, Fraser TR. Serum-insulin in acromegaly. Lancet. 1969;2(7621):615–618. doi:10.1016/s0140-6736(69)90327-4
41. Javed A, Balagopal PB, Vella A, et al. Association between thyrotropin levels and insulin sensitivity in euthyroid obese adolescents. Thyroid. 2015;25(5):478–484. doi:10.1089/thy.2015.0005
42. Hansen I, Tsalikian E, Beaufrere B, Gerich J, Haymond M, Rizza R. Insulin resistance in acromegaly: defects in both hepatic and extrahepatic insulin action. Am J Physiol. 1986;250(3 Pt 1):E269–E273. doi:10.1152/ajpendo.1986.250.3.E269
43. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi:10.1038/nrm1837
44. Del Rincon JP, Iida K, Gaylinn BD, et al. Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes. 2007;56(6):1638–1646. doi:10.2337/db06-0299
45. Pedersen MH, Svart MV, Lebeck J, et al. Substrate metabolism and insulin sensitivity during fasting in obese human subjects: impact of GH blockade. J Clin Endocrinol Metab. 2017;102(4):1340–1349. doi:10.1210/jc.2016-3835
46. Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30(2):152–177. doi:10.1210/er.2008-0027
47. Hjelholt AJ, Charidemou E, Griffin JL, et al. Insulin resistance induced by growth hormone is linked to lipolysis and associated with suppressed pyruvate dehydrogenase activity in skeletal muscle: a 2 × 2 factorial, randomised, crossover study in human individuals. Diabetologia. 2020;63(12):2641–2653. doi:10.1007/s00125-020-05262-w
48. Al-Massadi O, Parini P, Ferno J, Luquet S, Quinones M. Metabolic actions of the growth hormone-insulin growth factor-1 axis and its interaction with the central nervous system. Rev Endocr Metab Disord. 2022;23(5):919–930. doi:10.1007/s11154-022-09732-x
49. Rogers SA, Miller SB, Hammerman MR. Growth hormone stimulates IGF I gene expression in isolated rat renal collecting duct. Am J Physiol. 1990;259(3 Pt 2):F474–F479. doi:10.1152/ajprenal.1990.259.3.F474
50. O’Connell T, Clemmons DR. IGF-I/IGF-binding protein-3 combination improves insulin resistance by GH-dependent and independent mechanisms. J Clin Endocrinol Metab. 2002;87(9):4356–4360. doi:10.1210/jc.2002-020343
51. Moses AC, Young SC, Morrow LA, O’Brien M, Clemmons DR. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes. 1996;45(1):91–100. doi:10.2337/diab.45.1.91
52. Boulware SD, Tamborlane WV, Rennert NJ, Gesundheit N, Sherwin RS. Comparison of the metabolic effects of recombinant human insulin-like growth factor-I and insulin. Dose-response relationships in healthy young and middle-aged adults. J Clin Invest. 1994;93(3):1131–1139. doi:10.1172/jci117065
53. Elahi D, McAloon-Dyke M, Fukagawa NK, et al. Effects of recombinant human IGF-I on glucose and leucine kinetics in men. Am J Physiol. 1993;265(6 Pt 1):E831–E838. doi:10.1152/ajpendo.1993.265.6.E831
54. Russell-Jones DL, Bates AT, Umpleby AM, et al. A comparison of the effects of IGF-I and insulin on glucose metabolism, fat metabolism and the cardiovascular system in normal human volunteers. Eur J Clin Invest. 1995;25(6):403–411. doi:10.1111/j.1365-2362.1995.tb01721.x
55. Bondy C, Werner H, Roberts CT, LeRoith D. Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: comparison with insulin-like growth factors I and II. Neuroscience. 1992;46(4):909–923. doi:10.1016/0306-4522(92)90193-6
56. Muzumdar RH, Ma X, Fishman S, et al. Central and opposing effects of IGF-I and IGF-binding protein-3 on systemic insulin action. Diabetes. 2006;55(10):2788–2796. doi:10.2337/db06-0318
57. Fujita S, Honda K, Yamaguchi M, Fukuzo S, Saneyasu T, Kamisoyama H. Role of insulin-like growth factor-1 in the central regulation of feeding behavior in chicks. J Poult Sci. 2019;56(4):270–276. doi:10.2141/jpsa.0180127
58. Choi YM, Kim MK, Kwak MK, Kim D, Hong EG. Association between thyroid hormones and insulin resistance indices based on the Korean National Health and Nutrition Examination Survey. Sci Rep. 2021;11(1):21738. doi:10.1038/s41598-021-01101-z
59. Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013;16(3):294–302. doi:10.1007/s11102-012-0420-x
60. Sanders K, Kooistra HS, Galac S. Treating canine Cushing’s syndrome: current options and future prospects. Vet J. 2018;241:42–51. doi:10.1016/j.tvjl.2018.09.014
61. Barbot M, Ceccato F, Scaroni C. Diabetes mellitus secondary to Cushing’s disease. Front Endocrinol. 2018;9:284. doi:10.3389/fendo.2018.00284
62. Scaroni C, Zilio M, Foti M, Boscaro M. Glucose Metabolism abnormalities in Cushing syndrome: from molecular basis to clinical management. Endocr Rev. 2017;38(3):189–219. doi:10.1210/er.2016-1105
63. Tanabe H, Yokota K, Shibata N, Satoh T, Watari J, Kohgo Y. Alcohol consumption as a major risk factor in the development of early esophageal cancer in patients with head and neck cancer. Intern Med. 2001;40(8):692–696. doi:10.2169/internalmedicine.40.692
64. Agzarian J, Visscher SL, Knight AW, et al. The cost burden of clinically significant esophageal anastomotic leaks-a steep price to pay. J Thorac Cardiovasc Surg. 2019;157(5):2086–2092. doi:10.1016/j.jtcvs.2018.10.137
65. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572–587. doi:10.1038/nrendo.2017.80
66. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126(1):12–22. doi:10.1172/jci77812
67. Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92(4):2065–2072. doi:10.1172/jci116803
68. Chen TC, Kuo T, Dandan M, et al. The role of striated muscle Pik3r1 in glucose and protein metabolism following chronic glucocorticoid exposure. J Biol Chem. 2021;296:100395. doi:10.1016/j.jbc.2021.100395
69. Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47(1):3–6. doi:10.1016/s0026-0495(98)90184-6
70. Gupta A, Gupta Y. Glucocorticoid-induced myopathy: pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17(5):913–916. doi:10.4103/2230-8210.117215
71. Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. J Endocrinol. 2008;197(1):1–10. doi:10.1677/joe-07-0606
72. Löfberg E, Gutierrez A, Wernerman J, et al. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur J Clin Invest. 2002;32(5):345–353. doi:10.1046/j.1365-2362.2002.00993.x
73. Short KR, Bigelow ML, Nair KS. Short-term prednisone use antagonizes insulin’s anabolic effect on muscle protein and glucose metabolism in young healthy people. Am J Physiol Endocrinol Metab. 2009;297(6):E1260–E1268. doi:10.1152/ajpendo.00345.2009
74. Krebs M, Krssak M, Bernroider E, et al. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes. 2002;51(3):599–605. doi:10.2337/diabetes.51.3.599
75. Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am. 2014;43(1):75–102. doi:10.1016/j.ecl.2013.10.005
76. Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest. 2002;32(Suppl 3):14–23. doi:10.1046/j.1365-2362.32.s3.3.x
77. Peckett AJ, Wright DC, Riddell MC. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism. 2011;60(11):1500–1510. doi:10.1016/j.metabol.2011.06.012
78. Fain JN. Effect of dibutyryl-3’,5’-AMP, theophylline and norepinephrine on lipolytic action of growth hormone and glucocorticoid in white fat cells. Endocrinology. 1968;82(4):825–830. doi:10.1210/endo-82-4-825
79. Ramshanker N, Jessen N, Voss TS, et al. Effects of short-term prednisolone treatment on indices of lipolysis and lipase signaling in abdominal adipose tissue in healthy humans. Metabolism. 2019;99:1–10. doi:10.1016/j.metabol.2019.06.013
80. Bilotta FL, Arcidiacono B, Messineo S, et al. Insulin and osteocalcin: further evidence for a mutual cross-talk. Endocrine. 2018;59(3):622–632. doi:10.1007/s12020-017-1396-0
81. Chen SM, Peng YJ, Wang CC, Su SL, Salter DM, Lee HS. Dexamethasone down-regulates osteocalcin in bone cells through leptin pathway. Int J Med Sci. 2018;15(5):507–516. doi:10.7150/ijms.21881
82. Lee NK, Sowa H, Hinoi E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi:10.1016/j.cell.2007.05.047
83. Mizokami A, Kawakubo-Yasukochi T, Hirata M. Osteocalcin and its endocrine functions. Biochem Pharmacol. 2017;132:1–8. doi:10.1016/j.bcp.2017.02.001
84. Mizokami A, Yasutake Y, Gao J, et al. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS One. 2013;8(2):e57375. doi:10.1371/journal.pone.0057375
85. Mizokami A, Mukai S, Gao J, et al. GLP-1 signaling is required for improvement of glucose tolerance by osteocalcin. J Endocrinol. 2020;244(2):285–296. doi:10.1530/joe-19-0288
86. Brennan-Speranza TC, Henneicke H, Gasparini SJ, et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest. 2012;122(11):4172–4189. doi:10.1172/jci63377
87. Diakogiannaki E, Gribble FM, Reimann F. Nutrient detection by incretin hormone secreting cells. Physiol Behav. 2012;106(3):387–393. doi:10.1016/j.physbeh.2011.12.001
88. Meier JJ, Nauck MA. Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes Metab Res Rev. 2005;21(2):91–117. doi:10.1002/dmrr.538
89. Kappe C, Fransson L, Wolbert P, Ortsäter H. Glucocorticoids suppress GLP-1 secretion: possible contribution to their diabetogenic effects. Clin Sci. 2015;129(5):405–414. doi:10.1042/cs20140719
90. Jensen DH, Aaboe K, Henriksen JE, et al. Steroid-induced insulin resistance and impaired glucose tolerance are both associated with a progressive decline of incretin effect in first-degree relatives of patients with type 2 diabetes. Diabetologia. 2012;55(5):1406–1416. doi:10.1007/s00125-012-2459-7
91. Eriksen M, Jensen DH, Tribler S, Holst JJ, Madsbad S, Krarup T. Reduction of insulinotropic properties of GLP-1 and GIP after glucocorticoid-induced insulin resistance. Diabetologia. 2015;58(5):920–928. doi:10.1007/s00125-015-3522-y
92. Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia. 2012;55(4):1048–1057. doi:10.1007/s00125-011-2422-z
93. Linssen MM, van Raalte DH, Toonen EJ, et al. Prednisolone-induced beta cell dysfunction is associated with impaired endoplasmic reticulum homeostasis in INS-1E cells. Cell Signal. 2011;23(11):1708–1715. doi:10.1016/j.cellsig.2011.06.002
94. Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab. 2011;22(12):499–506. doi:10.1016/j.tem.2011.09.001
95. Rafacho A, Gonçalves-Neto LM, Santos-Silva JC, et al. Pancreatic alpha-cell dysfunction contributes to the disruption of glucose homeostasis and compensatory insulin hypersecretion in glucocorticoid-treated rats. PLoS One. 2014;9(4):e93531. doi:10.1371/journal.pone.0093531
96. Marco J, Calle C, Hedo JA, Villanueva ML. Enhanced glucagon secretion by pancreatic islets from prednisolone-treated mice. Diabetologia. 1976;12(4):307–311. doi:10.1007/bf00420973
97. van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest. 2009;39(2):81–93. doi:10.1111/j.1365-2362.2008.02067.x
98. Derrou S, Bouziane T, Salhi H, El Ouahabi H. Pheochromocytoma and glucoregulation disorders. Ann Afr Med. 2021;20(1):42–45. doi:10.4103/aam.aam_13_20
99. Hamaji M. Pancreatic alpha- and beta-cell function in pheochromocytoma. J Clin Endocrinol Metab. 1979;49(3):322–325. doi:10.1210/jcem-49-3-322
100. Komada H, Hirota Y, So A, et al. Insulin secretion and insulin sensitivity before and after surgical treatment of pheochromocytoma or paraganglioma. J Clin Endocrinol Metab. 2017;102(9):3400–3405. doi:10.1210/jc.2017-00357
101. Wiesner TD, Blüher M, Windgassen M, Paschke R. Improvement of insulin sensitivity after adrenalectomy in patients with pheochromocytoma. J Clin Endocrinol Metab. 2003;88(8):3632–3636. doi:10.1210/jc.2003-030000
102. Chiasson JL, Shikama H, Chu DT, Exton JH. Inhibitory effect of epinephrine on insulin-stimulated glucose uptake by rat skeletal muscle. J Clin Invest. 1981;68(3):706–713. doi:10.1172/jci110306
103. Harbeck B, Danneberg S, Rahvar AH, et al. Exploring the impact of short- and long-term hydrocortisone replacement on cognitive function, quality of life and catecholamine secretion: a pilot study. Appl Psychophysiol Biofeedback. 2016;41(3):341–347. doi:10.1007/s10484-016-9338-9
104. Zilberter T. Appetite, reward, and obesity: the causes and consequences of eating behaviors. Front Psychol. 2015;6:411. doi:10.3389/fpsyg.2015.00411
105. Perry RJ, Resch JM, Douglass AM, et al. Leptin’s hunger-suppressing effects are mediated by the hypothalamic-pituitary-adrenocortical axis in rodents. Proc Natl Acad Sci U S A. 2019;116(27):13670–13679. doi:10.1073/pnas.1901795116
106. Shimizu H, Arima H, Watanabe M, et al. Glucocorticoids increase neuropeptide Y and agouti-related peptide gene expression via adenosine monophosphate-activated protein kinase signaling in the arcuate nucleus of rats. Endocrinology. 2008;149(9):4544–4553. doi:10.1210/en.2008-0229
107. Xu J, Bartolome CL, Low CS, et al. Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature. 2018;556(7702):505–509. doi:10.1038/s41586-018-0049-7
108. Ishida-Takahashi R, Uotani S, Abe T, et al. Rapid inhibition of leptin signaling by glucocorticoids in vitro and in vivo. J Biol Chem. 2004;279(19):19658–19664. doi:10.1074/jbc.M310864200
109. Giordano C, Guarnotta V, Pivonello R, et al. Is diabetes in Cushing’s syndrome only a consequence of hypercortisolism? Eur J Endocrinol. 2014;170(2):311–319. doi:10.1530/eje-13-0754
110. Yamada S, Ohyama K, Taguchi M, et al. A study of the correlation between morphological findings and biological activities in clinically nonfunctioning pituitary adenomas. Neurosurgery. 2007;61(3):580–584; discussion 584–585. doi:10.1227/01.Neu.0000290906.53685.79
111. Caretto A, Lanzi R, Piani C, Molgora M, Mortini P, Losa M. Ovarian hyperstimulation syndrome due to follicle-stimulating hormone-secreting pituitary adenomas. Pituitary. 2017;20(5):553–560. doi:10.1007/s11102-017-0817-7
112. Ntali G, Capatina C, Grossman A, Karavitaki N. Clinical review: functioning gonadotroph adenomas. J Clin Endocrinol Metab. 2014;99(12):4423–4433. doi:10.1210/jc.2014-2362
113. Djerassi A, Coutifaris C, West VA, et al. Gonadotroph adenoma in a premenopausal woman secreting follicle-stimulating hormone and causing ovarian hyperstimulation. J Clin Endocrinol Metab. 1995;80(2):591–594. doi:10.1210/jcem.80.2.7852525
114. Banerjee AA, Mahale SD. Role of the extracellular and intracellular loops of follicle-stimulating hormone receptor in its function. Front Endocrinol. 2015;6:110. doi:10.3389/fendo.2015.00110
115. Liu XM, Chan HC, Ding GL, et al. FSH regulates fat accumulation and redistribution in aging through the Gαi/Ca(2+)/CREB pathway. Aging Cell. 2015;14(3):409–420. doi:10.1111/acel.12331
116. Onori P, Mancinelli R, Franchitto A, et al. Role of follicle-stimulating hormone on biliary cyst growth in autosomal dominant polycystic kidney disease. Liver Int. 2013;33(6):914–925. doi:10.1111/liv.12177
117. Song Y, Wang ES, Xing LL, et al. Follicle-stimulating hormone induces postmenopausal dyslipidemia through inhibiting hepatic cholesterol metabolism. J Clin Endocrinol Metab. 2016;101(1):254–263. doi:10.1210/jc.2015-2724
118. Sun L, Peng Y, Sharrow AC, et al. FSH directly regulates bone mass. Cell. 2006;125(2):247–260. doi:10.1016/j.cell.2006.01.051
119. Qi X, Guo Y, Song Y, et al. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia. 2018;61(5):1180–1192. doi:10.1007/s00125-018-4562-x
120. Kulaksizoglu M, Ipekci SH, Kebapcilar L, et al. Risk factors for diabetes mellitus in women with primary ovarian insufficiency. Biol Trace Elem Res. 2013;154(3):313–320. doi:10.1007/s12011-013-9738-0
121. Wang X, Zhang H, Chen Y, Du Y, Jin X, Zhang Z. Follicle stimulating hormone, its association with glucose and lipid metabolism during the menopausal transition. J Obstet Gynaecol Res. 2020;46(8):1419–1424. doi:10.1111/jog.14297
122. Bertone-Johnson ER, Virtanen JK, Niskanen L, et al. Association of follicle-stimulating hormone levels and risk of type 2 diabetes in older postmenopausal women. Menopause. 2017;24(7):796–802. doi:10.1097/gme.0000000000000834
123. Schmidt J, Brännström M, Landin-Wilhelmsen K, Dahlgren E. Reproductive hormone levels and anthropometry in postmenopausal women with polycystic ovary syndrome (PCOS): a 21-year follow-up study of women diagnosed with PCOS around 50 years ago and their age-matched controls. J Clin Endocrinol Metab. 2011;96(7):2178–2185. doi:10.1210/jc.2010-2959
124. Park SK, Harlow SD, Zheng H, et al. Association between changes in oestradiol and follicle-stimulating hormone levels during the menopausal transition and risk of diabetes. Diabet Med. 2017;34(4):531–538. doi:10.1111/dme.13301
125. Young KA, Chaffin CL, Molskness TA, Stouffer RL. Controlled ovulation of the dominant follicle: a critical role for LH in the late follicular phase of the menstrual cycle. Hum Reprod. 2003;18(11):2257–2263. doi:10.1093/humrep/deg467
126. Krishnan A, Muthusami S. Hormonal alterations in PCOS and its influence on bone metabolism. J Endocrinol. 2017;232(2):R99–r113. doi:10.1530/joe-16-0405
127. Shorakae S, Ranasinha S, Abell S, et al. Inter-related effects of insulin resistance, hyperandrogenism, sympathetic dysfunction and chronic inflammation in PCOS. Clin Endocrinol. 2018;89(5):628–633. doi:10.1111/cen.13808
128. Li H, Zhang G, Guo Y, et al. Autoimmune activation of the GnRH receptor induces insulin resistance independent of obesity in a female rat model. Physiol Rep. 2021;8(24):e14672. doi:10.14814/phy2.14672
129. Eisner JR, Dumesic DA, Kemnitz JW, Colman RJ, Abbott DH. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003;11(2):279–286. doi:10.1038/oby.2003.42
130. Solano ME, Sander VA, Ho H, Motta AB, Arck PC. Systemic inflammation, cellular influx and up-regulation of ovarian VCAM-1 expression in a mouse model of polycystic ovary syndrome (PCOS). J Reprod Immunol. 2011;92(1–2):33–44. doi:10.1016/j.jri.2011.09.003
131. Zhang Y, Li C, Zhang W, Zheng X, Chen X. Decreased Insulin Resistance by Myo-Inositol Is Associated with Suppressed Interleukin 6/Phospho-STAT3 Signaling in a Rat Polycystic Ovary Syndrome Model. J Med Food. 2020;23(4):375–387. doi:10.1089/jmf.2019.4580
132. Banaś M, Olszanecka-Glinianowicz M, Zahorska-Markiewicz B. Rola czynnika martwicy nowotworów i interleukiny-6 w zespole policystycznych jajników[The role of tumor necrosis factor and interleukin-6 in polycystic ovary syndrome]. Pol Merkur Lekarski. 2006;21(125):489–491. Polish.
133. Zhou Y, Lv L, Liu Q, Song J. Total flavonoids extracted from Nervilia Fordii function in polycystic ovary syndrome through IL-6 mediated JAK2/STAT3 signaling pathway. Biosci Rep. 2019;39(1). doi:10.1042/bsr20181380
134. Beck-Peccoz P, Giavoli C, Lania AA. 2019 update on TSH-secreting pituitary adenomas. J Endocrinol Invest. 2019;42(12):1401–1406. doi:10.1007/s40618-019-01066-x
135. Yamada S, Fukuhara N, Horiguchi K, et al. Clinicopathological characteristics and therapeutic outcomes in thyrotropin-secreting pituitary adenomas: a single-center study of 90 cases. J Neurosurg. 2014;121(6):1462–1473. doi:10.3171/2014.7.Jns1471
136. Li Y, Wang L, Zhou L, et al. Thyroid stimulating hormone increases hepatic gluconeogenesis via CRTC2. Mol Cell Endocrinol. 2017;446:70–80. doi:10.1016/j.mce.2017.02.015
137. Zhu P, Liu X, Mao X. Thyroid-stimulating hormone levels are positively associated with insulin resistance. Med Sci Monit. 2018;24:342–347. doi:10.12659/msm.905774
138. Zhang YJ, Zhao W, Zhu MY, Tang SS, Zhang H. Thyroid-stimulating hormone induces the secretion of tumor necrosis factor-α from 3T3-L1 adipocytes via a protein kinase A-dependent pathway. Exp Clin Endocrinol Diabetes. 2013;121(8):488–493. doi:10.1055/s-0033-1347266
139. Zhang Y, Feng L. Thyroid-stimulating hormone inhibits insulin receptor substrate-1 expression and tyrosyl phosphorylation in 3T3-L1 adipocytes by increasing NF-κB DNA-binding activity. Dis Markers. 2022;2022:7553670. doi:10.1155/2022/7553670
140. Del Aguila LF, Claffey KP, Kirwan JP. TNF-alpha impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Physiol. 1999;276(5):E849–E855. doi:10.1152/ajpendo.1999.276.5.E849
141. Weinstein SP, O’Boyle E, Fisher M, Haber RS. Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: evidence for hormonal regulation of the hepatic glucose transport system. Endocrinology. 1994;135(2):649–654. doi:10.1210/endo.135.2.8033812
142. Gierach M, Gierach J, Junik R. Insulin resistance and thyroid disorders. Endokrynol Pol. 2014;65(1):70–76. doi:10.5603/ep.2014.0010
143. Feng X, Jiang Y, Meltzer P, Yen PM. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol. 2000;14(7):947–955. doi:10.1210/mend.14.7.0470
144. Dimitriadis G, Baker B, Marsh H, et al. Effect of thyroid hormone excess on action, secretion, and metabolism of insulin in humans. Am J Physiol. 1985;248(5 Pt 1):E593–E601. doi:10.1152/ajpendo.1985.248.5.E593
145. Lenzen S, Kücking H. Inhibition of insulin secretion by L-thyroxine and D-thyroxine treatment in rats under the influence of drugs affecting the adrenergic nervous system. Acta Endocrinol. 1982;100(4):527–533. doi:10.1530/acta.0.1000527
146. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5(9):1066–1070. doi:10.1038/12506
147. Butler AA, Kesterson RA, Khong K, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–3521. doi:10.1210/endo.141.9.7791
148. Hainer V, Aldhoon hainerová I, Kunešová M, Taxová Braunerová R, Zamrazilová H, Bendlová B. Melanocortin pathways: suppressed and stimulated melanocortin-4 receptor (MC4R). Physiol Res. 2020;69(Suppl 2):S245–s254. doi:10.33549/physiolres.934512
149. Girardet C, Butler AA. Neural melanocortin receptors in obesity and related metabolic disorders. Biochim Biophys Acta. 2014;1842(3):482–494. doi:10.1016/j.bbadis.2013.05.004
150. Schneeberger M, Gómez-Valadés AG, Altirriba J, et al. Reduced α-MSH Underlies Hypothalamic ER-Stress-Induced Hepatic Gluconeogenesis. Cell Rep. 2015;12(3):361–370. doi:10.1016/j.celrep.2015.06.041
151. Lee YS, Challis BG, Thompson DA, et al. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab. 2006;3(2):135–140. doi:10.1016/j.cmet.2006.01.006
152. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
153. Lipton JM, Catania A, Ichiyama T. Marshaling the Anti-Inflammatory Influence of the Neuroimmunomodulator alpha-MSH. News Physiol Sci. 2000;15:192–195. doi:10.1152/physiologyonline.2000.15.4.192
154. Hill C, Dunbar JC. The effects of acute and chronic alpha melanocyte stimulating hormone (alphaMSH) on cardiovascular dynamics in conscious rats. Peptides. 2002;23(9):1625–1630. doi:10.1016/s0196-9781(02)00103-1
155. Baldelli R, Battista C, Leonetti F, et al. Glucose homeostasis in acromegaly: effects of long-acting somatostatin analogues treatment. Clin Endocrinol. 2003;59(4):492–499. doi:10.1046/j.1365-2265.2003.01876.x
156. Mazziotti G, Floriani I, Bonadonna S, Torri V, Chanson P, Giustina A. Effects of somatostatin analogs on glucose homeostasis: a metaanalysis of acromegaly studies. J Clin Endocrinol Metab. 2009;94(5):1500–1508. doi:10.1210/jc.2008-2332
157. Henry RR, Ciaraldi TP, Armstrong D, Burke P, Ligueros-Saylan M, Mudaliar S. Hyperglycemia associated with pasireotide: results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab. 2013;98(8):3446–3453. doi:10.1210/jc.2013-1771
158. Colao A, Bronstein MD, Freda P, et al. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab. 2014;99(3):791–799. doi:10.1210/jc.2013-2480
159. Giustina A. Optimal use of pegvisomant in acromegaly: are we getting there? Endocrine. 2015;48(1):3–8. doi:10.1007/s12020-014-0462-0
160. Jonas C, Maiter D, Alexopoulou O. Evolution of glucose tolerance after treatment of acromegaly: a study in 57 patients. Horm Metab Res. 2016;48(5):299–305. doi:10.1055/s-0035-1569277
161. Auriemma RS, Pirchio R, De Alcubierre D, Pivonello R, Colao A. Dopamine agonists: from the 1970s to today. Neuroendocrinology. 2019;109(1):34–41. doi:10.1159/000499470
162. Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27(5):485–534. doi:10.1210/er.2005-9998
163. Cincotta AH, Schiller BC, Meier AH. Bromocriptine inhibits the seasonally occurring obesity, hyperinsulinemia, insulin resistance, and impaired glucose tolerance in the Syrian hamster, Mesocricetus auratus. Metabolism. 1991;40(6):639–644. doi:10.1016/0026-0495(91)90057-4
164. Kamath V, Jones CN, Yip JC, et al. Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care. 1997;20(11):1697–1701. doi:10.2337/diacare.20.11.1697
165. Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care. 2000;23(8):1154–1161. doi:10.2337/diacare.23.8.1154
166. Pivonello R, Ferrigno R, De Martino MC, et al. Medical treatment of Cushing’s disease: an overview of the current and recent clinical trials. Front Endocrinol. 2020;11:648. doi:10.3389/fendo.2020.00648
167. Gilis-Januszewska A, Bogusławska A, Rzepka E, Ziaja W, Hubalewska-Dydejczyk A. Individualized medical treatment options in Cushing disease. Front Endocrinol. 2022;13:1060884. doi:10.3389/fendo.2022.1060884
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.