")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Effects of a novel ACE inhibitor, 3-(3-thienyl)-L-alanyl-ornithyl-proline, on endothelial vasodilation and hepatotoxicity in L-NAME-induced hypertensive rats
Authors Seth M, Hussain ME, Pasha S, Fahim M
Received 19 November 2014
Accepted for publication 12 August 2015
Published 20 April 2016 Volume 2016:10 Pages 1533—1542
DOI https://doi.org/10.2147/DDDT.S77761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Mahesh Kumar Seth,1–3 M Ejaz Hussain,2 Santosh Pasha,1 Mohammad Fahim3
1Peptide Synthesis Laboratory, CSIR, Institute of Genomics and Integrative Biology, Delhi, India; 2Centre for Physiotherapy and Rehabilitation Sciences, Jamia Millia Islamia, New Delhi, India; 3Department of Physiology, Jamia Hamdard Deemed University, New Delhi, India
Abstract: Nitric oxide (NO) is a widespread biological mediator involved in many physiological and pathological processes, eg, in the regulation of vascular tone and hypertension. Chronic inhibition of NO synthase by NG-nitro-L-arginine methyl ester (ʟ-NAME) hydrochloride results in the development of hypertension accompanied by an increase in vascular responsiveness to adrenergic stimuli. Recently, we developed a novel sulfur-containing angiotensin-converting enzyme inhibitor: 3-(3-thienyl)-ʟ-alanyl-ornithyl-proline (TOP). Our previous studies indicated a superior nature of the molecule as an antihypertensive agent in spontaneously hypertensive rats (showing the involvement of renin–angiotensin–aldosterone system) in comparison to captopril. The aim of the present study was to investigate the effect of TOP on NO pathway in ʟ-NAME-induced hypertensive rats, and captopril was included as the standard treatment group. Treatment with both TOP (20 mg/kg) and captopril (40 mg/kg) prevented the development of hypertension in ʟ-NAME model, but TOP showed better restoration of NO and normal levels of angiotensin-converting enzyme. In addition, in vitro vasorelaxation assay showed an improvement in endothelium-dependent vasodilation in both the cases. Further, the biochemical (malondialdehyde, alanine aminotransferase, and aspartate aminotransferase) and the histopathological effects of TOP on rat liver tissues revealed a protective nature of TOP in comparison to captopril in the ʟ-NAME model. In conclusion, TOP at 50% lesser dose than captopril was found to be better in the ʟ-NAME model.
Keywords: nitric oxide, ACE inhibitor
Introduction
Hypertension affects ~33% of the population of the USA aged ≥20 years and more than one billion people throughout the world with increasing prevalence among children. It brings, along with it, an increasing risk of concomitant diseases such as cardiovascular diseases, stroke, and kidney disorders.1,2 Renin–angiotensin system (RAS) plays a vital role in the regulation of vascular function and blood pressure3 and, therefore, provides potential target sites for the development of antihypertensive drugs. An example of such a target site is angiotensin-converting enzyme (ACE), ACE which is a membrane-anchored zinc metallopeptidase whose levels are increased in hypertension and cardiovascular disorders such as myocardial infarction and atherosclerosis. ACE inhibitors such as captopril have been adopted as a preferred therapy to treat the aforementioned disorders. In addition to the inhibition of ACE, it prevents the breakdown of bradykinin, a potent endothelium-dependent vasodilator,4 further decreasing the blood pressure. Although the elevated bradykinin levels were not always found during ACE inhibitor treatment, the increased bradykinin production was accompanied in most of the cases by a potential enhancement of subsequent nitric oxide (NO)5 and prostaglandin synthesis,6 leading to vasodilator effect. Among the currently available ACE inhibitor, captopril inhibits ACE by chelating Zn in the active site, which is involved in the hydrolysis of angiotensin I. However, the oxidation of the free mercapto group limits its usability due to a number of side effects such as dry cough, skin irritation, angioedema, and multiple dosing due to less bioavailability.7 Hence, a novel peptidomimetic ACE inhibitor, 3-thienylalanine-ornithine-proline (TOP), devoid of free sulfhydryl group was designed and synthesized in our laboratory.8 The presence of a cyclic sulfur-containing ring and an unnatural amino acid ornithine confer resistance against proteolysis.
Despite the RAS system, L-arginine NO9 pathway plays a major role in the regulation of blood pressure. It is an important regulatory system in the circulation and a well-established regulator of cardiovascular system.10,11 Endogenous NO is synthesized in vascular endothelial cells by the conversion of L-arginine to L-citrulline using the enzyme NO synthase.11 NO is continuously released from the endothelium and participates in the regulation of vascular tone.12 In addition to its direct effect on the vasculature, NO also modulates sympathetic neurotransmission.13 In fact, it is an acetylcholine (ACh) receptor that relies on NO mechanism to cause vasorelaxation. In addition, there are several other agents (histamine, bradykinin, 5-hydroxytryptamine, thrombin, etc) that produce relaxation of arteries either by direct action on the smooth muscles or by indirect action mediated by the activation of endothelial receptor, the indirect action being stronger than the direct one.14 Chronic administration of L-arginine analog, NG-nitro-L-arginine methyl ester (L-NAME), blocks all three NO synthase isoforms of the pathway, resulting in increased blood pressure and oxidative stress in rats,5 and hence forms a well-established model of experimental NO-deficient hypertension.
Oxidative stress has been reported to be involved in the pathophysiology of various forms of hypertension.15 The principle free radicals, such as superoxide (O2), hydroxyl (OH), and peroxyl (COO–) radicals, play major roles in tissue damage. The damage may be qualitatively determined by the measurement of the levels of malondialdehyde (MDA), which is a product of membrane lipid peroxidation. Further, to assess liver damage, the levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) can also be monitored.
In our previous studies on spontaneously hypertensive rats (SHRs), TOP showed a significant and an extended antihypertensive action than captopril. In spite of decreasing the ACE levels that are highly elevated in SHRs,7 it increased the levels of various oxidative parameters such as NO levels and antioxidants such as superoxide dismutase, catalase, and glutathione, thereby indicating the involvement of TOP in another pathway known as L-arginine NO pathway.9 Further, in 1996, Bernátová et al16 reported that the chronic administration of ACE inhibitor such as captopril prevents the development of L-NAME-induced hypertension indicating the role of ACE inhibitors in the NO pathway.
The aim of the present study was to 1) investigate the role of thiophene-ring-containing peptidomimetic TOP in the L-arginine NO pathway using L-NAME-induced hypertension model in rats by various biochemical parameters (NO and ACE levels), endothelial nitric oxide synthase (eNOS) expression studies, and in vitro functional assays (thoracic aortas) and 2) compare its hepatotoxicity with the standard sulfur-containing compound captopril using various biochemical (MDA, ALT, and AST) and histopathological studies.
Materials and methods
Peptidomimetic synthesis
TOP was synthesized by the solid phase method using ACT-90 peptide synthesizer (Advance ChemTech, Louisville, KY, USA) with the standard chemistry of 9-fluorenylmethoxycarbonyl amino acids (Novabiochem; Laufelfigen, Switzerland) and 1-hydroxybenzotriazole/diisopropylcarbodiimide activation method on the chlorotrityl resin.8 The peptide was purified on semi-preparative reverse phase high-performance liquid chromatography (Waters 600) by using a reverse phase C18 column (mBondapak 10 mm, 7.8×300 mm; Waters Corporation, Miliford, MA, USA with a 40-minute linear gradient from 10% to 50% acetonitrile; containing 0.05% trifluoroacetic acid) in water. The mass analysis was performed in a linear positive ion mode by (matrix-assisted laser desorption/ionization-time of flight)/time of flight (Bruker Optik GmbH, Ettlingen, Germany) with 2,5-dihydroxybenzoic acid as the matrix. The peptide sequence was confirmed by automated peptide sequencing (Procise 491; Thermo Fisher Scientific, Waltham, MA, USA).
Experimental animals
Male Wistar rats, 180–200 g, were housed two per cage in polyethylene cages and kept on a 12-hour light/dark cycle at 25°C±2°C. They were given standard rat chow and water ad libitum. They were kept in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals, India, and the Animal Ethical Committee of the Institute of Genomics and Integrative Biology (New Delhi, India). The rats were divided into six groups of six animals each. The first group served as the control. The second group was given L-NAME at a dose of 40 mg/kg per day (L-NAME group). The third group was simultaneously treated with L-NAME at a dose of 40 mg/kg per day and TOP at 20 mg/kg per day (L-NAME + TOP group). The fourth group was treated with L-NAME at a dose of 40 mg/kg per day and captopril at 40 mg/kg per day (L-NAME + captopril group). The fifth group was treated with TOP at a dose of 20 mg/kg per day (TOP group), and the sixth group (n=6) was given captopril at a dose of 40 mg/kg per day (captopril group). The substances were dissolved in tap water and given orally via gavage for 5 weeks. Systolic blood pressure (SBP) was measured by noninvasive tail-cuff plethysmography (ADInstruments, Sydney, NSW, Australia) at the end of first, third, and fifth weeks. At the end of the fifth week, the animals were sacrificed by intraperitoneal administration of thiopentone (300 mg/kg), and the heart, aorta, brain, and kidney were excised and used for biochemical, molecular, organ bath, and histopathological studies. Blood sample was collected for biochemical assays.
Estimation of biomarkers
NO levels were measured in rat plasma using Nitric Oxide Detection Kit (iNtRON Biotechnology, Inc., Seongnam-si, Gyeonggi-do, Korea), which is based on the diazotization assay (Griess method).17 Standard protocols were used to measure the ACE activity18 and enzymatic antioxidants such as MDA levels.19 The levels of (MDA) were determined using Teco Diagnostics kit (Teco Diagnostics, Anaheim, CA, USA). All the measurements were performed using SpectraMax Plus384 Spectrophotometer (Molecular Devices LLC, Sunnyvale, CA, USA).
In vitro experiments: vascular responses
Rats weighing between 200 and 300 g were sacrificed under deep anesthesia (thiopentone; 300 mg/kg body weight intraperitoneally). Thoracic aorta was carefully dissected, cleaned of adhering connective tissue, and cut into rings of 3–4 mm length. Tissues were mounted vertically under 2 g tension in a 10 mL organ bath chamber containing Krebs–Henseleit solution (pH 7.4, 37°C) and bubbled with 95% O2 and 5% CO2. The tissues were connected to an isotonic force transducer that was linked to eight-channel organ baths (ADInstruments) and allowed to equilibrate for 1 hour with washes every 10 minutes before exposure to drugs. Only the tissue preparations that responded to 2×10−4 mol/L of ACh by producing contractions of >1.5 g tension were used. Concentration–response curves of ACh (10−12–10−4 M) were obtained in endothelium-intact aortic rings that were precontracted with 10−7 M phenylephrine for different treatment groups.
Real-time polymerase chain reaction
Single-strand cDNA was reverse transcribed using a First Strand cDNA Synthesis Kit (Amersham Biosciences, Buckinghamshire, UK) and gene-specific antisense primers (the Centre for Genomic Application, New Delhi, India). Primers for real-time reverse transcription polymerase chain reaction (RT-PCR) were designed for eNOS and cyclophilin A using Primer Express™ Software (PerkinElmer Inc., Waltham, MA, USA) and are listed in Table 1. Real-time RT-PCR reactions were performed using SYBR Green PCR Master Mix (Thermo Fisher Scientific) on ABI Prism 7300 Sequence Detection System (Thermo Fisher Scientific). For each 25 μL of PCR reaction, 1.0 μL of cDNA, 1.0 μL of sense and antisense primers (2.5 μM each), 12.5 μL of SYBR Green PCR Master Mix, and 10.5 μL of PCR grade water were used. The cycling conditions were 94°C for 10 minutes, followed by 40 cycles of 94°C for 15 seconds and 59°C for 30 seconds. The PCR reactions were performed in five replicates for each gene. Relative transcript quantity was calculated using the ΔΔCt method, with cyclophilin A as the endogenous reference gene amplified from the samples. A validation experiment for ΔΔCt calculation was performed for each target by plotting the ΔCt (Ct target – Ct reference) values versus log input amounts to create a semilog regression line. The slope of the resulting semilog regression line was used as a general criterion for the validation experiment. The absolute value of the slope was <0.1. Finally, the amount of target (drug treatment) normalized to the endogenous reference and relative to a calibrator (saline-treated control) was calculated by 2−ΔΔCt.20
 | Table 1 Details of primers used for real-time RT-PCR |
Histology
Liver tissues were isolated and fixed in 10% formalin buffer overnight. The tissues were embedded in paraffin, sliced into 4 μm-thick sections, and stained with hematoxylin–eosin for the light microscopic study. The cross-sectional area of liver was evaluated from the photographs of whole tissue section taken at ×40 magnification.
Statistical analysis
The data are presented as mean ± standard error of the mean. Statistical analysis was carried out by the analysis of variance followed by Turkey’s multiple comparison tests and Student’s t-test (Origin Version 7.1). P-values <0.05 were considered significant.
Result
Systolic blood pressure
SBP was measured at the end of first, third, and fifth weeks of drug administration. The L-NAME-treated rats showed a progressive increase in the mean SBP compared with the control rats (P<0.05; Figure 1). After 5 weeks of L-NAME treatment, SBP reached ~187 mmHg. Concomitant treatment with TOP and captopril reduced the blood pressure in L-NAME-induced hypertensive rats to 136 and 153.5 mmHg, respectively (P<0.05). TOP and captopril treatment did not show any significant change in SBP when administered alone.
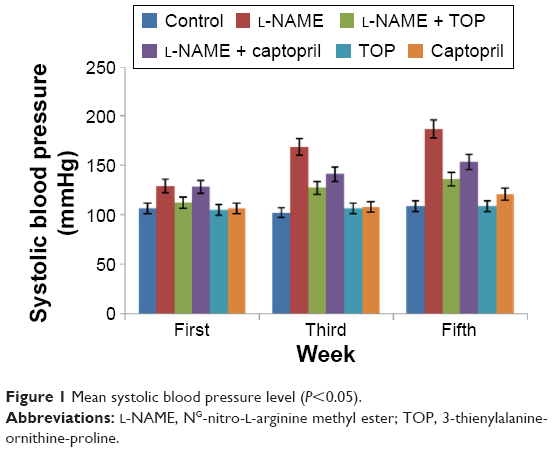 | Figure 1 Mean systolic blood pressure level (P<0.05). |
ACE activity and NO levels
ACE activity was measured in the rat serum. As shown in Figure 2, the ACE levels were significantly higher in the L-NAME group (94.3±4.7 mU/L) than in the control group (63.8±1.8 mU/L). Oral supplementation of TOP (10 mg/kg, twice a day) to the L-NAME-treated rats resulted (68.2±0.9 mU/L) in a complete reversal of the L-NAME-induced increase in ACE levels, while a less decrease was observed with captopril (74.9±3.5 mU/L). Chronic L-NAME administration for 5 weeks resulted in a significant depletion of plasma NO levels (55.4±5.4 to 20.8±1.9 μM; Figure 3). The treatment with TOP and captopril in combination with L-NAME resulted in the elevated plasma NO levels (40±2.4 and 32±3.7 μM). TOP and captopril treatment did not show any significant effect on the ACE activity and NO when administered alone.
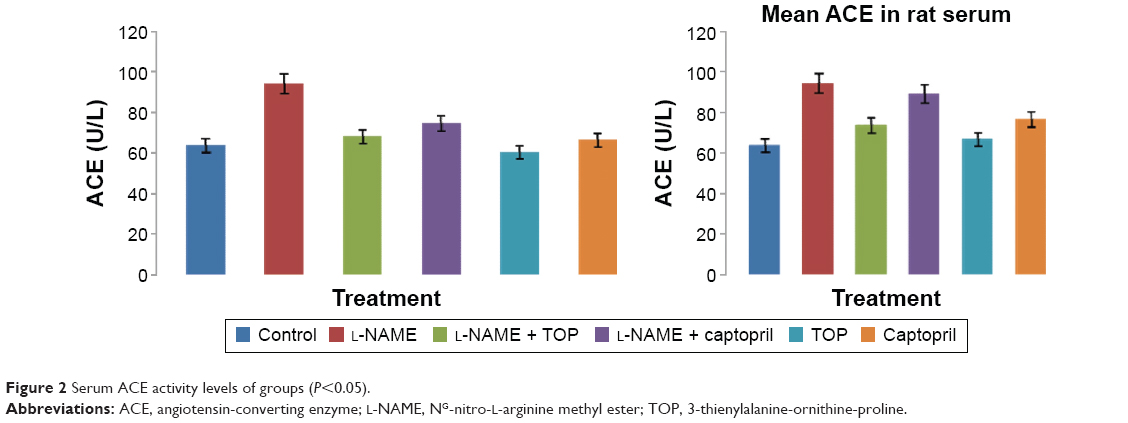 | Figure 2 Serum ACE activity levels of groups (P<0.05). |
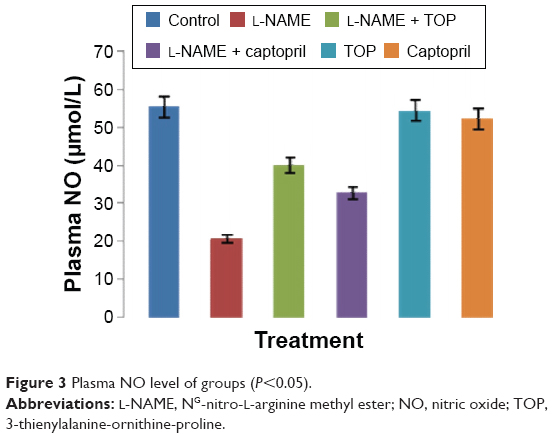 | Figure 3 Plasma NO level of groups (P<0.05). |
In vitro vasorelaxation assay
Precontracted endothelium-intact aortic rings were used to study the concentration-dependent vasorelaxation response of ACh (Figure 4). ACh induced a significantly weak relaxation in the L-NAME group (16% at 10−4 M) compared with the control group (58% at 10−4 M). When administered along with L-NAME, both TOP and captopril improved the relaxation response of Ach with TOP (40%) being slightly better than captopril (36%). No significant difference in vasorelaxation was observed when TOP and captopril were administered alone.
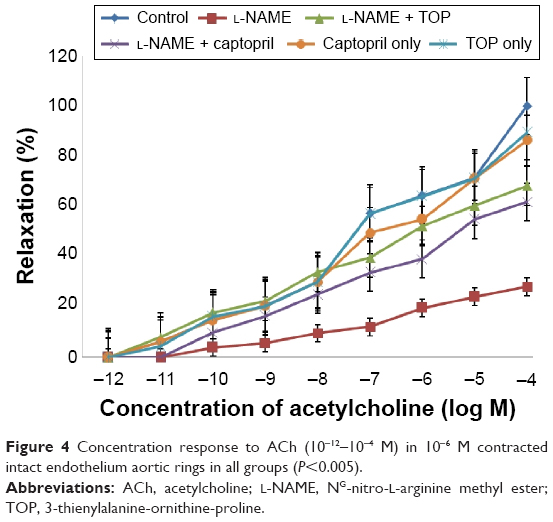 | Figure 4 Concentration response to ACh (10−12–10−4 M) in 10−6 M contracted intact endothelium aortic rings in all groups (P<0.005). |
eNOS mRNA expression studies
eNOS mRNA expression (Figure 5) was studied in aorta, heart, and kidney using cyclophilin A as the internal standard. During the treatment with L-NAME, eNOS mRNA expression decreased significantly in all the three organs, whereas it increased during the treatment with TOP and captopril in L-NAME-induced hypertensive rats with TOP showing better results in kidney. In kidney, no significant difference was observed between the mRNA levels of TOP and the control group, while the decrease was significant in the case of the captopril group.
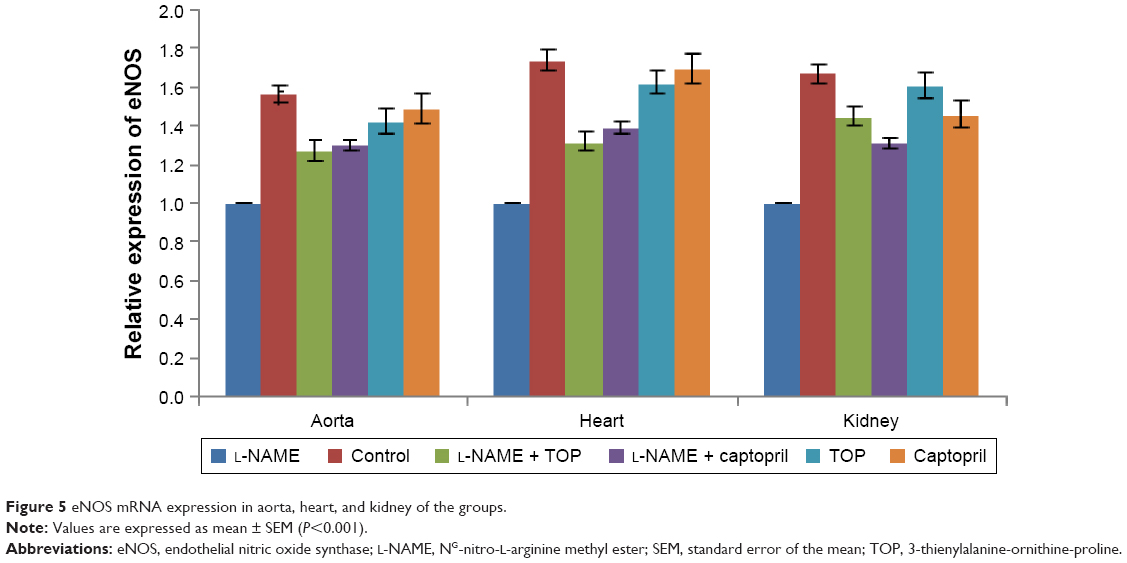 | Figure 5 eNOS mRNA expression in aorta, heart, and kidney of the groups. |
Toxicity biomarkers
The levels of MDA in the rat serum increased significantly in the L-NAME-treated rats compared with those of the control group (P<0.05; Figure 6). Simultaneously, captopril reduced the levels of serum MDA from 0.692 to 0.52 μM, and TOP reduced the levels of serum MDA (0.405 μM) to the levels of the control group (0.387 μM) in L-NAME-induced hypertensive rats. There was a slight decrease in the levels of MDA when TOP (0.31 μM) and captopril (0.33 μM) were given alone. As shown in Table 2, an approximately three- and twofold increase in the levels of ALT and AST was observed in the L-NAME group, with respect to the control group, thereby disturbing the AST/ALT ratio (1.20) from the normal levels (1.77). The concomitant treatment with TOP lowered the ALT levels to the normal levels, hence increasing the ratio to 1.66 in the L-NAME-induced hypertensive rats, whereas the co-treatment with captopril led to a further decrease in the ratio to 1.04. No significant changes in the levels were observed when TOP was administered alone. On the other hand, captopril led to a significant increase in the levels and decrease in the ratio.
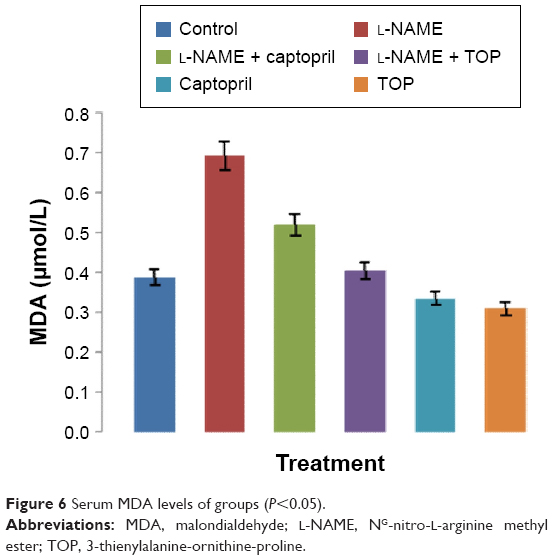 | Figure 6 Serum MDA levels of groups (P<0.05). |
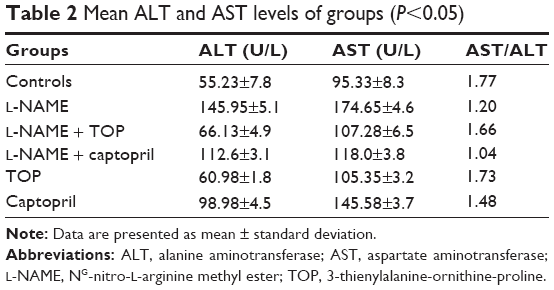 | Table 2 Mean ALT and AST levels of groups (P<0.05) |
Histopathology
Control groups (Figure 7A) showed normal hepatic tissue, while a slight mononuclear infiltration was observed in the L-NAME-only group (Figure 7B), and the samples from the L-NAME + TOP (Figure 7C) and TOP-only (Figure 7D) groups showed normal hepatic tissue. In L-NAME + captopril groups (Figure 7E) and captopril only groups (7F), prominent periportal lymphocytic collection with infiltration of the inflammatory cells into adjacent hepatic parenchyma showed significant histological changes in liver tissue.
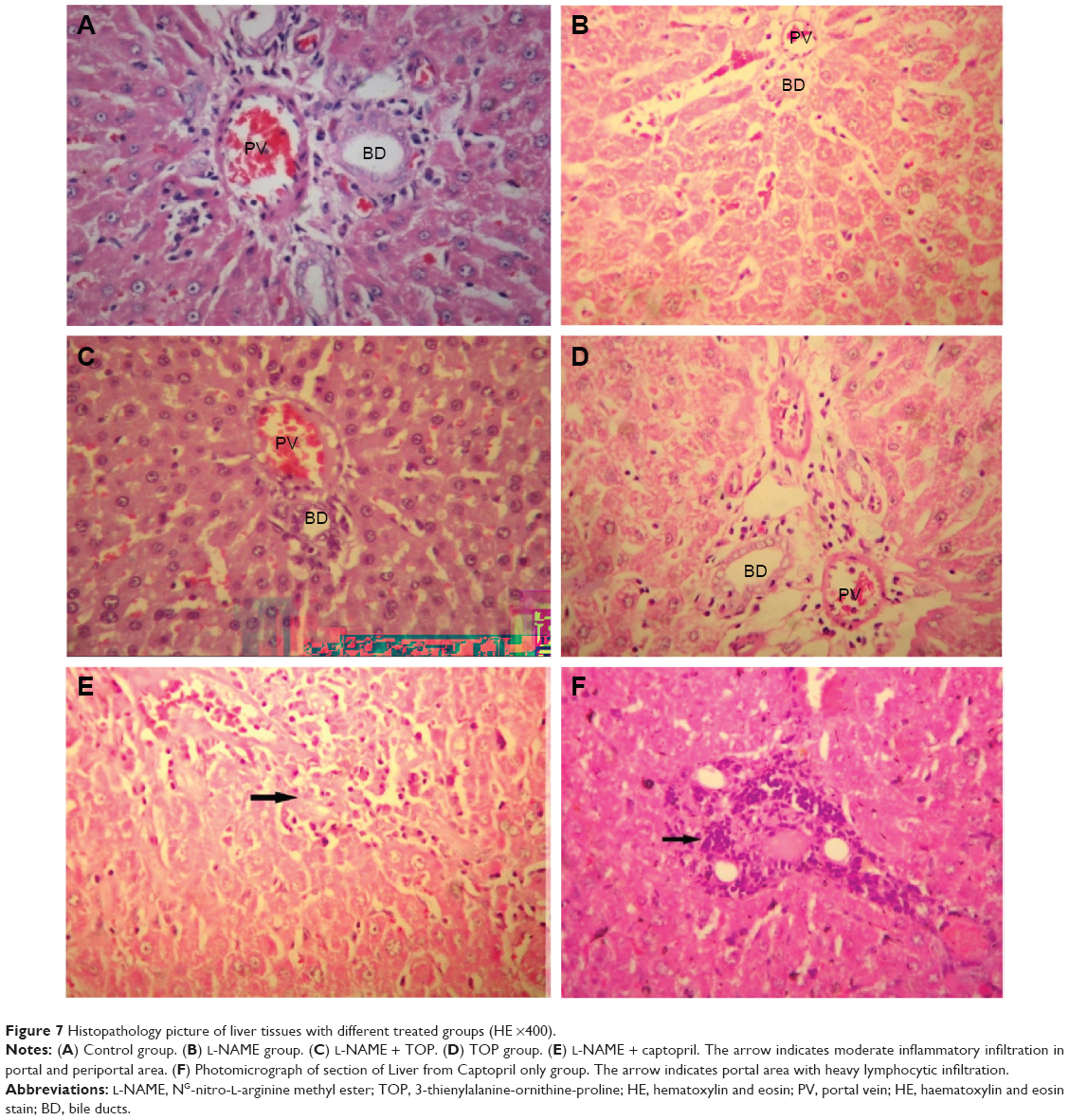 | Figure 7 Histopathology picture of liver tissues with different treated groups (HE ×400). |
Discussion
The endothelium-derived vasodilatory factor, NO, is a crucial regulator of vascular tone and a widespread biological mediator involved in many physiological and pathological processes.21–24 A complex set of interactions between the neural and endothelial mechanisms plays a critical role in maintaining the vasculature. Any long-term disturbance in the balance of these interactions may lead to the development of cardiovascular diseases.25 L-NAME, an established NO synthase inhibitor, is known to induce hypertension, which is characterized by endothelial dysfunction and perivascular inflammation26–28 along with marked deficiency of NO. The present study was carried out to investigate the effect of TOP on the L-NAME-induced hypertension model of rats in comparison to captopril, existing sulfur-containing ACE inhibitor.
Six groups of animals, namely control, L-NAME-, L-NAME + TOP-, L-NAME + captopril-, TOP-, and captopril-treated animals, were used to assess the effect of TOP on L-NAME-induced hypertension, taking the control (without any treatment) and L-NAME-treated groups as negative and positive controls. It is well documented that the inhibition of NO synthase results in an increase in the blood pressure.11,29,30 As expected, 5 weeks of chronic treatment with L-NAME showed a significant increase in SBP, while the treatment with TOP and captopril alone did not show any significant increase in SBP compared with the control group. Blood pressure was more potently reversed with the concomitant treatment of TOP with L-NAME than captopril with L-NAME. Demonstration of advanced antihypertensive activity at half the dose suggests better efficacy of TOP in comparison to captopril.
In accordance with the literature, the level of biochemical parameter NO was significantly decreased in the serum of L-NAME-treated group. Captopril given with L-NAME showed a significant recovery in NO level, but TOP given with L-NAME showed much better results in normalizing NO levels. There is sufficient data available which reveal the reciprocal relation between ACE inhibition and NO level.24,31,32 ACE inhibition effectively prevents left ventricular hypertrophy, even without a reduction in afterload33–35 and coronary artery hypertrophy.36 Reports suggest that both local and systemic RAS may be involved in the proliferative effects of Ang II or anti-proliferative effects of ACE inhibition.16 However, the mechanism of the proliferation and growth-modifying effect of NO and Ang II is not yet satisfactorily understood and varies depending on the model of the hemodynamic overload, period of overload, and the particular organ investigated.16
RAS system is known to play a predominant role in SHRs, a model of genetic hypertension. In spite of that, there exists an ambiguity about the role of RAS- and NO-dependent ACh-mediated endothelium-receptor activation in SHRs. In our previous studies on the SHR model, we found that persistently high levels of ACE and treatment with TOP resulted in a significant decrease in the ACE activity. Our results have also shown moderate changes in NO levels which may be due to the accumulation of bradykinin37,38 or may be due to homeostasis. These observations were further supported by organ bath experiments using SHR aortic rings, where concomitant treatment of TOP with L-NAME showed 100% relaxation at much lower concentration of ACh (10−7 M; unpublished data).
In the L-NAME-induced hypertensive rats, the involvement of RAS system is bare minimum as not much increase in the ACE activity is observed when compared with SHRs. However, a significant fall in NO level indicates that NO-dependent ACh-mediated endothelium-receptor activation is predominant for this type of hypertension. TOP was also found equally effective in this case, as it demonstrated a complete reversal of NO level to normal when compared with the L-NAME-treated group.
Furthermore, the results of the in vitro vasorelaxation assay performed using the aortic rings from animals of the various experimental groups suggest that NO production affects the vasorelaxation of aorta receptor through ACh as the L-NAME group showed 100% relaxation response at a much higher concentration of Ach (10−4 M) when compared with SHRs (10−7 M). ACh concentration-dependent vasorelaxation in animals treated with only TOP and captopril showed no change in response to its effect in rat aorta when compared with the control group. The results also demonstrate an improvement in endothelium-dependent vasodilation during the concomitant treatment of TOP and captopril with L-NAME when compared with L-NAME-treated animals, which showed significantly reduced vasorelaxation. Our results corroborate with previously reported data on the involvement of ACh-mediated receptor in vasorelaxation in the NO model.
Decreased functional activity of NOS in the long-term L-NAME-treated rats is well established.16,39 TOP and captopril almost homogeneously increased the levels of eNOS mRNA expression in the tissues of the L-NAME-induced hypertension model, which was contradictory to the effect of L-NAME. These findings are in well agreement with the previously reported ACE inhibitors.39 Additionally, our present findings were also validated by the potency of TOP and the standard compound captopril to restore eNOS mRNA expression in rat aorta, heart, and kidney with the effect being more prominent in kidney tissues.
In addition to a decrease in NO levels, L-NAME-induced hypertension also leads to the development of oxidative injury in various tissues and organs such as kidney and liver.40,41 The generation of reactive oxygen species is one of the major mechanisms involved in the tissue damage with enhanced superoxide anion activity.42 Oxidative stress facilitates damage of the membrane lipids, resulting in decreased membrane fluidity. MDA is a breakdown product of the major chain reactions leading to the oxidation of polyunsaturated fatty acids and, hence, serves as a reliable marker of lipid-peroxidation-mediated oxidative stress.43 A number of previous reports suggest an association between increased liver peroxidation and hypertension.43–45 Our data support these findings, as it clearly shows the increased levels of serum MDA in the L-NAME-induced hypertensive rats. Further, it also indicates that the administration of TOP reduces lipid peroxidation and brings the levels of MDA down to normal; hence, it must be quite effective in minimizing ROS-mediated oxidative injury in tissues.
To further investigate the liver tissue damage caused by L-NAME, TOP, and captopril, ALT and AST levels were recorded. AST and ALT are the members of the transaminase family of enzymes which facilitate various reactions within the cells. Under damaged conditions, these enzymes are leaked out of the cells into the blood. Thus, they serve as important markers for tissue damage. ALT is produced mainly in liver, while AST in heart, kidney, and muscle tissues. An increase in the level of these enzymes along with the ratio of the two (AST/ALT) helps in the prediction of the type and extent of tissue damage with liver tissue damage being closely associated with the elevated serum ALT levels.46,47 The present study clearly associates hepatotoxicity with L-NAME-induced hypertension, as a threefold increase in the levels of serum ALT was observed with a significant drop in the AST/ALT ratio. Captopril lowered the levels of ALT in hypertensive rats, but the ratio was further decreased indicating severe damage in organs other than liver such as heart and kidney. In the hypertensive rats, TOP normalized the levels of the both the factors, further supporting the previous results. Although TOP alone did not show any significant variation in the levels of AST and ALT, captopril resulted in an increase in both indicating tissue damage.
Additionally, hepatic changes were also studied to assess the extent of liver damage using hematoxylin–eosin-stained liver sections. Mononuclear cell infiltration in periportal areas and hepatocellular degeneration are common findings associated with hepatotoxicity.48 In the present study, periportal mononuclear cell infiltration was the only significant histological finding. Moderate mononuclear cell infiltration was observed in the L-NAME group, while higher levels were observed in the L-NAME + captopril group confirming that captopril causes damage to liver tissues. Although captopril alone was found to be hepatotoxic, TOP alone was not affecting liver tissues. Furthermore, TOP reduces the levels of infiltration in the hypertensive rats, again indicating its protective effect against L-NAME-induced hepatotoxicity.
Such a difference in the hepatotoxicity profile of captopril and TOP may be attributed to the nature of the present sulfhydryl group49 or their different modes of involvement in the NO pathway. Another suggested mechanism is the variation in the activation of bradykinin, resulting in an increased conversion of arachidonic acid to prostaglandins.49 Therefore, it can be concluded that TOP prevents various structural and biochemical changes in hepatic tissues in the L-NAME-induced hypertensive rats with several underlying mechanisms playing a role in it.
Conclusion
In conclusion, TOP at 50% less dose than captopril was found better in all the tested aspects in the L-NAME-induced hypertension model of rats suggesting its active role in improving endothelial vasodilation.
Acknowledgment
This study was supported by the Council of Scientific and Industrial Research, India.
Disclosure
The authors report no conflicts of interest in this work.
References
Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation. 2003;107:2864–2869. | ||
Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287. | ||
Bader M, Ganten D. Update on tissue renin–angiotensin systems. J Mol Med. 2008;86:615–621. | ||
Erdös EG, Tan F, Skidgel RA. Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension. 2010;55(2):214–220. | ||
Dabiré H, Barthélémy I, Blanchard-Gutton N, et al. Vascular endothelial dysfunction in Duchenne muscular dystrophy is restored by bradykinin through upregulation of eNOS and nNOS. Basic Res Cardiol. 2012;107(1):1–9. | ||
Sigusch HH, Campbell SE, Weber KT. Angiotensin II-induced myocardial fibrosis in rats: role of nitric oxide, prostaglandins and bradykinin. Cardiovasc Res. 1996;31(4):546–554. | ||
Hanif K, Snehlata, Pavar MC, et al. Effect of 3-thienylalanine-ornithine-proline, new sulfur-containing angiotensin-converting enzyme inhibitor on blood pressure and oxidative stress in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2009;53(2):145–150. | ||
Pavar MC, Hanif K, Azam A, Lata S, Qadar Pasha MA, Pasha S. Structure-activity relationship study between Ornithyl-Proline and Lysyl-Proline based tripeptidomimics as angiotensin-converting enzyme inhibitors. Bioorg Med Chem Lett. 2006;16(8):2112–2117. | ||
Lüscher TF, Vanhoutte PM. The Endothelium: Modulator of Cardiovascular Functions. Boca Raton, FL: CRC Press; 1991:1–228. | ||
Kuneš J, Hojná S, Kadlecová M, et al. Altered balance of vasoactive systems in experimental hypertension: the role of relative NO deficiency. Physiol Res. 2004;53(1):S23–S34. | ||
Moncada S. Nitric oxide gas: mediator, modulator, and pathophysiologic entity. J Lab Clin Med. 1992;120:187–191. | ||
Nava E, Farre A, Moreno C, et al. Alterations to the nitric oxide pathway in the spontaneously hypertensive rat. J Hypertens. 1998;16:609–615. | ||
Macarthur H, Westfall TC, Wilken GH. Oxidative stress attenuates NO-induced modulation of sympathetic neurotransmission in the mesenteric arterial bed of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;294:H183–H189. | ||
Török J. Participation of nitric oxide in different models of experimental hypertension. Physiol Res. 2008;57:813–825. | ||
Romero JC, Reckelhoff JF. Role of angiotensin and oxidative stress in essential hypertension. Hypertension. 1999;34:943–949. | ||
Bernátová I, Pecháňová O, Šimko F. Effect of captopril in L-NAME-induced hypertension on the rat myocardium, aorta, brain and kidney. Exp Physiol. 1999;84:1095–1105. | ||
Green LC, Wagner DA, Glagowski J. Analysis of nitrate, nitrite and [15N] nitrate in biological fluids. Anal Biochem. 1982;126:131–138. | ||
Bala M, Qadar Pasha MA, Bhardwaj DK. Novel peptidomimics as angiotensin converting enzyme inhibitors: a combinatorial approach. Bioorg Med Chem. 2002;10:3685–3691. | ||
Wright JR, Colby HD, Miles PR. Cytosolic factors that affect microsomal lipid peroxidation in lung and liver. Arch Biochem Biophys. 1981;206:296–304. | ||
Vats ID, Snehlata, Nath M, Pasha MA, Pasha S. Effect of chronic intra-peritoneally administered chimeric peptide of met-enkephalin and FMRFa-[D-Ala2] YFa on antinociception and opioid receptor regulation. Eur J Pain. 2009;14(3):295.e1–295.e9. | ||
Förstermann U, Kleinert H. Nitric oxide synthase: expression and expressional control of the three isoforms. Naunyn Schmiedebergs Arch Pharmacol. 1995;352(4):351–364. | ||
Marín J, Rodríguez-Martínez MA. Role of vascular nitric oxide in physiological and pathological conditions. Pharmacol Ther. 1997;75(2):111–134. | ||
Maxwell AJ, Schauble E, Bernstein D, Cooke JP. Limb blood flow during exercise is dependent on nitric oxide. Circulation. 1998;98(4):369–374. | ||
Pecháňová O, Bernátová I, Pelouch V, Simko F. Protein remodelling of the heart in NO-deficient hypertension: the effect of captopril. J Mol Cell Cardiol. 1997;29:3365–3374. | ||
Lincoln J, Burnstock G. Neural-endothelial interactions in control of local blood flow. In: Warren J, editor. The Endothelium: An Introduction to Current Research. New York, NY: Wiley-Liss; 1990:21–31. | ||
Török J, Gerová M. Vascular responses after long-term inhibition of nitric oxide synthesis in newborn dogs. Physiol Res. 1996;45:323–328. | ||
Holécyová A, Török J, Bernátová I, Pechánová O. Restriction of nitric oxide rather than elevated blood pressure is responsible for alterations of vascular responses in nitric oxide-deficient hypertension. Physiol Res. 1996;45:317–321. | ||
Hsieh NK, Wang JY, Liu JC, Wang SD, Chen HI. Nitric oxide inhibition accelerates hypertension and induces perivascular inflammation in rats. Clin Exp Pharmacol Physiol. 2004;31:212–218. | ||
Grossman HJ, Grossman VL, Bhatal PS. Intrahepatic vascular resistance in cirrhosis. In: Bosch J, Groszmann RJ, editors. Portal Hypertension: Pathophysiology and Treatment. London: Blackwell Scientific Publications; 1994:1–16. | ||
Zicha J, Dobešová Z, Kuneš J. Relative deficiency of nitric oxide-dependent vasodilation in salt-hypertensive Dahl rats: the possible role of superoxide anions. J Hypertens. 2001;19:247–254. | ||
Zicha J, Dobešová Z, Kuneš J. Antihypertensive mechanisms of chronic captopril or N-acetylcysteine treatment in L-NAME hypertensive rats. Hypertens Res. 2006;29:1021–1027. | ||
Pecháňová O, Zicha J, Paulis L, et al. The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur J Pharmacol. 2007;561:129–136. | ||
Linz W, Wiemer G, Schaper J, et al. Angiotensin converting enzyme inhibitors, left ventricular hypertrophy and fibrosis. Mol Cell Biochem. 1995;147(1–2):89–97. | ||
Nakamura Y, Yamamoto Y, Muraoka I. Autonomic control of heart rate during physical exercise and fractal dimension of heart rate variability. J Appl Physiol. 1993;74(2):875–881. | ||
Šimko F. Left ventricular hypertrophy regression as a process with variable biological implications. Can J Cardiol. 1996;12(5):507–513. | ||
Takemoto M, Egashira K, Usui M, et al. Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in rats. J Clin Invest. 1997;99:278–287. | ||
Linz W, Wiemer G, Schölkens BA. ACE-inhibition induces NO-formation in cultured bovine endothelial cells and protects isolated ischemic rat hearts. J Mol Cell Cardiol. 1992;24(8):909–919. | ||
Ritchie RH, Marsh JD, Lancaster WD, Diglio CA, Schiebinger RJ. Bradykinin blocks angiotensin II-induced hypertrophy in the presence of endothelial cells. Hypertension. 1998;31(1):39–44. | ||
Kobayashi N, Hara K, Watanabe S, Higashi T, Matsuoka H. Effect of imidapril on myocardial remodeling in L-NAME–induced hypertensive rats is associated with gene expression of NOS and ACE mRNA. Am J Hypertens. 2000;13:199–207. | ||
Khattab MM, Mostafa A, Al-Shabanah O. Effects of captopril on cardiac and renal damage, and metabolic alterations in the nitric oxide-deficient hypertensive rat. Kidney Blood Press Res. 2005;28(4):243–250. | ||
Usui M, Egashira K, Kitamoto S, et al. Pathogenic role of oxidative stress in vascular angiotensin-converting enzyme activation in long-term blockade of nitric oxide synthesis in rats. Hypertension. 1999;34:546–551. | ||
Kopkan L, Majid DS. Enhanced superoxide activity modulates renal function in NO-deficient hypertensive rats. Hypertension. 2006;47(3):568–572. | ||
Ozguner F, Oktem F, Ayata A, Koyu A, Yilmaz HR. A novel antioxidant agent caffeic acid phenethyl ester (CAPE) prevents long term mobile phone exposure-induced renal impairment in rat: prognostic value of malondialdehyde, N-acetyl-β-D-glucosaminidase and nitric oxide determination. Mol Cell Biochem. 2005;277:73–80. | ||
Oktem F, Kirbas A, Armagan A, et al. Lisinopril attenuates renal oxidative injury in L-NAME-induced hypertensive rats. Mol Cell Biochem. 2011;352(1–2):247–253. | ||
Nabha L, Garbern JC, Buller CL, Charpie JR. Vascular oxidative stress precedes high blood pressure in spontaneously hypertensive rats. Clin Exp Hypertens. 2005;27(1):71–82. | ||
Jeserich M, Ihling C, Allgaier HP, Berg PA, Heilmann C. Acute liver failure due to enalapril. Herz. 2000;25(7):689–693. | ||
Gonzalez de la Puente MA, Calderon E, Espinosa R, Rincón M, Varela JM. Fatal hepatotoxicity associated with enalapril. Ann Pharmacother. 2001;35:1492. | ||
Gokcimen A, Kocak A, Kilbas S, et al. Effect of lisinopril on rat liver tissues in L-NAME induced hypertension model. Mol Cell Biochem. 2007;296:159–164. | ||
Hagley MT, Hulisz DT, Burns CM. Hepatotoxicity associated with angiotensin-converting enzyme inhibitors. Ann Pharmacother. 1993;27(2):228–231. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.