")
Back to Journals » International Journal of Nanomedicine » Volume 16
Effective Antitumor of Orally Intestinal Targeting Penetrating Peptide-Loaded Tyroserleutide/PLGA Nanoparticles in Hepatocellular Carcinoma
Authors Ma C, Wei T, Hua Y, Wang Z, Zhang L
Received 23 April 2021
Accepted for publication 16 June 2021
Published 2 July 2021 Volume 2021:16 Pages 4495—4513
DOI https://doi.org/10.2147/IJN.S315713
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Chenjun Ma, Tiantian Wei, Yingying Hua, Zhongjie Wang, Liefeng Zhang
School of Food and Pharmaceutical Engineering, Nanjing Normal University, Nanjing, 210046, People’s Republic of China
Correspondence: Liefeng Zhang Wenyuan Road 1#, Qixia District, Nanjing, 210046, People’s Republic of China
Tel +86-25-85898184
Fax +86-25-85898184
Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) is a common malignant tumor that seriously threatens human life and health. Currently, the majority of antitumor drugs are administered in an injectable manner, which can cause pain and side effects to patients. Objective of this study is to establish an effective oral drug delivery system for anti hepatoma drugs.
Methods: In this study, intestinal targeting cell penetrating peptide (R6LRVG) was obtained by binding cell penetrating peptide (R6) with the polypeptide of LRVG (targeting intestinal epithelial cells). Next, R6LRVG-modified tyroserleutide-poly(lactic-co-glycolic acid) (PLGA) nanoparticles (YSL-PLGA/R6LRVG NPs) were prepared. After that, the nanoparticles were characterized and their stability was evaluated. The cellular uptake, in vitro bioactivity and in vivo antitumor activity of the nanoparticles were investigated. In addition, the mechanism, including the endocytic pathway and respiratory rate detection of mitochondria, was further investigated.
Results: YSL-PLGA/R6LRVG NPs were successfully prepared. Characterization revealed YSL-PLGA/R6LRVG NPs to be globular particles with smooth surfaces and an average diameter of 222.6 nm. The entrapment efficiency and drug loading of tyroserleutide were 70.27% and 19.69%, respectively. Furthermore, the YSL-PLGA/R6LRVG NPs group exhibited the largest amount of YSL uptake. We also found that cell uptake of YSL-PLGA/R6LRVG NPs could be related to the endocytosis pathways mediated by reticulin and caveolae/lipid rafts. Additionally, the YSL-PLGA/R6LRVG NPs could interfere with mitochondrial function. In vivo experiments revealed that orally administered YSL-PLGA/R6LRVG NPs exerted excellent anticancer effects in tumor-bearing mice. Hematoxylin-eosin staining did not show any histological changes in the major organs.
Conclusion: To summarize, YSL-PLGA/R6LRVG NPs could be a useful oral delivery system of YSL and may provide a new platform for the oral delivery of anticancer drugs.
Keywords: nanoparticles, target penetrating peptide, tyroserleutide, hepatocellular carcinoma, cellular respiration rate
Introduction
Cancer is one of the most common chronic diseases in the world that is characterized by cell proliferation and spread.1 According to data released by the International Agency for Research on Cancer, cancer is the deadliest disease in both China and the United States, and its incidence is expected to continue to rise in the next decade.2,3 Hepatocellular carcinoma (HCC) is the sixth most common cancer in the world and the third leading cause of cancer-related deaths.4–7 It is a common malignant tumor that seriously threatens human life and health.8 The treatment of HCC is challenging. The side effects of chemotherapy limit its use in liver cancer, especially in underweight patients. Therefore, it is necessary to identify drugs with low toxicity and side effects in the treatment of liver cancer.9
Tyroserleutide (YSL) (C18H27N3O6, MW 381.42 Da) is a tripeptide extracted from pig spleen, which is composed of L-tyrosine, L-serine, and L-leucine. It can inhibit the invasion and adhesion of mouse melanoma cell lines, and the more encouraging result is its anti-HCC effect.10 YSL is mainly distributed in the cytoplasm and is also found in the mitochondria of tumor cells. It directly acts on the mitochondria of tumor cells, reduces the mitochondrial membrane potential, affects membrane permeability, and leads to mitochondrial swelling.11 YSL can significantly prolong the survival of mice transplanted with H22 hepatoma cells and inhibit the growth of human BEL-7402 tumors in nude mice.12 Moreover, YSL is a polypeptide associated with very few side effects.
Chemotherapy is often used in the management of cancers in a clinical setting. The intravenous route is typically used to administer anticancer drugs directly into the systemic circulation. This mode of administration causes a rapid increase in drug levels in the blood, which exceeds the maximum tolerable concentration (MTC) of the drug in humans. YSL is a tripeptide that is cleared or rapidly degraded after intravenous injection. Therefore, frequent drug administration is often necessary, which could inconvenience patients. In contrast, during oral chemotherapy, drugs gradually enter the systemic circulation through intestinal absorption. Subsequently, appropriate drug concentrations in the blood are maintained, avoiding drug levels in the blood exceeding the MTC, thereby prolonging the interaction of the drug with cancer cells. Thus, the side effects of drugs are reduced and their efficacy is improved. Moreover, the ease and convenience of oral chemotherapy are associated with better patient compliance, which results in a significant saving in time and expenses during treatment.13,14 However, improving the oral bioavailability of YSL continues to pose a challenge in drug design.
Nanoparticles (NPs) are considered as promising platforms in enhancing the absorption of anticancer drugs.15,16 Overall, nanoparticles protect drugs from adverse conditions and increase their absorption from the gastrointestinal tract (GIT).17–22 Poly(lactic-co-glycolic acid) (PLGA) is a lactic acid/glycolic acid copolymer synthesized using lactic and glycolic acids and other monomers. PLGA can enhance drug stability and absorption, providing faster dissolution and higher adsorption for surface loading, preventing the effect of proteolytic enzymes, improving mucus adhesion, and increasing gastrointestinal retention. Besides, PLGA is non-toxic, and its final degradation products are water and carbon dioxide, which are environmentally friendly and safe for humans.23
To further improve the permeability of orally bioavailable drugs, it is also necessary to enhance the absorption of anticancer drugs.24 Cell-penetrating peptides (CPPs) have attracted increasing attention as carriers and are a promising class of biomolecules in the intestinal mucosal system.25–27 Oligo-arginine is an artificially synthesized cell-penetrating peptide that is safe for human use.28 The concept of arginine-rich CPPs was first proposed in 2001. After determining membrane-penetrating ability, it was found that within a certain range (R4–R8), the transmembrane permeability of arginine-rich CPPs increased with the number of arginine units and reached the maximum at R6 and R8. Cell permeability was found to decrease when the arginine units in the molecule were increased to beyond eight.29 Further studies show that arginine-rich CPPs are effective carriers with low toxicity and can be easily synthesizeds. Arginine-rich CPPs are widely used in drug delivery and constitute a hot research topic in biomedical research.30,31 Moreover, arginine-rich residues as terminal groups could likely interact with tumor membranes by multiple hydrogen bonding and electrostatic interactions. Targeted penetrating peptide consists of a polypeptide sequence that has inherent targeting abilities and can penetrate cell membranes through receptor interaction on the cell surface upon reaching a specific site.32,33 Researchers have used phage display technology to screen in vitro models of the human follicle-associated epithelium (FAE).34 The LRVG peptide sequence has a high frequency through the cross cell screening of intestinal cells and FAE. Several research groups have synthesized vaccine-loaded nanoparticles. LRVG peptide-modified nanoparticles significantly increase nanoparticle transport compared to the unmodified nanoparticles.35 These findings indicate that LRVG peptides can target intestinal epithelial cells. LRVG peptides can transport drugs through the epithelial cells of the small intestine, thereby enabling the uptake of macromolecules by improving oral absorption.
In this study, we developed a new penetrating peptide targeting intestinal epithelial cells. Further modification of YSL-PLGA NPs revealed that it could enhance not only the cellular uptake but also the inhibitory effect of YSL on HCC. The schematic diagram of the preparation of YSL-PLGA/R6LRVG NPs and the mechanistic representation of the uptake of nanoparticles is depicted in Scheme 1. Specifically, CPP (R6) was combined with the polypeptide of LRVG (targeting intestinal epithelial cells) to yield intestinal targeting penetrating peptide (R6LRVG). We prepared R6LRVG-modified YSL-PLGA NPs (YSL-PLGA/R6LRVG NPs) to enhance the absorption of YSL. First, YSL-PLGA NPs were prepared using double emulsion volatilization (W/O/W), giving the nano platform high drug-carrying capacity. Next, R6LRVG was modified on its surface using electrostatic adsorption. Lastly, the synthesized NPs were characterized using transmission electron microscopy (TEM), Fourier transform-infrared (FTIR) spectroscopy, and zeta potential determination. Furthermore, the cell uptake, in vitro bioactivity and in vivo anti-tumor activity of the NPs were determined. Moreover, the mechanisms, including internalization modes and the respiration rate of mitochondria, were elucidated.
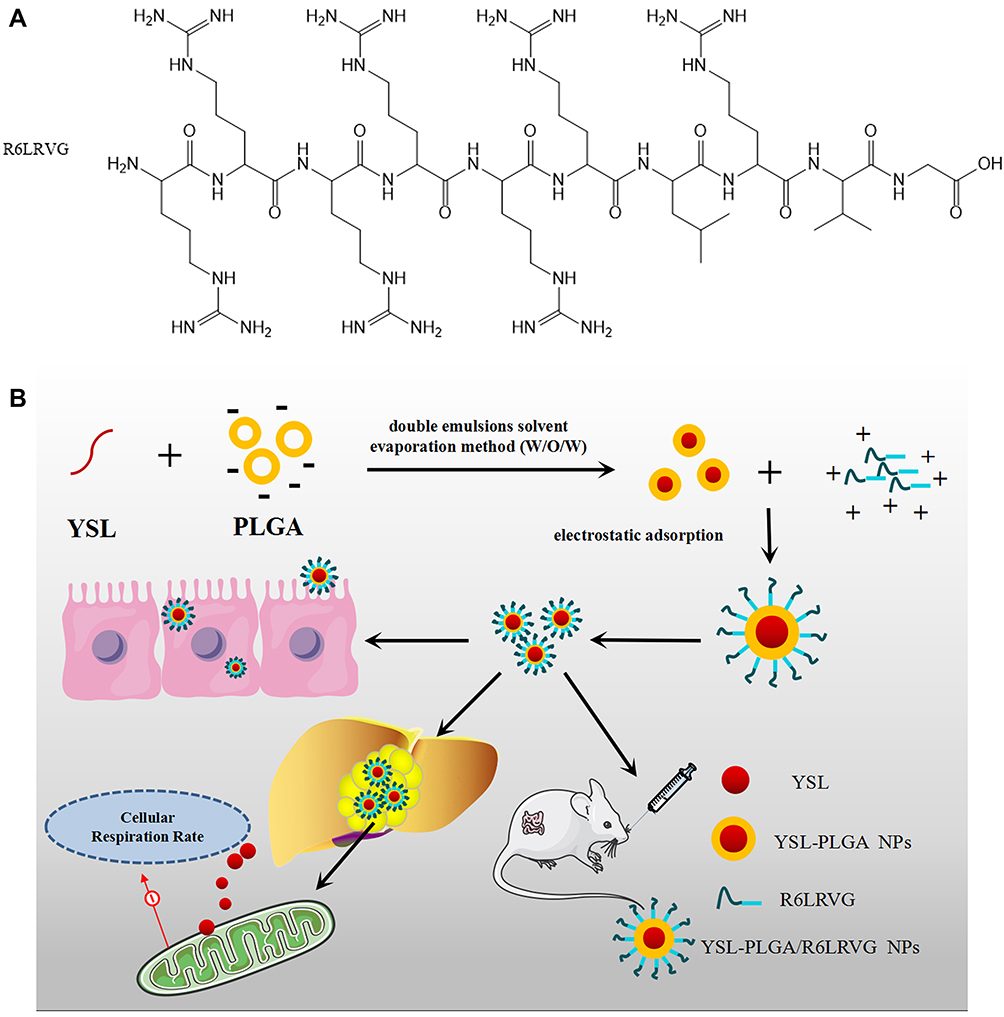 |
Scheme 1 (A) The chemical structure of R6LRVG and (B) schematic diagram of YSL-PLGA/R6LRVG NPs preparation technology and anti-cancer mechanism diagram of nanoparticles. |
Materials and Methods
Materials
YSL and target penetrating peptide (R6LRVG) were purchased from GenScript Co. Ltd (Nanjing, China). PLGA (LA/GA ratio 50:50, average MW 30000) was purchased from Jinan Daigang Biomaterial Co., Ltd (Jinan, China). Sodium azide was obtained from Beyotime (Nantong, China). Fluorescein isothiocyanate (FITC), Hoechst 33,342, methyl-β-cyclodextrin (M-β-CD), chlorpromazine and amiloride hydrochloride were acquired from Aladdin (Shanghai, China). All other solvents were of analytical or chromatographic grades and were procured commercially.
Preparation of YSL-PLGA/R6LRVG NPs
First, YSL-PLGA NPs were prepared using double emulsion solvent evaporation (W/O/W). Briefly, 0.1 mL of aqueous phase containing 20 mg/mL of YSL was slowly dripped into the organic phase containing 0.020 g of PLGA dissolved in 1 mL dichloromethane at 37 °C using magnetic stirring. The mixture was emulsified using ultrasound (160 W, 30 sec) on ice to form a first-order emulsion. Next, the emulsion was slowly added into 2 mL of aqueous sodium cholate (0.5%, w/v) under stirring and subjected to ultrasound (160 W, 60 sec) on ice. Dichloromethane was removed using a rotary evaporator under reduced pressure at 30 °C for 15 min, and the solution was centrifuged at 13,500 rpm at 4 °C for 40 min to obtain a precipitate, which was washed with deionized water. This centrifugation-wash cycle was performed three times. The nanoparticle dispersions that were obtained were pre-frozen overnight at −20 °C and freeze-dried to obtain the final product, which was stored at 4 °C until further use.
Next, YSL-PLGA/R6LRVG NPs were prepared by adding an equal volume of 6 mg/mL R6LRVG solution to the solution containing unmodified nanoparticles. The reaction was allowed to progress for 1 h before centrifuging. Then the supernatant was centrifuged to measure the concentration of R6LRVG by UV spectrophotometer. The precipitate was freeze-drying to obtain the final product, which was stored in 4 °C until further use. Fluorescent-labeled NPs were prepared by dissolving PLGA and FITC in the organic phase using in the method described earlier.
Characterization of the NPs
The morphologies of PLGA NPs, YSL-PLGA NPs, PLGA/R6LRVG NPs and YSL-PLGA/R6LRVG NPs were observed using TEM (Jeol JEM-1400, Jeol Ltd., Japan). Particle size (Z-average), polydispersity index (PDI), and zeta potential (ζ-potential) of the NPs were determined using a Malvern Zetasizer Nano ZS instrument (DLS, Zetasizer Nano, Malvern Instruments Ltd, UK). Before these measurements, the samples were dispersed in deionized water. FTIR spectra (NEXUS670, Nicolet, USA) of samples were collected over a scanning range of 400 to 4000 cm−1 and the resolution of 2 cm−1. The elemental composition of nanoparticles was determined using an elemental analyzer (Vario EL III, Elementar, Germany).
Encapsulation Efficiency (EE) and Drug Loading (DL)
The supernatant was obtained after centrifugation of the nanoparticle preparation and the free YSL content was determined using UV spectrometry (UV-2450, Shimadzu, Japan). At least three replicates were performed to calculate the amount of YSL in the with NPs. EE and DL were calculated using the following formulas:
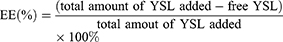
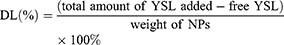
Stability and Drug Release of the NPs in vitro
The stability of NPs was assessed in pepsin-containing simulated gastric fluid (SGF) (0.3%, w/v), pancreatin-containing simulated intestinal fluid (SIF) (1.0%, w/v), and phosphate-buffered saline (PBS) in a shaker at 100 rpm and 37 °C. Samples were measured at defined time intervals (0, 1, 2, and 4 h) or (0, 1, 3, and 6 h) using a Malvern Zetasizer NanoZS90 analyzer (Malvern Instruments Ltd., UK).
The in vitro drug release of NPs was measured using dialysis (MWCO 2 kDa). The dialysis bag was immersed in 20 mL of simulated gastric fluid (SGF) for 1 h, followed by immersion in 20 mL of simulated intestinal fluid (SIF) for 3 h and 20 mL of PBS for 12 h, at 37 °C with gentle shaking. The pH of SGF and SIF was 1.2 and 6.8, respectively. The composition of SGF was 35 mM NaCl, 80 mM HCl, and 0.3% (w/v) pepsin; and the composition of SIF was 50 mM KH2PO4, 15 mM NaOH, and 1.0% (w/v) pancreatin at pH 6.8. Drug concentrations in the dialysis buffer were measured using UV spectrometry at predetermined time intervals.
Cell Culture
Caco-2 cells (KeyGen Biotech Inc.; Nanjing, China) have morphology and function similar to that of the human intestinal epithelium. Human hepatocellular carcinoma BEL-7402 cells were purchased from the Institute of Cell Biology (Academia Sinica; Shanghai, China). Caco-2 cells and BEL-7402 were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 100 IU/mL penicillin, 100μg/mL streptomycin, 2 mM L-glutamine, 1% non-essential amino acids, and 10% fetal bovine serum to prevent contamination. The medium was replaced every two days. Caco-2 cells were cultured in an incubator at 37 °C in an atmosphere of 5% CO2. Cells were passaged using trypsin-EDTA (0.25%, 0.53 mM) at a density of 2.5×105 cells/25 cm2 flask.
Cytotoxicity and Anti-Tumor Effects in vitro
Caco-2 cells were used to measure the biosafety of the synthesized nanoparticles. The viability of NP-treated Caco-2 cells was evaluated using a Cell Counting Kit-8 (CCK-8) assay (Nanjing Keygen Biotech Corp. Ltd, China). Briefly, Caco-2 cells, in the logarithmic growth phase, were seeded in 96-well plates at a density of 1.5×105 cells/well and incubated for 1–2 days. The medium was replaced by serum-free medium containing different concentrations of the NPs (0.1, 0.2, 0.4, 0.8, 1, and 2 mg/mL) as calculated based on the DL rate. The concentration of nanoparticles was selected by referring to previous studies.36 After 24 h and 48 h, the washed cells were treated with CCK-8 reagent. Cells that were not treated with NPs were used as a negative controls. Cell viability was determined using spectrometry by measuring the absorbance at 450 nm and calculated using the following formula:
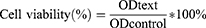
BEL-7402 cells were used to investigate the role of the synthesized nanoparticles in the proliferation of anticancer cells. Briefly, BEL-7402 cells in the logarithmic growth phase were seeded in 96-well plates at a density of 1×105 cells/well and cultured for 24 h. Then, NPs were added to each well separately at predetermined concentrations. After 24 and 48 h, cytotoxicity was measured using a CCK-8 assay kit. Cell viability was determined by calculating the ratio of the absorbance values of the experimental groups to the negative controls. Furthermore, the half-maximal inhibitory concentration (IC50) was calculated for each group based on survival analysis using GraphPad Prism 5 (GraphPad Software Inc., USA).
Transportation of Nanoparticles Across the Caco-2 Cell Monolayer
Caco-2 cells were seeded on transwell diffusion cells (pore size 0.4 μm, 1.12 cm2 growth area, Corning Life Sciences, Acton, MA, USA) and maintained in DMEM for 3 weeks. Transepithelial electrical resistance was measured across the Caco-2 cells growing on the 1.12-cm2 polycarbonate filters of the transwell diffusion cells using an Epithelial Volt-Ohm Meter (MillicellERS-2, USA) to evaluate tight junction barrier. FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs were added on the apical side, and 0.1 mL of samples were collected from the basolateral chamber at predetermined time intervals. The percentage of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs transported through the cell monolayers was calculated using a Multimode Plate Reader (PerkinElmer, Germany).
Cellular Uptake Studies
The cellular uptake of FITC-PLGA NPs, FITC-PLGA/R6LRVG NPs, FITC-PLGA/R6 NPs, and FITC-PLGA/LRVG NPs in Caco-2 and BEL-7402 cells was determined using laser confocal scanning microscopy (LCSM, Fluoview FV 1000, Olympus, Japan). FITC-loaded NPs were prepared following the method described previously. Caco-2 cells in the logarithmic growth phase were seeded on the laser confocal dish at a density of 1×104 cells/well and cultured overnight. FITC-loaded NPs were added to each well at the indicated concentrations and incubated for 1, 3, and 6 h. FITC-PLGA/R6 NPs and FITC-PLGA/LRVG NPs were incubated for 3 h to determine the role of the arginine-rich peptide and LRVG peptide in the cellular uptake of the nanoparticles. Next, the cells were stained with Hoechst 33342 and the cellular uptake of NPs was observed using LCSM. To assess the cellular uptake in cells other than the intestinal-derived cells, BEL-7402 cells were used as a control. A similar method was used to treat BEL-7402 cells with nanoparticles for 3 h to determine cell selectivity.
Flow cytometry (FCS, Beckman Coulter, Inc.) was used to quantify the cellular uptake capacity of NPs. Caco-2 cells were seeded in a 6-well plate and cultured overnight. Then, the cells incubated with FITC-loaded NPs were suspended in DMEM. After incubation for 3 and 6 h, the cells were digested from the 6-well plates and centrifuged to collect single-cell suspensions. The suspensions were diluted and the number of cells in each group was controlled at about 10,000. Lastly, the X-mean intracellular fluorescence intensity of cells was measured using FLowJo 7.6 software. Cells incubated with PBS were used as a negative control.
Endocytosis Pathways
The possible mechanisms of endocytosis of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs were studied by incubating Caco-2 cells with specific endocytosis inhibitors. Caco-2 cells were cultured as described Cell Culture and treated according to the method described previously. Caco-2 cells were pre-incubated with 100 mM sodium azide, 10 µg/mL chlorpromazine, 5 µg/mL colchicine, 0.3 mg/mL amiloride hydrochloride, or 5 mg/mL methyl-β-cyclodextrin (M-β-CD) in serum-free medium at 37 °C for 30 min, and then incubated with a suspension of FITC-loaded NPs at 37 °C for 3 h. To determine the effect of temperature on cellular uptake, cells were incubated with the nanoparticle suspension at 4 °C for 4 h. The results of the inhibition tests are represented as the cellular uptake ratio of the FITC-loaded NPs relative to the untreated control group.
Cellular Respiration Rate
The rate of cellular respiration was measured using an XF96 cell Mito stress test kit (Seahorse Bioscience, Billerica, MA). First, we optimized the experimental conditions to determine the ideal cell density of BEL-7402 cells and the reagent concentrations to be used in our study. Next, BEL-7402 cells (4 × 105 cells/mL) were seeded on 96-well plates (Seahorse Bioscience, Billerica, MA) and cultured overnight based on the optimized experimental results. In the concentration-dependent study, BEL-7402 cells were treated with YSL-PLGA/R6LRVG NPs (containing YSL at a concentration of 0.1 and 0.2 mg/mL) and YSL (0.1 and 0.2 mg/mL) for 1 h, whereas in the time-dependent study, cells were treated with YSL-PLGA/R6LRVG NPs (containing YSL at a concentration of 0.1 mg/mL) and YSL (0.1 mg/mL) for 1 and 3 h.37,38 Next, the original cell culture medium was replaced with Seahorse detection medium (Seahorse Bioscience, Billerica, MA) and cultured at 37 °C for 1 h without CO2 to elicit a response. The basal oxygen consumption rate (OCR) was measured at the beginning of the experiment. Next, we added the respiratory inhibitors (0.5 μM oligomycin, 0.3 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and a mixture of 0.5 μM antimycin A and 0.5 μM rotenone) sequentially to detect changes in the OCR in cells under different conditions using XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA).
In vivo Anti-Tumor Activity
All animal experiments were performed following the guidelines approved by the Ethics Committee of Nanjing Normal University, Nanjing, China (Ethics review number: IACUC-200312). Four six-week-old female BALB/c mice were used for our in vivo studies. To establish the orthotopic tumor model, approximately 2×106 BEL-7402 cells were subcutaneously injected into the right or left flanks of mice.
The antitumor effects of NPs on hepatocellular carcinoma were determined using the BEL-7402-induced tumor-bearing mouse model. The therapeutic effect of free YSL was evaluated after BEL-7402 cells were transplanted subcutaneously into the right axillary subcutaneous of nude mice.
Mice bearing BEL-7402 cells were randomly grouped for drug administration. YSL-PLGA/R6LRVG NPs (160 μg/kg/d) were injected intraperitoneal. The free YSL group (320 μg/kg/d), YSL-PLGA/R6LRVG NPs (320 μg/kg/d) and the saline group (0.2 mL/d) were all given by gavage. Before the oral administration of NPs, the gastric acid was neutralized by the intragastric administration of 0.5 mL of 3% NaHCO3 solution. Intraperitoneal injection and gavage were commenced the next day after tumor implantation, and the drug was administered twice a day for 28 days. The tumor size and weight of mice were measured using a Vernier caliper and balances every 2 days, and the study was completed after 28 days. The tumor size was calculated as 1/6·π·L·W2. On the second day after the last injection, the tumors of each mouse were dissected and the weights recorded. Three tumor diameters (A, B, C) were measured using Vernier caliper, and the tumor volume (V) was calculated using the formula V = 1/6·ρ·A·B·C. Tumor-growth inhibition was calculated as follows:

The mice were weighed every two days to determine drug toxicity. To evaluate NP-induced organ toxicity, the main organs of mice (heart, liver, spleen, lung, and kidney) were harvested, fixed with 4% paraformaldehyde, and sectioned. Hematoxylin and eosin (H&E) staining was used for histological analysis.
Statistical Analysis
Results are presented as mean values and standard error (mean ± SE). Data were evaluated using a one-way analysis of variance (ANOVA) to determine statistical significance. In all cases, the difference was considered statistically significant at P < 0.05. All experiments were performed in three replicates.
Results and Discussion
Preparation and Characterization of the NPs
YSL-PLGA/R6LRVG NPs were prepared for oral delivery based on the double emulsion solvent-evaporation method (W/O/W). The molecular formula of R6LRVG is presented in Scheme 1A. The preparation scheme and anticancer mechanism of YSL-PLGA/R6LRVG NPs is illustrated in Scheme 1B. The results showed that there was no significant difference in R6LRVG content between the two nanoparticles. According to the experimental results, the penetration/targeting peptide has been adsorbed on the surface of nanoparticles by electrostatic adsorption at approximately 33.64%. The results indicated that a considerable amount of R6LRVG had been successfully adsorbed onto the surface of the nanoparticles.
TEM Study
The primary morphologies of the prepared NPs were characterized using TEM. As shown in Figure 1, PLGA NPs, YSL-PLGA NPs, PLGA/R6LRVG NPs, YSL-PLGA/R6LRVG NPs were mostly spherical with smooth surfaces and uniform size distribution. Meanwhile, dynamic light scattering (DLS) studies showed that the particle size of YSL-PLGA/R6LRVG NPs was 222.6 ± 10.4 nm and had a PDI of 0.354 ± 0.069 (Table 1), which was in accordance with the findings of TEM. The zeta potential of YSL-PLGA NPs was −10.34 mV, whereas that of YSL-PLGA/R6LRVG NPs was 18.23 mV. The change in the zeta potential of YSL-PLGA/R6LRVG NPs from negative to positive may be owing to the addition of positively charged R6LRVG. We found that R6LRVG was present on the surface of the nanoparticles. Furthermore, the average diameter of YSL-PLGA/R6LRVG NPs was higher than that of the YSL-PLGA NPs, likely because of the adsorption of R6LRVG on the surface of YSL-PLGA/R6LRVG NPs. (Table 1).
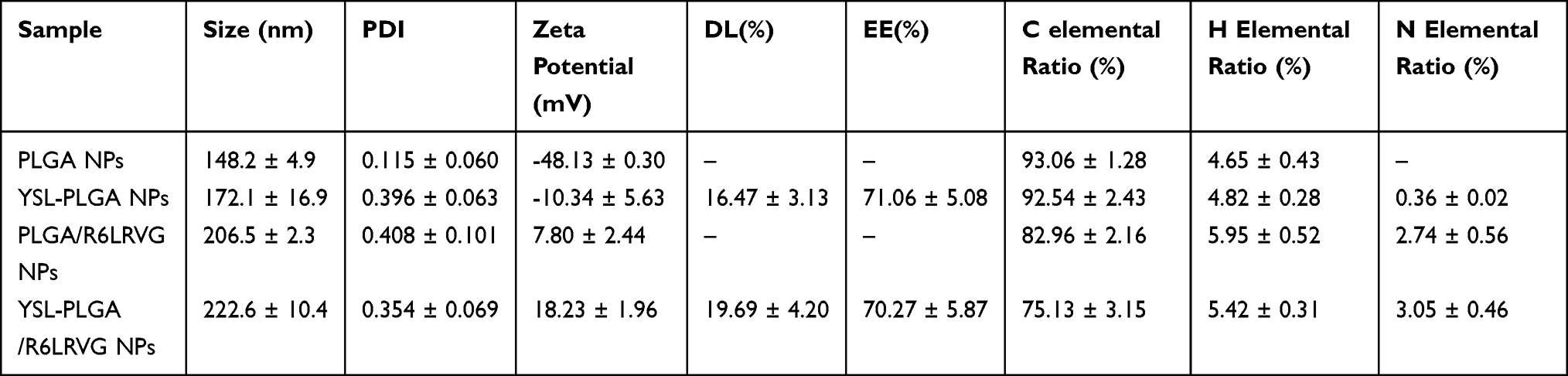 |
Table 1 Mean Size, PDI, ζ-Potential, Encapsulation Efficiency (EE) and Drug Loading (DL) of Nanoparticles and Elemental Analysis of the Peptide Bulk and Nanoparticle Surface |
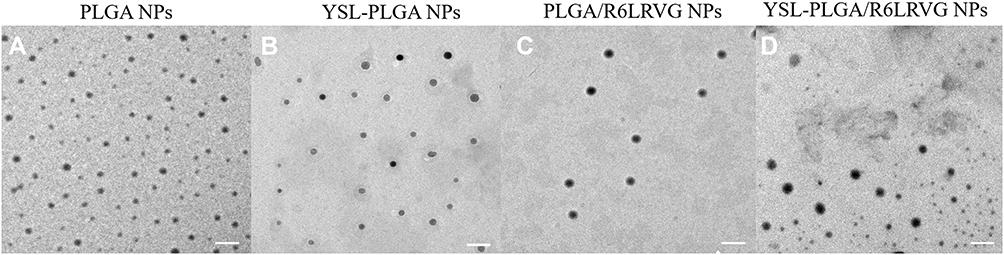 |
Figure 1 The TEM images of (A) PLGA NPs, (B) YSL-PLGA NPs, (C) R6LRVG/PLGA NPs and (D) YSL-R6LRVG/PLGA NPs. Scale bar = 200 nm. |
The Encapsulation Efficiency (EE) and Drug Loading (DL)
The EE and DL of YSL-PLGA/R6LRVG NPs were 70.27 ± 5.87% and 19.69 ± 4.20%, respectively. These findings were not significantly different from the corresponding values determined for YSL-PLGA NPs (71.06 ± 5.08% and 16.47 ± 3.13%, respectively) (Table 1).
FTIR Spectrum Study
FTIR spectra are shown in Figure 2. The characteristic peaks of YSL were the stretching vibration peak of the =CH of the benzene ring at 2962 cm−1 and the stretching vibration peaks of the C=C skeleton in the benzene ring at 1668 cm−1 and 1517 cm−1. However, in the nanoparticles, these characteristic peaks were either weakened or not detected, indicating the encapsulation of YSL in the nanoparticles. The PLGA and YSL-PLGA NPs were found to have typical characteristic absorption peaks of PLGA at 1759 cm−1 (-CO-), 1186 cm−1 (C-O-C), and 1456 cm−1 and 1386 cm−1 (-CH). These findings indicated that the molecular structure of PLGA had not changed significantly after the preparation of nanoparticles. The FTIR spectrum of R6LRVG shows the absorption spectrum of amino acids appearing at 3413 cm−1 and a peak at 1650 cm−1 corresponding to the amide group (Figure 2). Similarly, the YSL-PLGA/R6LRVG NPs showed a significant increase in the intensity of the carbonyl group (-CO-) stretching band at 1649 cm−1, and the presence of a broad secondary amine (-NH-) stretching band at 3200–3700 cm−1. This increase can be attributed to the large number of peptide bonds and guanidine groups in R6LRVG, suggesting the successful modification of R6LRVG on the surface of the nanoparticles.
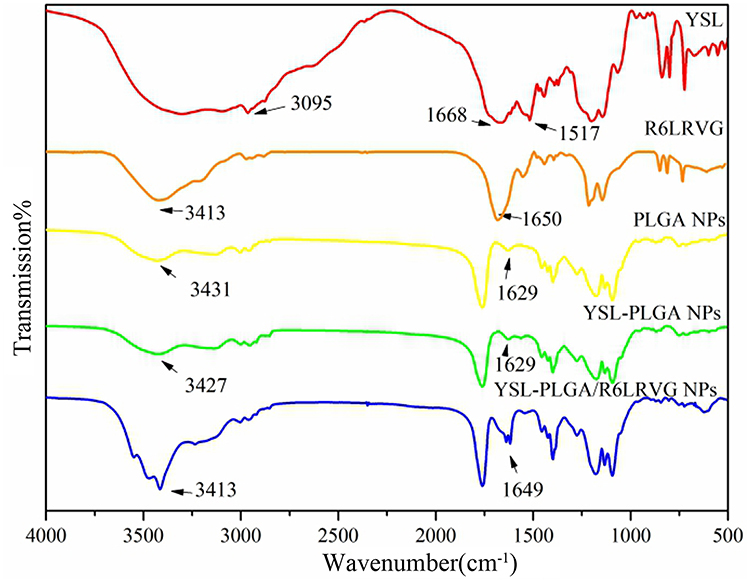 |
Figure 2 The Fourier Transform Infrared Spectrum of YSL; R6LRVG; PLGA NPs; YSL-PLGA NPs; YSL-R6LRVG/PLGA NPs. |
Elemental Analysis
The C/H/N content of nanoparticles is shown in Table 1. The elemental N level in PLGA NPs was undetectable, whereas its levels in YSL-PLGA NPs and YSL-PLGA/R6LRVG NPs were determined to be 0.361% and 3.050%, respectively, and significantly higher in the latter. At the same time, in YSL-PLGA/R6LRVG NPs, the elemental composition percentage of H increased and that of C decreased in comparison with YSL-PLGA NPs. In terms of chemical structure, the main components of R6LRVG were N and H, further confirming that R6LRVG was successfully modified into YSL-PLGA NPs.
The above characterization indicated the successful preparation of YSL-PLGA/R6LRVG NPs.
Enzymatic Stability and Drug Release of the NPs in vitro
The stability of NPs in the GIT is one of the main problems during oral drug delivery. To study the stability of the NPs under various conditions of the GIT, the stability of the NPs was evaluated in the presence of SGF and SIF at 37 °C at 4 and 6 h. Particle-size analysis revealed that, overall, the mean particle size and PDI of all formulations tended to increase. In the presence of SIF, the mean particle size of YSL-PLGA NPs did not show a significant change after four hours, but rather increased after the sixth hour. On the other hand, the mean particle size of YSL-PLGA/R6LRVG NPs did not change significantly after 6 h (Figure 3A). Our results indicated that both nanoparticles were stable in SIF and that YSL-PLGA/R6LRVG NPs were more stable than the YSL-PLGA NPs. Moreover, a significant increase in the size of YSL-PLGA/R6LRVG NPs and YSL-PLGA NPs was observed after treatment with SGF, which may be ascribed to the degradation of nanoparticles in the strongly acidic environment. The change in particle size of the YSL-PLGA/R6LRVG NPs was smaller compared to the YSL-PLGA NPs after 4 h (Figure 3B), indicating poor stability of the two NPs in SGF; however, YSL-PLGA/R6LRVG NPs were more stable than the YSL-PLGA NPs. In conclusion, R6LRVG modification could improve the stability of the nanoparticles in the GIT.
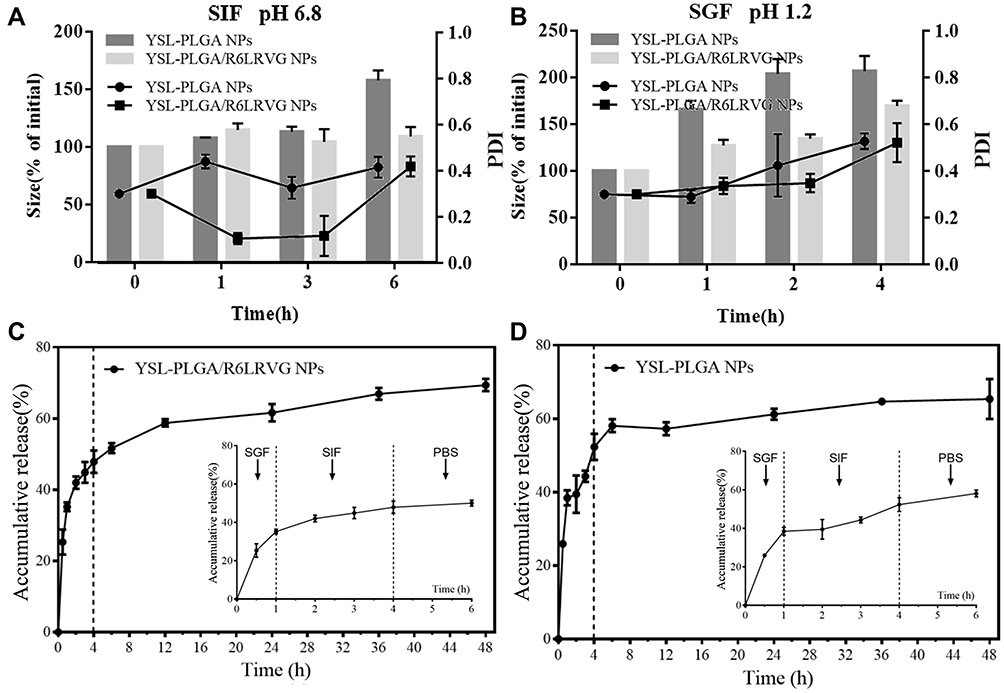 |
Figure 3 Stability and in vitro drug release of YSL-PLGA/R6LRVG NPs and YSL-PLGA NPs. Particle size and PDI variation of YSL-PLGA/R6LRVG NPs and YSL-PLGA NPs during the incubation in SIF (35 mM NaCl, 80 mM HCl, and 0.3% (w/v) pepsin, ph 1.2) (A) or SGF (50 mM KH2PO4, 15 mM NaOH, and 1.0% (w/v) pancreatin, ph 6.8) (B). The accumulative release profile of YSL-PLGA/R6LRVG NPs (C) and YSL-PLGA NPs (D) in SGF, SIF and PBS, respectively. The two vertical dashed lines indicate the two time points of buffer changing (1h and 4h). The inset shows the the expanded curve in the first 6 h. Data represent mean s.d. (n = 3). All experiments were conducted at 37 °C. |
The drug-release characteristics of YSL-PLGA/R6LRVG NPs and YSL-PLGA NPs in the gastrointestinal environment were studied based on the transport time of the nanoparticles in the GIT under normal physiological conditions (1 h in the stomach and 3 h in the small intestine).39,40 As shown in Figure 3C and D, YSL was rapidly released in the SGF solution during the first hour and gradually released over the next three hours. This finding was consistent with results of the stability experiment. The two NPs were found to be poorly stable in SGF but highly stable in SIF. After 4 h, the cumulative release of YSL from YSL-PLGA/R6LRVG NPs (50.02%) was slightly lower than that from YSL-PLGA NPs (58.10%), suggesting that YSL-PLGA/R6LRVG NPs had a better controlled-release profile than YSL-PLGA NPs in the GIT. Overall, our findings revealed that there was no initial burst release in the acidic SGF and SIF, and about 70% cumulative release was observed within 2 days, highlighting the favorable drug-release characteristics of the nanoparticles.
Cytotoxicity and Antitumor Effect in vitro
We used colon adenocarcinoma Caco-2 cells to evaluate the systemic cytotoxicity of the four nanoparticles. Cells were incubated with different concentrations of YSL-PLGA NPs, YSL-PLGA/R6LRVG NPs, PLGA NPs, and PLGA/R6LRVG NPs, and the cell viability was determined using a CCK-8 assay. As shown in Figure 4A and B, after 24 or 48 h of incubation, the cell viability of the drug-loaded groups (YSL-PLGA NPs, YSL-PLGA/R6LRVG NPs) was not significantly different from the material group (PLGA NPs, PLGA/R6LRVG NPs). The survival rate of Caco-2 cells in the treatment group was between 80% and 120% at each concentration. Overall, no significant cytotoxicity was detected at the tested concentrations in the range of 0.2–3.2 mg/mL when compared to the control group. Our findings revealed that the nanoparticles had good cell compatibility, which is consistent with the non-cytotoxicity of PLGA in previous studies.23 Therefore, YSL-PLGA/R6LRVG NPs could be considered safe for oral administration.
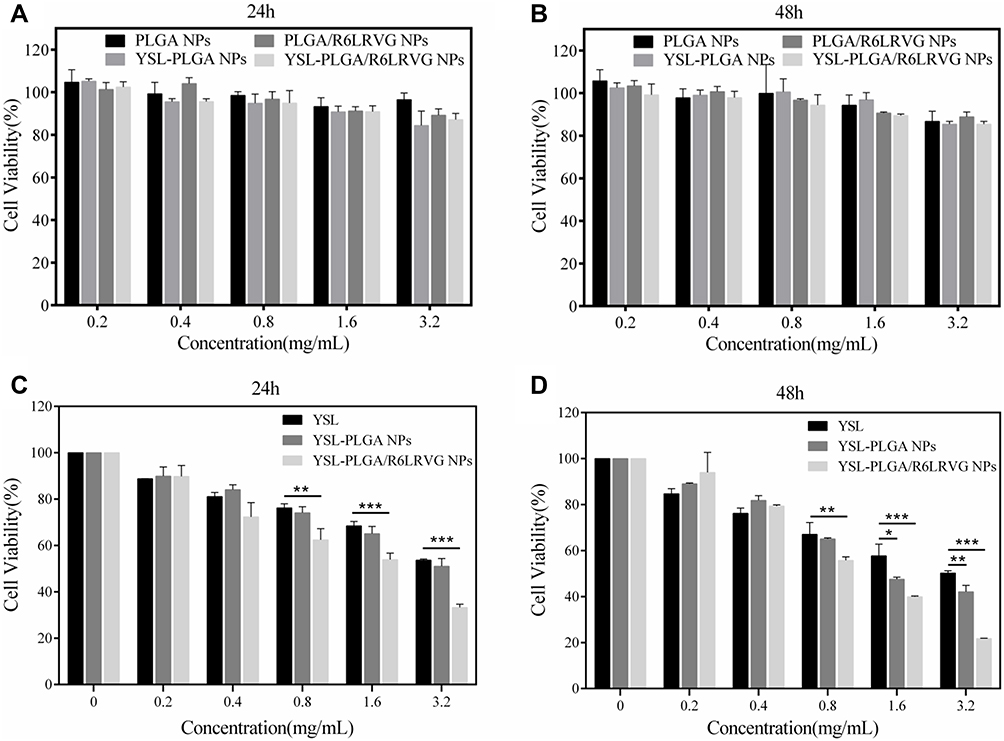 |
Figure 4 In vitro cell viability of Caco-2 cells against PLGA NPs, YSL-PLGA NPs, PLGA/R6LRVG NPs and YSL-PLGA/R6LRVG NPs after incubating 24 h (A) or 48 h (B) with different concentration by CCK-8 assay. In vitro cell viability of BEL-7402 against YSL, YSL-PLGA NPs, YSL-PLGA/R6LRVG NPs after incubating 24 h (C) or 48 h (D) with different concentration by CCK-8 assay. Data are presented as average ± standard deviation (n=3). Significant differences are denoted as follows: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with the control. |
The in vitro cytotoxicity of two YSL-loaded nanoparticles was evaluated using the human hepatoma cell line, BEL-7402. The viability of tumor cells after 24 and 48 h of incubation with different concentrations of free YSL and loaded YSL nanoparticles is shown in Figure 4C and D. The viability of cells treated with pure YSL and YSL-loaded nanoparticles (both concentrations of 3.2 mg/mL) was less than 50%. Compared to the 24-hour treatment, the 48-hour treatment of cells with YSL-PLGA/R6LRVG NPs significantly reduced the cell-survival rate (P < 0.01). Additionally, compared to the cytotoxicity observed after treatment with free YSL, treatment with YSL-PLGA/R6LRVG NPs was found to be extremely significantly cytotoxic to BEL-7402 cells (P < 0.0001) with a cell-survival rate of only 21% after 48 h. These findings could be attributed to the improved delivery of YSL to the cells by the YSL-PLGA/R6LRVG NPs. Collectively, our results indicated that BEL-7402 cells incubated with YSL-PLGA/R6LRVG NPs could induce the extensive inhibition of cell proliferation compared to the incubation with YSL and YSL-PLGA NPs.
Cellular Transport and Uptake
The permeability of nanoparticles across the Caco-2 monolayer cells was studied. As illustrated in Figure 5A, compared to FITC-PLGA NPs, the rate of transport of FITC-PLGA/R6LRVG NPs appears significantly increased by approximately 2-fold (P < 0.05). This finding demonstrates that the R6LRVG-modified nanoparticles were more efficiently absorbed across the intestinal epithelium.
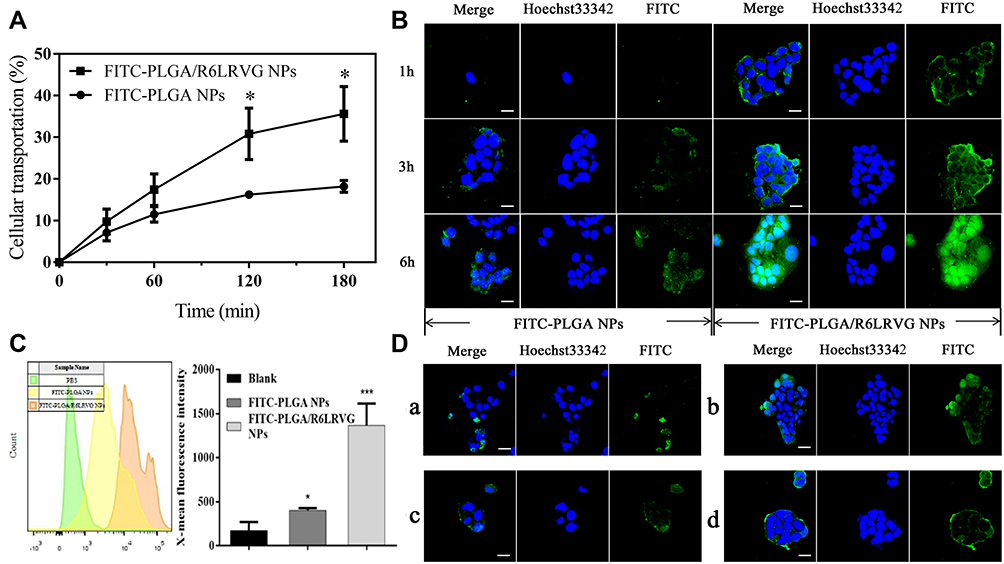 |
Figure 5 (A) Time course of apical-to-basolateral transportation of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs across the transwell-grown Caco-2 cell monolayer under 37°C incubation. (B) CLSM images of Caco-2 cells treated with FITC-PLGA and FITC-PLGA/R6LRVG NPs. The blue signal represents nucle stained with Hoechst33342 and the green signal represents FITC. Scale bar = 10 μm. (C) Intracellular internalization of particles in Caco-2 cells was detected by FCS. The cells were exposed to FITC-PLGA or FITC-PLGA/R6LRVG NPs at a concentration of 600 µg mL−1. The blank group was exposed to HBSS buffer without NPs. Data are presented as average ± standard deviation (n=3). (D) CLSM images of Caco-2 cells treated with (A) FITC-PLGA/R6 and (B) FITC-PLGA/LRVG NPs and BEL-7402 cells treated with (C) FITC-PLGA and (D) FITC-PLGA/R6LRVG NPs. The blue signal represents nucle stained with Hoechst33342 and the green signal represents FITC. Scale bar = 10 μm. Significant differences are denoted as follows: * P < 0.05, *** P < 0.001, compared with the control. |
The uptake of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs in Caco-2 cells was observed using CLSM. The nanoparticles and nuclei were labeled with FITC (exhibiting green fluorescence) and Hoechst 33,258 (exhibiting blue fluorescence), respectively. As shown in Figure 5B, it was found that the cellular uptake of the two NPs increased with treatment time. Furthermore, the intensity of FITC fluorescence exhibited by FITC-PLGA/R6LRVG NPs was significantly greater than that of FITC-PLGA NPs at all time points, indicating that Caco-2 cells could better absorb FITC-PLGA/R6LRVG NPs. The overlap of nuclear and FITC signals observed during CLSM indicated the consistency in results. R6LRVG-modified nanoparticles showed an enhanced cellular uptake. The results showed that the cellular uptake of FITC-PLGA/R6LRVG NPs was significantly increased compared to the FITC-PLGA NPs.
To determine the contribution of these peptide sequences in the cellular uptake of nanoparticles, we measured the absorbances of YSL-PLGA/R6 NPs and YSL-PLGA/LRVG NPs. As shown in Figure 5D a and b, FITC-PLGA/LRVG NPs showed stronger fluorescence intensity and wider distribution compared to FITC-PLGA/R6 NPs, indicating the cell-targeting effect post LRVG peptide modification after its cellular uptake in Caco-2 cells. However, the FITC fluorescence-signal intensity of FITC-PLGA/R6LRVG NPs was significantly stronger than that of FITC-PLGA/R6 NPs and FITC-PLGA/LRVG NPs treated for 3 h, which suggested that the binding of R6LRVG sequence contributed more to cell targeting and/or cellular uptake than the two peptide sequences, thereby effectively enhancing the uptake.
The selective uptake of R6LRVG was determined by studying the cellular uptake of the nanoparticles in BEL-7402 cells. As shown in Figure 5D c and d, the fluorescence intensity of FITC-PLGA NPs in BEL-7402 cells was similar to that in Caco-2 cells, whereas FITC-PLGA/R6LRVG NPs showed a significant decrease. These results indicated that R6LRVG-modified nanoparticles could target the intestinal epithelial cells and facilitate their uptake.
Results from flow cytometry showed that the average intracellular fluorescence intensity of FITC-PLGA/R6LRVG NPs was 3 times higher than that of FITC-PLGA NPs, suggesting the increase in cellular uptake (Figure 5C). This finding revealed that the modification of R6LRVG on the surface of NPs significantly improved their cellular uptake efficiency (P < 0.01), which may shorten the residence time of nanoparticles in the intestinal tract and reduce drug loss.
Endocytosis Pathways
To investigate the possible uptake mechanism of YSL-loaded nanoparticles, further studies were conducted using specific inhibitors. NaN3 is a comprehensive energy-consuming inhibitor of active transport. M-β-CD is used as a specific endocytosis inhibitor mediated by caveolae/lipid rafts. Chlorpromazine can inhibit reticulin-dependent endocytosis. Amiloride is a Na+/H+ exchange inhibitor during micropinocytosis, whereas colchicine inhibits macropinocytosis. Cells were treated at 4 °C to determine the energy-dependent uptake.
As shown in Figure 6A and B, incubation at 4 °C and treatment with sodium azide extremely significantly inhibited the uptake of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs in Caco-2 cells (P < 0.001), indicating that the mode of cellular uptake was by active transport. Meanwhile, co-incubation with M-β-CD (P < 0.01) and chlorpromazine (P < 0.001) significantly inhibited the uptake of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs, suggesting that caveolae and reticulin-mediated endocytosis play a significant role in their cellular uptake. Overall, amiloride affected the uptake of FITC-PLGA NPs and FITC-PLGA/R6LRVG NPs in Caco-2 cells (P < 0.05). On the contrary, the fluorescence intensity of the two variants of nanoparticles was similar to that of the control group in the colchicine-treated group, indicating that the endocytosis of nanoparticles was not closely related to macropinocytosis. Furthermore, there were no significant differences in the endocytosis pathway between the two types of nanoparticles. Therefore, our results suggested that the cellular uptake of YSL-PLGA NPs and YSL-PLGA/R6LRVG NPs may be mainly mediated by the endocytosis of reticulin and caveolae/lipid rafts. In a previous study,41,42 using inhibitor assays, octaarginine-modified nanoparticles were shown to be taken up by cells via the endocytosis of reticulons and caveolae/lipid rafts. These findings agree with the results of our study. However, findings using LRVG peptide-decorated nanoparticles have not been reported. In general, the endocytosis pathway of R6LRVG is similar to that of the arginine-rich peptide.
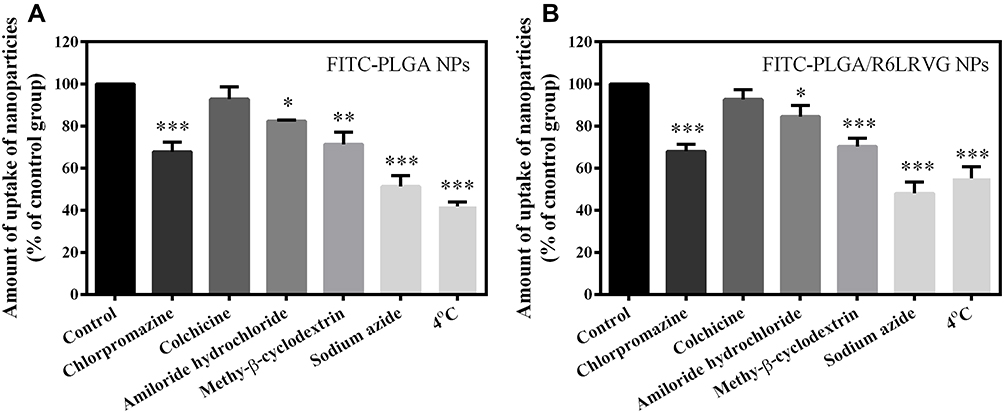 |
Figure 6 Cellular uptake mechanism by Caco-2 cells after incubation with (A) FITC-PLGA NPs and (B) FITC-PLGA/R6LRVG NPs under 100 mM NaN3, 5 mg/mL M-β-CD, 10 µg/mL Chlorpromazine, 0.3 mg/mL Amiloride, 5 µg/mL Colchicine (n = 3). Data are presented as average ± standard deviation (n=3). Significant differences are denoted as follows: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with the control. |
Cellular Respiration Rate
To further investigate the effects of YSL-PLGA/R6LRVG NPs on mitochondrial respiration in human hepatoma BEL-7402 cells, we used the XF cell Mito stress test kit. Oligomycin (ATP coupling agent), FCCP (uncoupling agent), mycin (antimicrobial), and rotenone (mitochondrial inhibitor) were added at predetermined time points, and the OCR of mitochondria at various stages was detected using an XF96 analyzer. This approach is frequently used to measure basal respiratory values, oxygen consumption for ATP synthesis, maximum oxygen consumption, and standby maximum respiratory values after mitochondrial inhibition, all of which are essential parameters representing mitochondrial respiratory values. BEL-7402 cells were treated with either NaN3 (positive control), DMEM medium without serum (negative control), different concentrations of YSL-PLGA/R6LRVG NPs, and pure YSL over different time points.
Figure 7 represents the changes in respiratory values in BEL-7402 after treatment with pure YSL and YSL-PLGA/R6LRVG NPs. As shown in Figure 7A, the respiratory value of BEL-7402 cells decreased with an increase in YSL concentration in both the pure YSL and YSL-PLGA/R6LRVG NP groups. Compared to the negative control, the spare respiratory capacity of cells treated with YSL-PLGA/R6LRVG NPs at YSL concentrations of 0.1 and 0.2 mg/mL was reduced by 30.88 and 52.63%, respectively. The inhibition of ATP synthesis by YSL-PLGA/R6LRVG NPs is related to blocking of the proton channel of the F0 component of ATP synthase and uncoupling of the electron transport chain. After the addition of FCCP, the reduction rates of coupling efficiency in YSL-PLGA/R6LRVG NPs treated groups were 42.14 and 59.02%, respectively, while the respiratory reserve capacity was reduced by 44.12 and 52.91%, respectively at the doses on 0.1 and 0.2 mg/mL respectively. As shown in Figure 7A, different concentrations of pure YSL inhibited mitochondrial respiration; however, similar concentrations of YSL-PLGA/R6LRVG NPs exhibited a more substantial inhibitory effect than the pure YSL. Compared to the results obtained using the same equivalent of pure YSL, the spare respiratory capacity of cells treated with YSL-PLGA/R6LRVG NPs at YSL concentrations of 0.1 and 0.2 mg/mL decreased by 13.03 and 42.61%, respectively, the coupling efficiency decreased by 23.71 and 33.09%, respectively, and the respiratory reserve capacity decreased by 30.13 and 36.86%, respectively. Collectively, these findings suggested that YSL-PLGA/R6LRVG NPs could significantly increase mitochondrial damage and that the reduction in effect was concentration dependent.
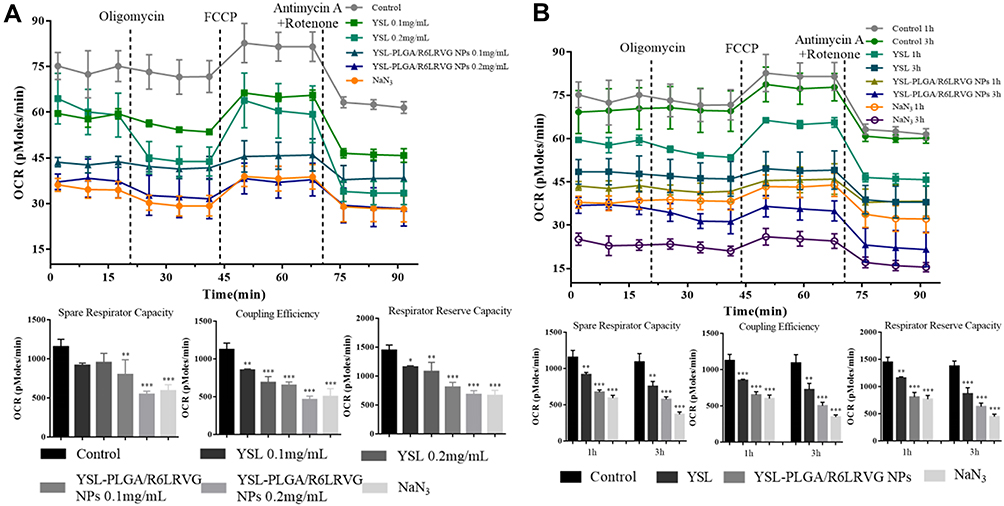 |
Figure 7 Concentration- and time-dependence effects of YSL-PLGA/R6LRVG NPs on (A and B) mitochondrial respiration of BEL-7402 cell were measured by the Seahorse extracellular flux analyzer. The line graph shows the oxygen consumption rate (OCR) of tested cells exposed sequentially to the modulator of mitochondrial respiration (oligomycin, FCCP, antimycin A, and rotenone) following treated with YSL-PLGA/R6LRVG NPs (at YSL concentration of 0.1 mg/mL and 0.2 mg/mL) and YSL (0.1 and 0.2 mg/mL) for 1 h, or with YSL-PLGA/R6LRVG NPs and YSL at the same concentration (0.1 mg/mL) for different times (1 or 3 h). FBS-free medium was a negative control, and NaN3 (10 μM) was a positive control. The column diagrams are spare respirator capacity, coupling efficiency and respirator reserve capacity by the mitochondrial OCR data analysis. Data are presented as average ± standard deviation (n=3). Significant differences are denoted as follows: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with the control. |
To further explore the relationship between mitochondrial respiratory function and the treatment time of YSL-PLGA/R6LRVG NPs, the respiration of cells treated with YSL-PLGA/R6LRVG NPs at a concentration of 0.1 mg/mL of YSL for 1 and 3 h was determined. It can be seen in Figure 7B that compared to the negative control group, YSL-PLGA/R6LRVG NPs reduced the spare respiratory capacity by 41.47% (1 h) and 47.23% (3 h), the coupling efficiency by 42.14% (1 h) and 54.08% (3 h), and the respiratory reserve capacity by 44.12% (1 h) and 54.11% (3 h). Meanwhile, we found that incubation with YSL over different time periods also inhibited mitochondrial respiration to a certain extent. Compared to that of the cells treated with pure YSL, the spare respiratory capacity of cells treated with YSL-PLGA/R6LRVG NPs for 1 and 3 h reduced by 26.35% (1 h) and 23.85% (3 h), the coupling efficiency decreased by 23.71% (1 h) and 30.80% (3 h), and the respiratory reserve capacity reduced by 30.13% (1 h) and 27.29% (3 h).
These findings revealed that free YSL could decrease the basal respiratory value, mitochondrial oxygen consumption, and maximal respiratory rate compared to the control group, indicating that free YSL could interfere with mitochondrial function. These results are consistent with the findings reported by Xuchun et al (2018),11 which indicate that YSL directly affected the mitochondria. Results of the mitochondrial respiration test showed that the inhibition of mitochondrial respiration in cancer cells by YSL-PLGA/R6LRVG NPs was more significant than that of free YSL. This result suggested that YSL-PLGA/R6LRVG NPs had an apparent mitochondrial accumulation, following which the circulatory function of cells was damaged, resulting in cell death. It has been reported that there was the considerable membrane potential in the inner membrane of mitochondrial, and the positive charge was conducive to target mitochondria.43–49 The YSL-PLGA/R6LRVG NPs were positively charged owing to the modification of the targeted membrane-penetrating peptide, R6LRVG, which enhanced mitochondrial targeting and their eventual accumulation in the mitochondria. Overall, YSL-PLGA/R6LRVG NPs had a significant effect on mitochondrial ATP synthesis and respiratory reserve capacity, thereby reducing cellular respiration in a concentration- and time-dependent manner.
Antitumor Activity in vivo
To determine the antitumor activity in vivo of YSL-PLGA/R6LRVG NPs in liver cancer, BEL-7402-induced tumor-bearing mice were intraperitoneally injected and gavaged with the prepared samples.
In Figure 8B, the tumor-volume growth curve of mice in the saline-treated and YSL (P.O.) groups increased significantly with time. However, the tumor volume decreased slightly in the YSL-PLGA NPs (P.O.), YSL-PLGA/R6LRVG NPs (P.O.), and YSL-PLGA/R6LRVG NPs (i.p.) groups (Figure 8A). As shown in Figure 8B and C, the tumor volumes of mice in the saline and YSL (P.O.) groups were 684.36 and 652.66 mm3 after 28 days of administration, with a mass of 0.78 and 0.73 g, respectively. Our results indicated that YSL (P.O.) had no inhibitory effect on tumor cells compared to the negative control, which could likely be attributed to the rapid degradation and inactivation of the oral polypeptide after entering the GIT, rendering it unsuitable to reach the tumor site. Meanwhile, tumor growth was significantly inhibited in the groups of mice administered intragastric YSL-PLGA NPs and YSL-PLGA/R6LRVG NPs, with tumor volumes of 97.72 and 60.01 mm3 and mass of 0.12 and 0.07 g, respectively. In the YSL-PLGA/R6LRVG NP-administered (i.p.) group, the tumor volume and mass were determined to be 55.13 mm3 and 0.05 g, respectively. The results showed that upon injection, YSL-PLGA/R6LRVG NPs could effectively reach the tumor site and inhibit tumor growth. As shown in Figure 8C, the tumor-inhibition rate of mice in the YSL-PLGA/R6LRVG NPs group (P.O.) was higher than those administered YSL-PLGA NPs (P.O.) by 2%. Compared to the injection group, the YSL-PLGA NPs group had a significant difference (P < 0.05), while the YSL-PLGA/R6LRVG NPs group had no significant difference. This suggests that the tumor mass of YSL-PLGA/R6LRVG NPs group is closer to that of injection group. These results indicated that YSL-PLGA/R6LRVG NPs had better anticancer efficacy in vivo. The R6LRVG-modified nanoparticles are more efficiently absorbed through the intestinal epithelial cells owing to the co-modification of the hexa-arginine in the molecule and the presence of the targeted membrane-penetrating peptide, LRVG. Moreover, PLGA nanoparticles can help prevent drug degradation in the GIT and improve drug stability, thus resulting in the accumulation of YSL in tumors and enhancing the anticancer effect after oral administration.
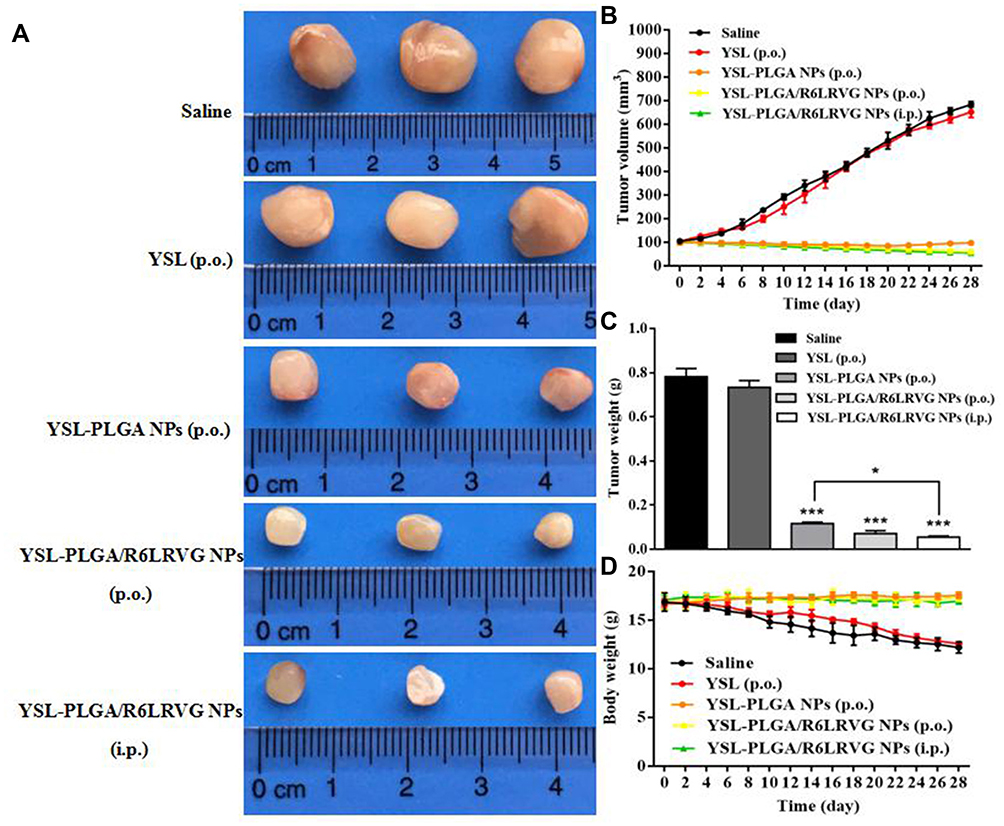 |
Figure 8 Images show tumor growth (A). Influence of different compounds on changes of antitumor effects (B) and body weight (D) of BEL-7402 xenograft-bearing nude mice. Weights of tumors (C) resected from each group of sacrificed mice on the last day. Data are presented as average ± standard deviation (n=3). Significant differences are denoted as follows: * P < 0.05, *** P < 0.001, compared with the control. |
Changes in the body weights of mice were recorded as an indicator of safety. With an increase in tumor volume, the nude mice in the saline and YSL (P.O.) groups gradually presented with dysfunctional manifestations, such as impaired movement, loss of appetite, emaciation, and weight loss. No significant weight loss was observed in mice treated with YSL-PLGA NPs (P.O.), YSL-PLGA/R6LRVG NPs (P.O.), and YSL-PLGA/R6LRVG NPs (i.p.) (Figure 8D), indicating the safety of the nanoparticles. Owing to the excellent inhibition of tumor growth, hematoxylin-eosin (H&E) staining was performed to differentiate the necropsied tumor tissues from the normal tissues (Figure 9). H&E analysis did not show damage to the heart, liver, spleen, lung, or kidney in any of the groups, suggesting that YSL-PLGA/R6LRVG NPs did not lead to noticeable adverse effects in normal tissue during antitumor therapy at the tested doses.
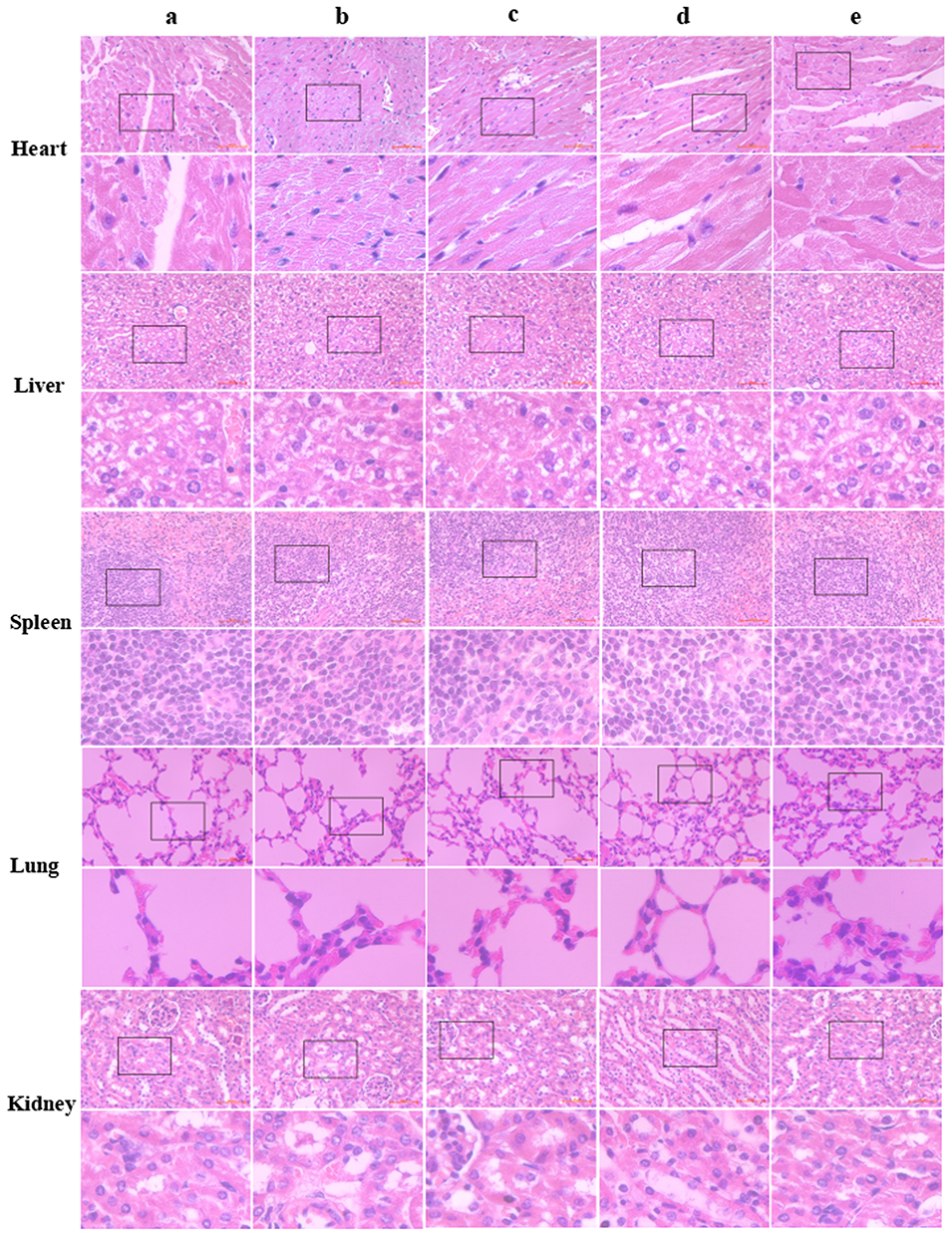 |
Figure 9 H&E staining images determined the toxicity of (A) saline; (B) YSL (P.O.); (C) YSL-PLGA NPs (P.O.); (D) YSL-PLGA/R6LRVG NPs (P.O.) and (E) YSL-PLGA/R6LRVG NPs (i.p.) to the main organs (heart, liver, spleen, lung and kidney) of BEL-7402 xenografted nude mice. |
In addition, self assembly of short peptides is a common phenomenon.50 YSL may self-assemble. In this study, the prepared nanoparticles had a good dispersion coefficient, which was not conducive to the aggregation of the short peptide itself. Meanwhile, YSL peptide loaded nanoparticles exhibited stronger in vitro hepatoma cell toxicity and better absorption. The results of pharmacodynamic study showed that the nanodelivery system with targeting effect could significantly improve the therapeutic efficacy of YSL polypeptides, which overcame its difficulty to be orally absorbed into the site of action by intestine.
Conclusions
We successfully prepared and evaluated R6LRVG-modified YSL-PLGA nanoparticles (YSL-PLGA/R6LRVG NPs) for oral anti-tumor therapy in the management of HCC. The multifunctional nanoparticles (YSL-PLGA/R6LRVG NPs) synthesized in this study had good biocompatibility, high cell permeability, and excellent efficacy in tumor cells. Further mechanistic studies revealed that the uptake of YSL-PLGA/R6LRVG NPs could be related to multiple pathways mediated by endocytosis of reticulin and caveolae/lipid rafts. Moreover, we found that YSL-PLGA/R6LRVG NPs could interfere with mitochondrial function. Specifically, YSL-PLGA/R6LRVG NPs could decrease the basal respiratory value, mitochondrial oxygen consumption, and maximal respiratory rate. Most importantly, antitumor effects were observed in vivo after oral drug delivery. H&E findings did not reveal any histological changes in the major organs. The results showed R6LRVG peptide modification played a vital role on oral YSL delivery. This may be mainly because R6LRVG not only has the function of intestinal targeting, but also has the ability of penetrating. Besides, R6LRVG has a certain function of targeting mitochondria. Therefore it is conducive to oral intestinal absorption and oral therapeutic effect. Taken together, it can be reasonably inferred that YSL-PLGA/R6LRVG NPs constitute an effective oral drug delivery system owing to their ease of intestinal absorption after oral delivery, which highlights their potential in the management of HCC.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81573337).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bilensoy E. Cationic nanoparticles for cancer therapy. Expert Opin Drug Delivery. 2010;7(7):795–809. doi:10.1517/17425247.2010.485983
2. Jemal A, Siegel R, Xu JQ, et al. Cancer Statistics. CA Cancer J Clin. 2010;60(5):277–300. doi:10.3322/caac.20073
3. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
4. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
5. Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi:10.1002/ijc.25516
6. Chen JG, Zhang SW. Liver cancer epidemic in China: past, present and future. Semin Cancer Biol. 2011;21(1):59–69. doi:10.1016/j.semcancer.2010.11.002
7. Hernandez-Alvarez MI, Zorzano A. Mitochondrial Dynamics and Liver Cancer. Cancers. 2021;13(11):2571. doi:10.3390/cancers13112571
8. Lan HY, An P, Liu QP, et al. Aidi injection induces apoptosis of hepatocellular carcinoma cells through the mitochondrial pathway. J Ethnopharmacol. 2021;274:114073. doi:10.1016/j.jep.2021.114073
9. Ruman U, Fakurazi S, Masarudin MJ, et al. Nanocarrier-Based Therapeutics and Theranostics Drug Delivery Systems for Next Generation of Liver Cancer Nanodrug. Int J Nanomedicine. 2020;15:1437–1456. doi:10.2147/IJN.S236927
10. Yao Z, Che XC, Lu R, et al. Inhibition by tyroserleutide (YSL) on the invasion and adhesion of the mouse melanoma cell. Mol Med. 2007;13(1–2):14–21. doi:10.2119/2006-00061.Yao
11. Che XC, Lu R, Fu Z, et al. Therapeutic effects of tyroserleutide on lung metastasis of human hepatocellular carcinoma SK-HEP-1 and its mechanism affecting ICAM-1 and MMP-2 and −9. Drug Des, Dev Ther. 2018;12:3357–3368. doi:10.2147/DDDT.S177126
12. Lu R, Jia J, Bao L, et al. Experimental study of the inhibition of human hepatocarcinoma Bel7402 cells by the tripeptide tyroserleutide(YSL). Cancer Chemother Pharmacol. 2006;57(2):248–256. doi:10.1007/s00280-005-0046-z
13. Date AA, Hanes J, Ensign LM. Nanoparticles for oral delivery: design, evaluation and state-of-the-art. J Control Release. 2016;240:504–526. doi:10.1016/j.jconrel.2016.06.016
14. Plapied L, Duhem N, Des Rieux A, et al. Fate of polymeric nanocarriers for oral drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):228–237. doi:10.1016/j.cocis.2010.12.005
15. Shan W, Zhu X, Liu M, et al. Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano. 2015;9(3):2345–2356. doi:10.1021/acsnano.5b00028
16. Kriegel C, Attarwala H, Amiji M. Multi-compartmental oral delivery systems for nucleic acid therapy in the gastrointestinal tract. Adv Drug Deliv Rev. 2013;65(6):891–901. doi:10.1016/j.addr.2012.11.003
17. Bakhru SH, Furtado S, Morello AP, et al. Oral Delivery of Proteins by Biodegradable Nanoparticles. Adv Drug Deliv Rev. 2013;65(6):811–821. doi:10.1016/j.addr.2013.04.006
18. Aguirre TAS, Teijeiro-Osorio D, Rosa M, et al. Current Status of Selected Oral Peptide Technologies in Advanced Preclinical Development and in Clinical Trials. Adv Drug Deliv Rev. 2016;106:223–241. doi:10.1016/j.addr.2016.02.004
19. Lonn P, Kacsinta AD, Cui X-S, et al. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci Rep. 2016;6(1):32301. doi:10.1038/srep32301
20. Hu X, Fan W, Yu Z, et al. Evidence does not support absorption of intact solid lipid nanoparticles via oral delivery. Nanoscale. 2016;8(13):7024–7035. doi:10.1039/C5NR07474F
21. Zhang H, Shahbazi MA, Makila EM, et al. Diatom silica microparticles for sustained release and permeation enhancement following oral delivery of prednisone and mesalamine. Biomaterials. 2013;34(36):9210–9219. doi:10.1016/j.biomaterials.2013.08.035
22. Chen MC, Sonaje K, Chen KJ, et al. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32(36):9826–9838. doi:10.1016/j.biomaterials.2011.08.087
23. Selmin F, Musazzi UM, Magri G, et al. Regulatory aspects and quality controls of polymer-based parenteral long-acting drug products: the challenge of approving copies. Drug Discov Today. 2020;25(2):321–329. doi:10.1016/j.drudis.2019.12.008
24. Boegh M, Nielsen HM. Mucus as a barrier to drug delivery–understanding and mimicking the barrier properties. Basic Clin Pharmacol Toxicol. 2015;116(3):179–186. doi:10.1111/bcpt.12342
25. Li L, Yang L, Li M, et al. A cell-penetrating peptide mediated chitosan nanocarriers for improving intestinal insulin delivery. Carbohyd Polym. 2017;174:182–189. doi:10.1016/j.carbpol.2017.06.061
26. Feger G, Angelov B, Angelova A. Prediction of Amphiphilic Cell-Penetrating Peptide Building Blocks from Protein-Derived Amino Acid Sequences for Engineering of Drug Delivery Nanoassemblies. J Phys Chem B. 2020;124(20):4069–4078. doi:10.1021/acs.jpcb.0c01618
27. Ye J, Shin MC, Liang Q, et al. 15 years of attempts: a macromolecular drug delivery system based on the CPP-mediated intracellular drug delivery and antibody targeting. J Control Release. 2015;205:58–69. doi:10.1016/j.jconrel.2014.12.002
28. Schmidt N, Mishra A, Lai GH, et al. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584(9):1806–1813. doi:10.1016/j.febslet.2009.11.046
29. Nakase I, Takeuchi T, Futaki S. Cell penetrating peptides for chemical biological studies. Methods Mol Biol. 2015;1324:387–396. doi:10.1007/978-1-4939-2806-4_26
30. Futaki S, Hirose H, Nakase I. Arginine-rich peptides: methods of translocation through biological membranes. Curr Pharm Des. 2013;19(16):2863–2868. doi:10.2174/1381612811319160003
31. Accardo A, Morelli G. Peptide-Targeted Liposomes for Selective Drug Delivery: advantages and Problematic Issues. Biopolymers. 2015;104(5):462–479. doi:10.1002/bip.22678
32. Suma T, Cui JW, Mullner M. Modulated Fragmentation of Proapoptotic Peptide Nanoparticles Regulates Cytotoxicity. J Am Chem Soc. 2017;139(11):4009–4018. doi:10.1021/jacs.6b11302
33. Liu D, Angelova A, Liu JW, et al. Self-assembly of mitochondria-specific peptide amphiphiles amplifying lung cancer cell death through targeting the VDAC1-hexokinase-II complex. J Mater Chem B. 2019;7(30):4706–4716. doi:10.1039/C9TB00629J
34. Yoo MK, Kang SK, Choi JH, et al. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials. 2010;31(30):7738–7747. doi:10.1016/j.biomaterials.2010.06.059
35. Fievez V, Plapied L, Plaideau C, et al. In vitro identification of targeting ligands of human M cells by phage display. Int J Pharm. 2010;394(1–2):35–42. doi:10.1016/j.ijpharm.2010.04.023
36. Li XL, Liu JY, Lu R, et al. Evaluation of the therapeutic efficacy of tripeptide tyroserleutide (YSL) for human hepatocarcinoma by in vivo hollow fiber assay. Invest New Drugs. 2008;26(6):525–529. doi:10.1007/s10637-008-9121-8
37. Attene-Ramos MS, Huang RL, Sakamuru S, et al. Systematic Study of Mitochondria Toxicity of Environmental Chemicals Using Quantitative High Throughput Screening. Chem. Res.Toxicol. 2013;26(9):1323–1332. doi:10.1021/tx4001754
38. Wang DF, Rong WT, Lu Y, et al. TPGS(2k)/PLGA Nanoparticles for Overcoming Multidrug Resistance by Interfering Mitochondria of Human Alveolar Adenocarcinoma Cells. ACS Appl Mater Interfaces. 2015;7:3888–3901. doi:10.1021/am508340m
39. Mittal G, Carswell H, Brett R, et al. Development and evaluation of polymer nanoparticles for oral delivery of estradiol to rat brain in a model of Alzheimer’s pathology. Controlled Release. 2011;150(2):220–228. doi:10.1016/j.jconrel.2010.11.013
40. Zhang X, Qi J, Lu Y, et al. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomedicine. 2014;10(1):167–176. doi:10.1016/j.nano.2013.07.011
41. Li MM, Sun YS, Ma CJ, et al. Design and Investigation of Penetrating Mechanism of Octaarginine-Modified Alginate Nanoparticles for Improving Intestinal Insulin Delivery. J Pharm Sci. 2021;110(1):268–279. doi:10.1016/j.xphs.2020.07.004
42. Zhou JL, Chau Y. Different oligoarginine modifications alter endocytic pathways and subcellular trafficking of polymeric nanoparticles. Biomater Sci. 2016;4(10):1462–1472. doi:10.1039/C6BM00371K
43. Spivak AY, Nedopekina DA, Gubaidullin RR, et al. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: selective Targeting Cancer Cell Mitochondria. J Pers Med. 2021;11(6):470. doi:10.3390/jpm11060470
44. Li L, Geisler I, Chmielewski J, et al. Cationic amphiphilic polyproline helix P11LRR targets intracellular mitochondria. J Control Release. 2010;142:259–266. doi:10.1016/j.jconrel.2009.10.012
45. Maghsoudnia N, Baradaran Eftekhari R, Naderi Sohi A, et al. Mitochondrial delivery of microRNA mimic let-7b to NSCLC cells by PAMAM-based nanoparticles. J Drug Target. 2020;28(7–8):818–830. doi:10.1080/1061186X.2020.1774594
46. Peng N, Yu H, Yu W, et al. Sequential-targeting nanocarriers with pH-controlled charge reversal for enhanced mitochondria-located photodynamic-immunotherapy of cancer. Acta Biomater. 2020;105:223–238. doi:10.1016/j.actbio.2020.01.005
47. Feng Y, Qin G, Chang S, et al. Antitumor Effect of Hyperoside Loaded in Charge Reversed and Mitochondria-Targeted Liposomes. Int J Nanomedicine. 2021;16:3073–3089. doi:10.2147/IJN.S297716
48. Zhang Y, Kong X, Li M, et al. The development of a biotin-guided and mitochondria-targeting fluorescent probe for detecting SO2 precisely in cancer cells. Talanta. 2021;225:121992. doi:10.1016/j.talanta.2020.121992
49. Fang L, Fan H, Guo C, et al. Novel Mitochondrial Targeting Multifunctional Surface Charge-Reversal Polymeric Nanoparticles for Cancer Treatment. J Biomed Nanotechnol. 2019;15(11):2151–2163. doi:10.1166/jbn.2019.2854
50. Chan KH, Xue B, Robinson RC, et al. Systematic single moiety variations of ultrashort peptides produce profound effects on self-assembly, nanostructure formation, hydrogelation, and phase transition. Sci Rep. 2017;7:12897. doi:10.1038/s41598-017-12694-9
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.