")
Back to Journals » Infection and Drug Resistance » Volume 13
Effect of the Short-Term Use of Fluoroquinolone and β-Lactam Antibiotics on Mouse Gut Microbiota
Authors Gu SL, Gong Y, Zhang J, Chen Y , Wu Z, Xu Q, Fang Y , Wang J, Tang LL
Received 22 September 2020
Accepted for publication 13 November 2020
Published 21 December 2020 Volume 2020:13 Pages 4547—4558
DOI https://doi.org/10.2147/IDR.S281274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Si-Lan Gu,1,* Yiwen Gong,1,* Jiaying Zhang,1,* Yunbo Chen,1 Zhengjie Wu,1 Qiaomai Xu,1 Yunhui Fang,1 Jingxia Wang,1 Ling-Ling Tang2
1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310003, People’s Republic of China; 2Shulan (Hangzhou) Hospital Affiliated to Zhejiang Shuren University, Shulan International Medical College, Hangzhou 310000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ling-Ling Tang
Shulan (Hangzhou) Hospital Affiliated to Zhejiang Shuren University, Shulan International Medical College, Hangzhou 310000, People’s Republic of China
Email [email protected]
Background: Antibiotics play an important role in the treatment of infectious diseases. However, the overuse of antibiotics increases the spread of drug-resistant bacteria and causes dysbiosis of the intestinal microbiota. Few studies have addressed the longitudinal effects of antibiotics on the microbiome and host immunity.
Materials and Methods: In this study, the short-term effect of fluoroquinolone (levofloxacin) and β-lactam antibiotics (meropenem, cefoperazone/sulbactam, and aztreonam) on the gut microbiota of mice was evaluated by 16S rRNA gene sequencing. The susceptibility of Bifidobacterium longum, Lactobacillus lactis, Enterococcus faecium, and Clostridium butyricum to these antimicrobial agents was assessed using the disc diffusion method.
Results: Our results showed that 4-day antibiotic exposure significantly reduced the alpha and beta diversity of gut bacteria and increased serum inflammatory cytokines, and these changes persisted long after antibiotic withdrawal and did not return to pre-treatment levels. Nonetheless, the bacterial community composition tended to return to pre-treatment levels after discontinuing treatment. The tested probiotic strains were resistant to aztreonam but were sensitive to meropenem and cefoperazone/sulbactam.
Conclusion: Short-term antibiotic treatment led to significant changes in the intestinal flora with a tendency to recover. The antibiotics had different effects on the intestinal microbial community and probiotic strains. This study provides guidance for the concomitant use of probiotics and antibiotics, and the results emphasize the importance of using broad-spectrum antibiotics responsibly to prevent the long-term disruption of the native microbiota.
Keywords: antibiotics, gut microbiota, 16S rRNA gene sequencing, probiotics
Introduction
The discovery of penicillin in 1928 opened the era of antibiotic therapy and decreased the mortality rate caused by severe infection.1 Antibiotics have been the pillar of medicine and are used worldwide on a large scale.2 However, these medicines kill commensal microorganisms and increase the susceptibility to infections.3 Coronavirus disease 2019 (COVID-19) has become a global health concern, and the ratio of Lactobacillus, Bifidobacterium, and C. butyricum is decreased in COVID-19 patients.4,5 A significant proportion of COVID-19 patients received empirical antibiotics,6 leading to further loss of beneficial symbionts and exacerbation of gut dysbiosis.
The intestinal microbiota is a complex ecosystem comprising diverse populations of commensal microorganisms that play a major role in human health.6,7 This ecosystem regulates nutrient metabolism and absorption, intestinal development, gut barrier integrity and function, and central nervous system and intestinal immune homeostasis.8,9 Moreover, commensal bacteria inhibit the proliferation of invading pathogens, and microbiota-mediated innate immune defenses provide colonization resistance.10,11
Host-microbial symbiosis has a central role in health and disease. Observational, clinical, and epidemiologic studies have demonstrated that antibiotics disrupt intestinal homeostasis and increase the risk of diseases, including obesity, diabetes, inflammatory bowel disease, allergies, asthma, and celiac disease.12–14 Broad-spectrum antibiotics have been shown to alter the abundance of 30% of intestinal bacteria and decrease microbial diversity, richness, and evenness.7,15 Clostridium difficile proliferates after antibiotic-mediated dysbiosis and affects immunocompromised individuals after receiving broad-spectrum antibiotics, such as cephalosporins.16 Nonetheless, it was proven that the normal fecal flora provides colonization resistance against C. difficile.17
Although controlling bacterial infections is essential, antibiotics kill both pathogenic and beneficial microorganisms, decrease microbial diversity, and have short- and long-term adverse effects on health.18 Furthermore, antibiotic-mediated changes in the microbiota can impair host immunity since host-microbe interactions are intricate and dynamic. For instance, the administration of antibiotics early in life increases the incidence of autoimmune disorders.19 Therefore, understanding the effect of antibiotics on the microbial ecosystem and host immunity and finding appropriate treatments to maintain immune homeostasis are crucial.
Few studies have evaluated the differential effect of antibiotics on beneficial and pathogenic bacteria, and the resistance of probiotic strains to antimicrobials. We selected three commonly used antibiotics (cefoperazone/sulbactam [CPZ], meropenem [MEM] and levofloxacin [LEV]) in the treatment of infections and aztreonam [ATM], which may cause minimal disruption of the anaerobic microflora.6,20,21 Our study assessed the extent of gut microbiome disturbance by different classes of antibiotics and compared the effect of treatment on microbial composition in mice. Data on the recovery of microbial composition over time and the susceptibility of probiotic strains to antibiotics may provide guidance for the clinical use of these medications.
The objectives of this study are to determine the vitro susceptibility of four probiotic bacterial strains to three β-lactams (ATM, CPZ, and MEM) and one fluoroquinolone (LEV) and the short-term effect of these antibiotics on the gut microbiota using 16S rRNA gene sequencing.
Materials and Methods
Bacterial Strains
The probiotic bacterial strains used in this study were isolated from Bifico (Bifidobacterium longum, Lactobacillus lactis, and Enterococcus faecium; Shanghai Sine Pharmaceutical Co. Ltd) and MIYA-BM (C. butyricum; Miyarisan Pharmaceutical Co. Ltd.) The purity of the strains was checked before performing antibiotic susceptibility tests. Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 25923, and Streptococcus pneumoniae ATCC 49619 were used as controls.
Antibiotic Susceptibility Test
The susceptibility of bacterial strains to LEV, MEM, CPZ, and ATM (Oxoid Ltd, Basingstoke, UK) was evaluated using the disc diffusion method according to the guidelines of the Clinical and Laboratory Standards Institute. B. longum, L. lactis, and E. faecium cultures were activated in MRS medium and incubated at 37°C for 18–24 h to obtain 7–8 log10 CFU/mL. Ten microliters of the bacterial suspension were spread on the surface of the MRS medium (Difco) supplemented with 1% agar to increase the diffusion of the evaluated substances. C. butyricum was cultured on blood agar under similar experimental conditions. The antibiotic discs were manually placed over the agar surface, the plates were incubated at 37°C for 24 h under aerobic or anaerobic conditions depending on the bacterial growth requirements, and the diameter of each clear zone was determined.22 The experiments were performed in triplicate and repeated independently three times.
Experimental Groups
Eight-week-old female C57BL/6J mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. Mice were allowed to acclimatize for 1–2 weeks and fed on specific pathogen-free conditions. The animals were randomly divided into five groups (seven animals per group) according to treatment: LEV, MEM, CPZ, ATM, and no treatment (NT) (control). The drugs were administered subcutaneously for 4 days at the following doses (mg/20 gm body weight): 1.2 (LEV), 4.5 (MEM), 6.0 (CPZ), and 3.0 (ATM). The study was approved by the Research Ethics Committee of the First Affiliated Hospital from the School of Medicine of Zhejiang University (2019–1085) and conformed to the 2011 National Institutes of Health Guide for the care and use of laboratory animals.23 Two mice whose weight presented the largest deviation from the median were removed from each group.
Sample Collection
Fresh fecal specimens were collected at four times points, before antibiotic treatment (D1), and 4, 8, and 60 days after treatment (D4, D8, and D60, respectively), transferred to sterile test tubes, and immediately stored at −80°C for microbiological testing for C. difficile, DNA extraction, and microbial analysis. Microbiological testing for C. difficile was performed as detailed previously.24
DNA Extraction and 16S Ribosomal RNA Gene Sequencing
Genomic DNA was extracted from 200 mg of frozen stool using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) and subjected to 16S rRNA gene sequencing. DNA concentration and purity were monitored on 1.5% agarose gels.
PCR amplification of the V3-V4 hypervariable region of 16S rRNA genes was performed using barcoded primers 338F and 806R. Amplification conditions consisted of an initial denaturation at 98°C for 1 min, followed by 30 amplification cycles (98°C for 10 s, 50°C for 30 s, and 72°C for 30 s), and one cycle at 72°C for 5 min. The amplified products were analyzed on a 2% agarose gel and purified using the GeneJETTM Gel Extraction Kit (Thermo Scientific). Sequence libraries were generated using the Ion Plus Fragment Library Kit 48 rxns (Thermo Scientific) according to the manufacturer’s recommendations. DNA concentration and purity were checked using a Qubit 2.0 fluorometer (Thermo Scientific). The libraries were sequenced on an Ion S5TM XL platform, and 400 bp single-end reads were generated. All sequence data were deposited in the NCBI Sequence Read Archive (SRA) database (Accession Number SRP 201996).
Data Processing
Single-end reads were separated by barcode, and barcodes and primers were removed. High-quality reads were obtained using CutAdapt. Chimeric sequences were detected and removed using UCHIME. Sequences were analyzed using UPARSE software. Sequences with ≥ 97% similarity were assigned to the same operational taxonomic units (OTUs). The taxonomic annotation of each representative sequence was performed using Mothur software and the SILVA ribosomal RNA gene database (https://www.arb-silva.de/). Phylogenetic relationships between OTUs and the abundance of dominant species across different groups were determined by multiple sequence alignment using MUSCLE software. OTU abundance data were normalized using the sample with the lowest number of sequences.
Analysis of Microbial Diversity
Community richness and diversity were determined using Chao1 and Shannon indices. These indices were calculated with QIIME version 1.7.0 and displayed using R software version 2.15.3. Cluster analysis was preceded by principal coordinate analysis (PCoA), which was performed to obtain principal coordinates and visualize complex, multidimensional data. PCoA was displayed using the packages WGCNA, stat, and ggplot2 in R software version 2.15.3.
Differences in taxonomic composition were determined using the linear discriminant analysis (LDA) effect size (LEfSe) analysis (http://huttenhower.sph.harvard.edu/galaxy/).
Serum Cytokines and Chemokines
Blood samples were collected from mice on D60. Serum cytokines were detected using the Bio-Plex Pro Mouse Cytokine 23-Plex Panel (Bio-Rad) and analyzed on a MAGPIX system (Luminex Corporation) using Bio-Plex Manager software version 6.1 (Bio-Rad). The following cytokines and chemokines were analyzed: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17, IFN-γ, eotaxin, G-CSF, GM-CSF, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α.
Statistical Analysis
Body weight and cytokine levels were measured in five mice from each group. Normally distributed data were analyzed by one-way analysis of variance, and non-normally distributed data were evaluated by the Mann–Whitney U-test. The diversity and relative abundance of fecal bacteria were determined using the Kruskal–Wallis and Mann–Whitney U-test. Data were analyzed using SPSS version 22.0 and GraphPad version 7.0. P-values or q-values (false discovery rate adjusted) less than 0.05 were considered statistically significant.
Results
Antibiotic Susceptibility Test
The susceptibility test results are presented in Table 1. All four probiotic bacterial strains were susceptible to MEM and CPZ but resistant to ATM. E. faecium and C. butyricum were sensitive to LEV, whereas L. lactis and B. longum were resistant to this antibiotic.
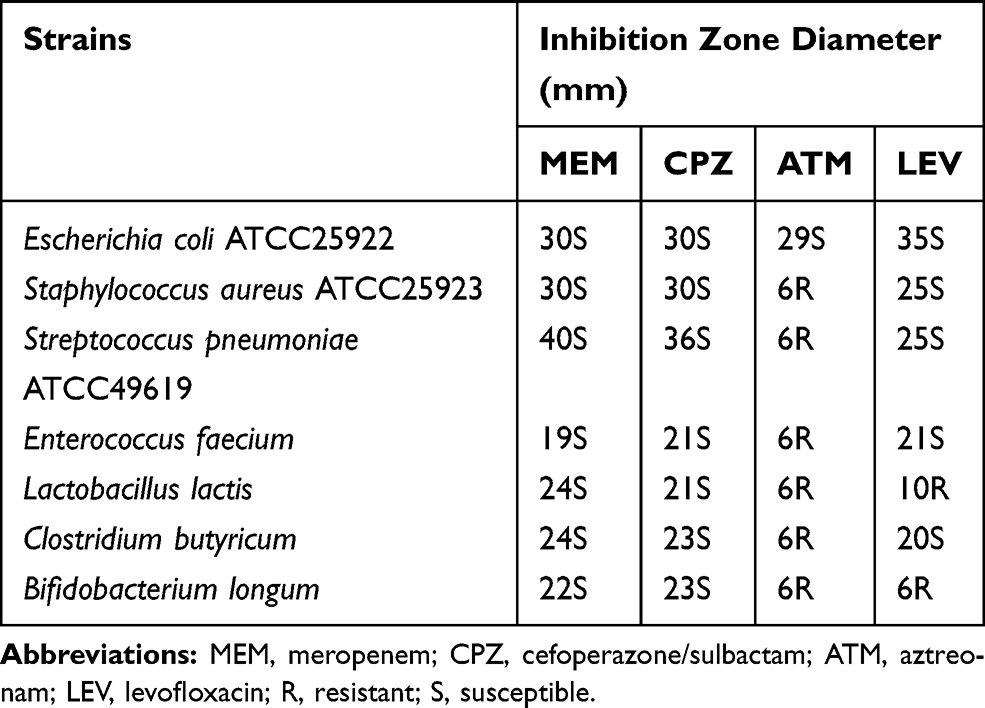 |
Table 1 Susceptibility of Bacterial Strains to Four Antibiotics in Disc Diffusion Assays |
Changes in Animal Body Weight During Treatment
On D1, there was no significant difference in body weight between the NT and treated groups. From D4, the weight of the groups treated with LEV, MEM, and ATM decreased significantly, compared with the NT group. The CPZ and LEV groups showed obvious weight loss on D8. The weight of the animals from the CPZ group was significantly lower than that of the other three groups on D60 (Figure 1A). Antibiotic treatment significantly decreased weight in the four intervention groups from D1 to D4.
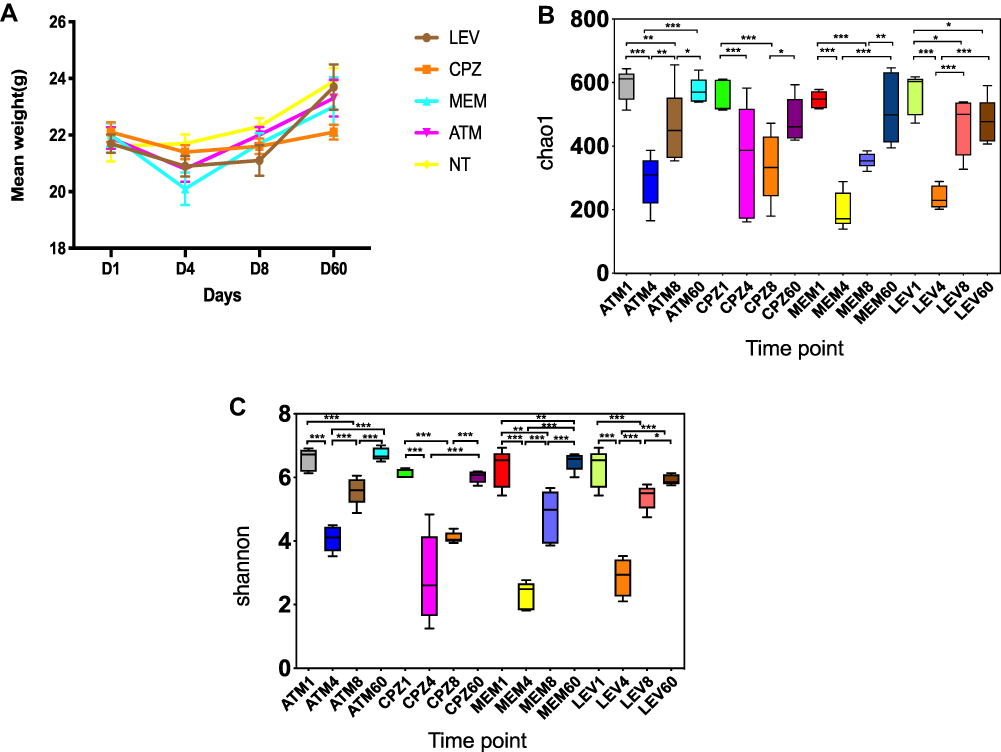 |
Figure 1 (A) Changes in the body weight of mice during antibiotic treatment. D1: before treatment. D4, D8, and D60: 4, 8, and 60 days after treatment. Microbial diversity measured by the Chao1 index (B) and Shannon index (C) was altered by antibiotic treatment across the treatment groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs the control group. Abbreviations: ATM, aztreonam; CPZ, cefoperazone/sulbactam; MEM, meropenem; LEV, levofloxacin; NT, no treatment (control). |
Culture of C. Difficile on D1 and D4
Most antibiotic-treated mice had wet and soft fecal characters on D4. To explore the involvement of toxigenic C. difficile in diarrhea symptoms, stool samples were collected and analyzed 4 days before and after antibiotic administration. The results showed that these samples were negative for C. difficile.
Inflammatory Cytokine Levels
Serum cytokines were measured on D60. IL-1β and TNF-α levels were increased in the CPZ, MEM, and ATM groups (Figure 2A and G). IL-12 and IL-17 levels were increased in the CPZ and MEM groups (Figure 2C and E). IL-13 and IFN-γ were increased in the MEM group (Figure 2D and F). The levels of IL-6 and IL-10 were significantly elevated in the MEM and ATM groups (Figure 2B and H). In addition, the level of GM-CSF was significantly higher in the CPZ and MEM groups (Figure 2I).
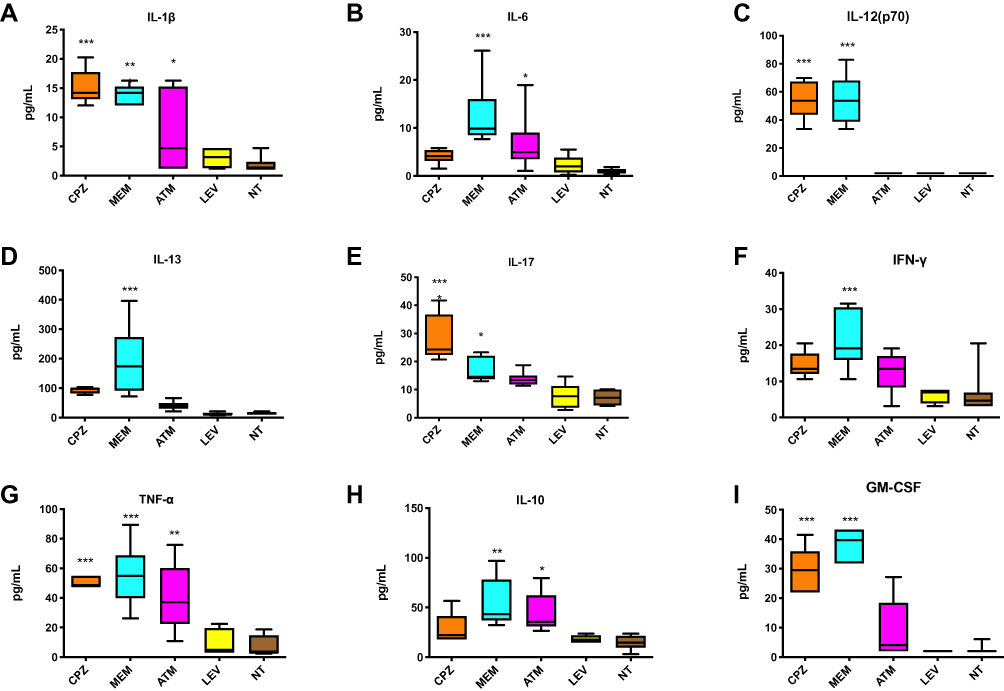 |
Figure 2 Serum cytokine expression in antibiotic exposed mice and a control group. (A) IL-1β, (B) IL-6, (C) IL-12 (p70), (D) IL-13, (E) IL-17, (F) IFN-γ, (G) TNF-α, (H) IL-10, (I) GM-CSF. *P < 0.05, **P < 0.01, ***P < 0.001 vs the control group. Abbreviations: CPZ, cefoperazone/sulbactam; MEM, meropenem; ATM, aztreonam; LEV, levofloxacin; NT, no treatment (control). |
The Composition of the Gut Microbiota Improved Over Time After Antibiotic Exposure
Alterations in the gut microbiota composition before, during, and after antibiotic treatment were evaluated by 16S rRNA gene sequencing. Alpha diversity between the groups was compared using Chao1 and Shannon indices (Figure 1B and C). Alpha diversity was dramatically reduced from D1 to D4, demonstrating a global change in microbial community structure, but gradually increased from D8 to D60, suggesting the recovery of surviving microorganisms.
The microbial composition between the groups was compared using the PCoA of Bray–Curtis dissimilarities (Figure 3). The results showed that from D4 to D8, the microbial composition of the four antibiotic treatment groups was different from that pre-antibiotic exposure (D1), but gradually returned to baseline levels on D60.
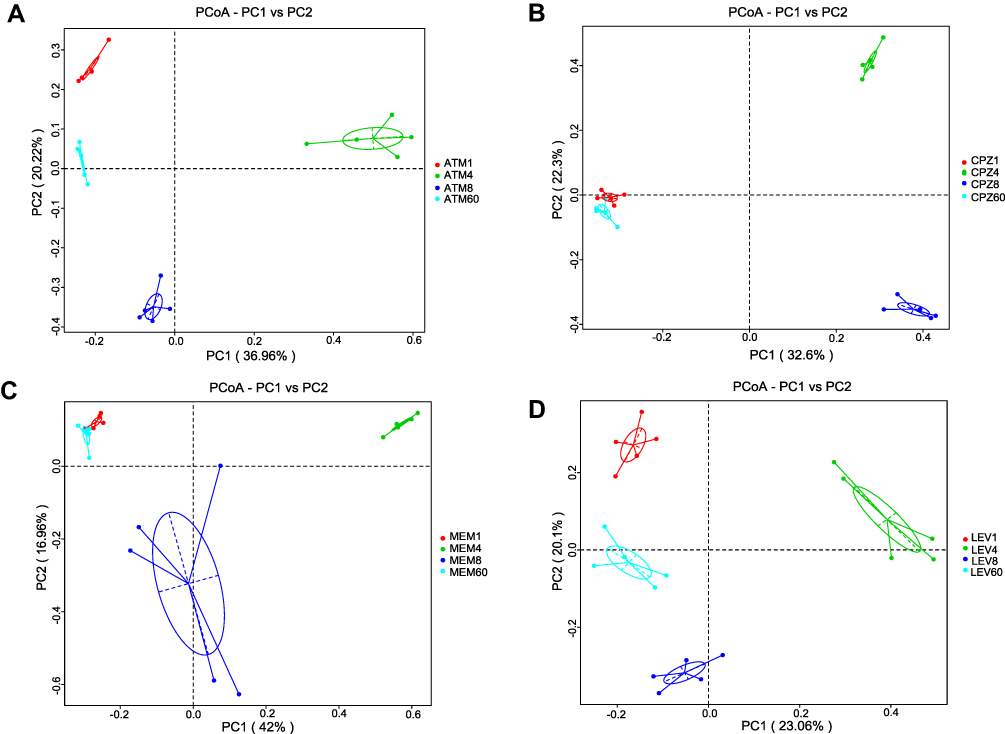 |
Figure 3 Analysis of microbial communities by principal coordinate analysis (PCoA) at different time points after antibiotic treatment. (A) ATM, (B) CPZ, (C) MEM, (D) LEV. Antibiotics disrupted the microbial community composition. Red dots, before treatment; green dots, immediately after treatment; blue and cyan dots, 8 and 60 days after cessation of treatment, respectively. Abbreviations: ATM, aztreonam; CPZ, cefoperazone/sulbactam; MEM, meropenem; LEV, levofloxacin. |
Impact of Antibiotics on the Gut Microbial Community Structure
Our results show that four days of antibiotic exposure greatly disturbed the microbial composition at the phylum, class, order, family and genus levels of distribution (Figure 4A–E). Thirty-one phyla were identified by sequencing analysis. As expected, phyla Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria accounted for more than 90% of the gut microbiota composition (Figure 4A). The relative abundance of phyla and genera varied between groups (Figures S1-S8). The most representative phylum in the intervention groups on D1 was Firmicutes. On D4, Firmicutes was enriched (q = 0.02) and Bacteroidetes was depleted (q < 0.001) in the CPZ and LEV groups, and abundance returned to baseline levels from D8 to D60. In the MEM and ATM groups on D4, there were no significant changes in the abundance of Firmicutes, Bacteroidetes, and Actinobacteria, whereas Proteobacteria was depleted (q < 0.001) (Figure 4A). In Firmicutes and Bacteroidetes, there was a significant decrease in the relative abundance of families Lactobacillaceae (q = 0.03 and 0.04), Ruminococcaceae (q = 0.01 and 0.04), Bacteroidaceae (q = 0.01 and 0.04), Muribaculaceae (q = 0.01 and 0.04), Prevotellaceae (q = 0.01 and 0.04), and Rikenellaceae (q = 0.01 and 0.03) in the CPZ and MEM groups on D4 (Figure 4D). Similarly, the representativeness of Muribaculaceae (q = 0.01), Rikenellaceae (q = 0.01), and Lachnospiraceae (q = 0.02) was decreased in the LEV group on D4 (Figure 4D). The abundance of Enterococcaceae (q = 0.03) was increased in the MEM group on D4.
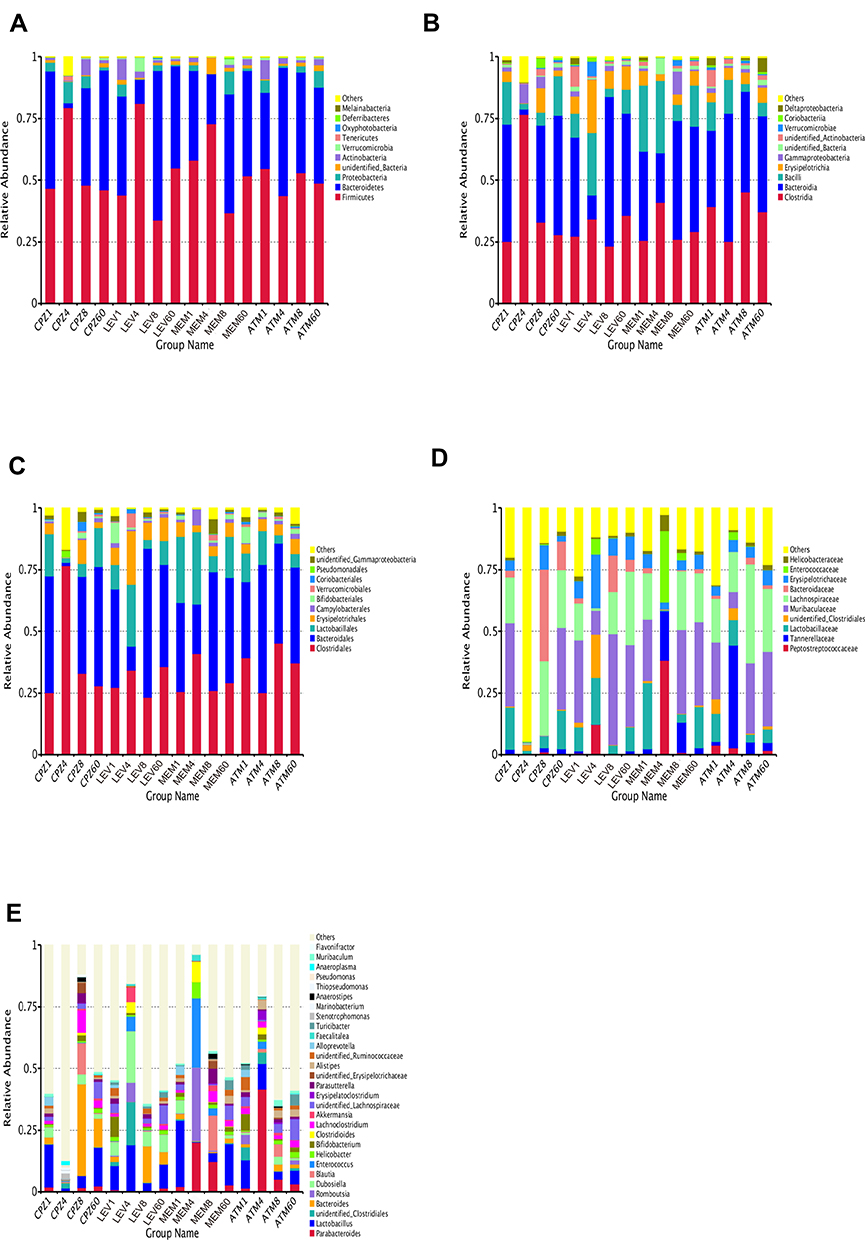 |
Figure 4 Changes in bacterial community composition by antibiotic treatment in mice at the phylum (A), class (B), order (C), family (D), and genus (E) levels. Antibiotics disrupted the gut microbial community and differentially affected community composition.Abbreviations: ATM, aztreonam; CPZ, cefoperazone/sulbactam; MEM, meropenem; LEV, levofloxacin. |
The 30 most abundant genera were analyzed (Figure 4E), and the most dominant groups on D1 were Bacteroides, Parabacteroides, Lactobacillus, Bifidobacterium, and Dubosiella. On D4, the abundance of Muribaculum, Rikenella, Desulfovibrio, and Candidatus Saccharimonas, and several butyrate producers from family Ruminococcaceae (q < 0.05) was significantly decreased in the intervention groups. On D4, there was a significant increase in the representativeness of Enterorhabdus (q = 0.02), Parabacteroides (q = 0.02), and Faecalitalea (q = 0.02) in the ATM group, and Enterococcus (q = 0.01) and Romboutsia (q = 0.03) in the MEM group.
The abundance of Bacteroides (phylum Bacteroidetes) increased in the CPZ and LEV groups from D4 to D8. In the ATM and LEV groups, the predominance of several genera from families Lachnospiraceae and Ruminococcaceae increased on D8. However, the abundance of the facultative anaerobic genus Enterococcus decreased significantly in the MEM group from D4 to D8 (Figure 4E).
The bacterial community structure returned to baseline levels within 60 days after the cessation of treatment. On D60, although the relative abundance of Bacteroides, Lactobacillus, and unidentified Lachnospiraceae increased substantially, the representativeness of other taxa was not restored (Figure 4E).
Changes in microbial abundance and biomarker levels were evaluated according to LDA values (Figure 5). The opportunistic pathogens Enterococcus and Clotridioides were significantly enriched in the treated groups, whereas butyrate-producing bacteria Blautia, Lachnoclostridium, and Roseburia were less abundant on D4, suggesting that antibiotics significantly affected the composition and alpha diversity of the gut microbiome.
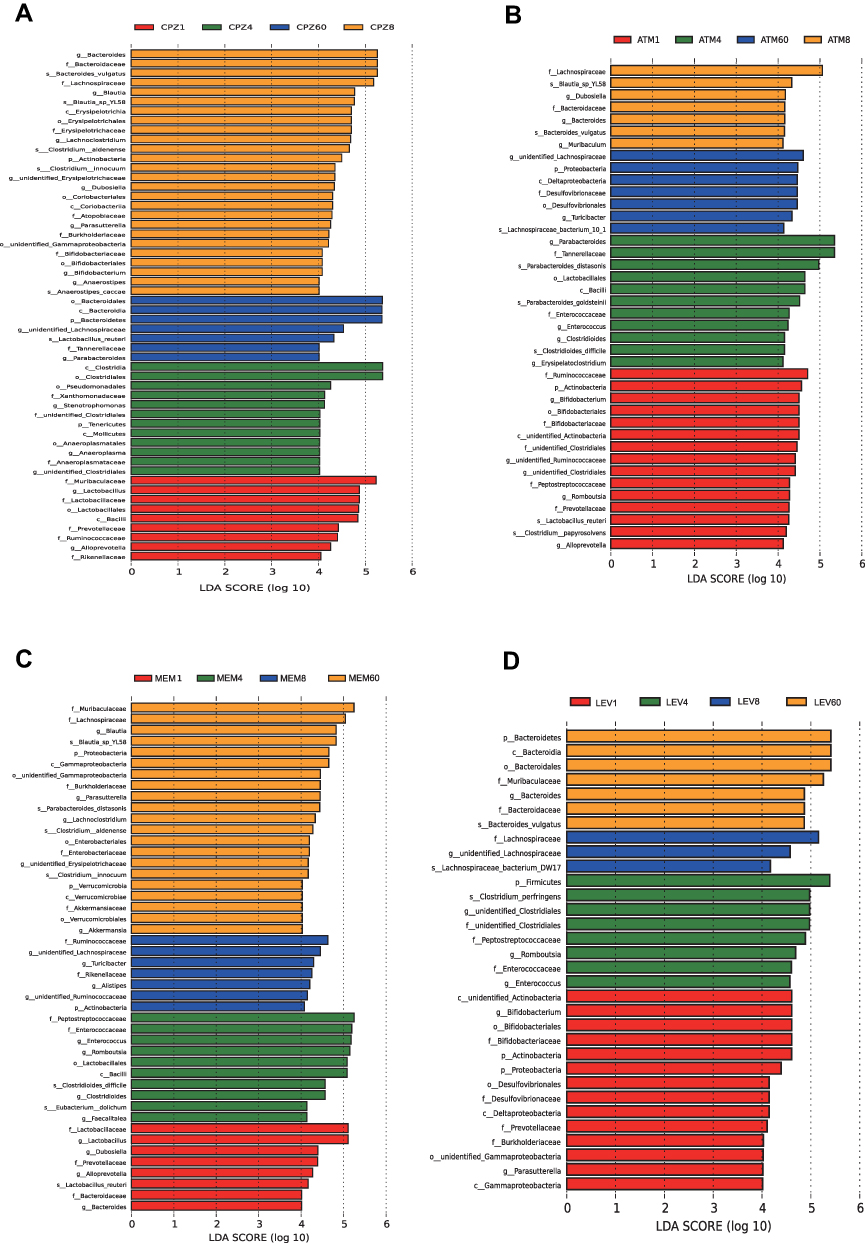 |
Figure 5 Time-dependent changes in the abundance of microbial taxa by antibiotic treatment according to linear discriminant analysis effect size analysis. (A) CPZ, (B) ATM, (C) MEM, (D) LEV. The length indicates the effect size associated with a taxon. Only taxa with an effect size >4 are shown. Each color corresponds to a sampling time. Abbreviations: CPZ, cefoperazone/sulbactam; ATM, aztreonam; MEM, meropenem; LEV, levofloxacin. |
Discussion
The use of broad-spectrum antibiotics to cure infectious diseases has improved human health.25,26 However, the widespread use of these medications can lead to significant spatiotemporal changes in the gut microbiota.7,27 Here we assessed the impact of four commonly used antibiotics on gut microbiota and immunity in mice. Consistent with the literature, our results demonstrated that the short-term use of antibiotics might disrupt the diversity and biomass of the intestinal microbiota in healthy mice and lead to long-term influence with deficient resilience of microbiota and inflammatory response.28 Probiotics are used clinically to counteract the potential effects of antibiotics on the gut microbiota.29,30 Similar to previous reports,21 the results of the drug sensitivity test showed that most of the isolated probiotic bacteria strains were highly resistant to ATM and LEV, suggesting that Bifico and MIYA could be used in combination with these drugs to improve dysbiosis. Conversely, all four probiotics strains were susceptible to MEM and CPZ, indicating that the combined use of related probiotics and these agents should be avoided. Moreover, many probiotics are sensitive to antibiotics and should be taken at regular intervals.31
High-throughput sequencing of the 16S ribosomal RNA V3-V4 region was performed to analyze the short-term effect of fluoroquinolone and β-lactam antibiotics on mouse gut microbiota. The gut microbiota of mice is less variable than that of humans under controlled environmental and dietary conditions.32 In our study, changes in microbial abundance were determined by alpha and beta diversity. Consistent with the literature,15,33 4-day antibiotic intervention significantly decreased stool alpha diversity and affected intestinal microbial composition, whereas microbial richness returned to pretreatment levels within 60 days after the termination of treatment.
β-lactams are widely used to treat infectious diseases and disrupt cell wall and peptidoglycan synthesis, leading to cell death.34 Butyrate-producing taxa (Roseburia, Lachnospiraceae, and Ruminococcaceae) and H2-consuming bacteria (Blautia and Bifidobacterium) predominated on D1. On D4, CPZ significantly decreased the abundance of phylum Bacteroidetes (represented by the genus Bacteroides) and increased the representativeness of Firmicutes. However, Bacteroides was dominant on D8, suggesting the development of antibiotic resistance, which agrees with a previous study, wherein resistance genes against cephalosporin were identified in this taxon.35 Other commensals were depleted in the β-lactam-treated groups, especially butyrate-producing microorganisms, which corroborates the decrease in short-chain fatty acid (SCFA) producers with antibiotic treatment. Previous studies have shown that butyrate, which has strong anti-inflammatory effects, may serve as an energy source for colonocytes, induce mucin synthesis, and improve intestinal integrity by decreasing tight junction permeability.36
The decrease in the abundance of anaerobic bacteria in the MEM group was compensated for by a slight increase in the representativeness of opportunistic families Enterococcaceae and Peptostreptococcaceae. The enrichment of intestinal pathogens after broad-spectrum antibiotic treatment has been observed in murine models and is due to increased carbohydrate availability after the depletion of native microbiota.37 Similar results were obtained by Lu et al after β-lactam treatment in humans.38 The increase in the abundance of Enterococcus may lead to urinary tract infections, endocarditis, and meningitis in humans.39 The increase in C. difficile (family Peptostreptococcaceae) is related to the use of broad-spectrum antibiotics, which reduce host resistance to pathogen colonization and expansion.40 In antibiotic-treated mice, the absence of competitive exclusion bacteria and decreased SCFA levels impaired colonization resistance against C. difficile.41 Randomized controlled trials showed that carbapenems increased C. difficile infections compared to other antibiotics.42 However, in our study, neither C. difficile nor its toxin was detected in antibiotic-treated mice.
Although the overall microbial composition returned to pretreatment levels within 60 days after discontinuation of β-lactam treatment, serum cytokines were increased on D60. Moreover, antibiotic exposure significantly changed the expression of pro-inflammatory cytokines and chemokines in serum, including IL-1β, IL-6, IL-12, IL-17, and TNF-α, which might promote inflammatory reactions and increase gut permeability.43,44 Ferrer et al found that the intestinal epithelial structure modulated bacterial community composition during β-lactam therapy.45 Nonetheless, immunological responses during antibiotic treatment need to be further studied.
Levofloxacin, a broad-spectrum fluoroquinolone, has activity against Gram-negative and Gram-positive bacteria and a wide margin of safety and efficacy.46 Our results found that there was a transient increase in Firmicutes and a decrease in Bacteroidetes after LEV treatment on D4 and a return to baseline levels on D8 and D60. In contrast to β-lactam treatment, the expression of serum inflammatory cytokines after LEV treatment was similar to that of the control group.
This study has limitations. First, the number of antibiotic classes and time points for the analysis of microbial changes was small. Second, the response of probiotic bacteria to antibiotics was not assessed in vivo. Third, although the changes in the microbiome were significant and reproducible, the mechanisms underlying these changes were not evaluated.
In summary, the results of the antimicrobial susceptibility test and animal experiments demonstrated that MEM and CPZ had similar disruptive effects on probiotic strains and intestinal flora in mice, and these effects were much greater than those of LEV. Furthermore, broad-spectrum antibiotics increased the serum levels of pro-inflammatory cytokines, underscoring the need to use these agents responsibly to prevent the long-term disruption of the native microbiome and host immunity.
Funding
This study was supported by grants from the National Science and Technology Major Project (No. 2017ZX10204401), the Medical Health Science Projects in Zhejiang Province (2018KY045), and the National Natural Science Foundation of China (No. 81800457, 81872672).
Disclosure
The authors report no conflicts of interest for this work and no competing financial interests relevant to the present study.
References
1. Tan SY, Tatsumura Y. Alexander Fleming (1881–1955): discoverer of penicillin. Singapore Med J. 2015;56(7):366–367. doi:10.11622/smedj.2015105
2. Gajdács M. The Concept of an Ideal Antibiotic: implications for Drug Design. Molecules. 2019;24(5):5. doi:10.3390/molecules24050892
3. Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801.
4. Gu S, Chen Y, Wu Z, et al. Alterations of the Gut Microbiota in Patients with COVID-19 or H1N1 Influenza. Clin Infect Dis. 2020.
5. Tang L, Gu S, Gong Y, et al. Clinical Significance of the Correlation between Changes in the Major Intestinal Bacteria Species and COVID-19 Severity. Engineering. 2020.
6. Feng Y, Ling Y, Bai T, et al. COVID-19 with Different Severities: A Multicenter Study of Clinical Features. Am J Respir Crit Care Med. 2020;201(11):1380–1388.
7. Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561.
8. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The Central Nervous System and the Gut Microbiome. Cell. 2016;167(4):915–932.
9. Takiishi T, Fenero CIM, Camara NOS. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5(4):e1373208.
10. Diehl GE, Longman RS, Zhang J-X, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature. 2013;494(7435):116–120. doi:10.1038/nature11809
11. Wingender G, Stepniak D, Krebs P, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology. 2012;143(2):418–428. doi:10.1053/j.gastro.2012.04.017
12. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573–584.
13. Arrieta M-C, Stiemsma LT, Dimitriu PA, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7(307):307ra152. doi:10.1126/scitranslmed.aab2271
14. Azad MB, Bridgman SL, Becker AB, Kozyrskyj AL. Infant antibiotic exposure and the development of childhood overweight and central adiposity. Int j Obesity. 2014;38(10):1290–1298. doi:10.1038/ijo.2014.119
15. Dethlefsen L, Huse S, Sogin ML, Relman DA, Eisen JA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi:10.1371/journal.pbio.0060280
16. Chang JY, Antonopoulos DA, Kalra A, et al. Decreased Diversity of the Fecal Microbiome in Recurrent Clostridium difficile –Associated Diarrhea. J Infect Dis. 2008;197(3):435–438. doi:10.1086/525047
17. Urbán E, Terhes G, Extraintestinal GM. Clostridioides difficile Infections: epidemiology in a University Hospital in Hungary and Review of the Literature. Antibiotics. 2020;9:1.
18. Ubeda C, Pamer EG. Antibiotics, microbiota, and immune defense. Trends Immunol. 2012;33(9):459–466. doi:10.1016/j.it.2012.05.003
19. Candon S, Perez-Arroyo A, Marquet C, et al. Correction: antibiotics in Early Life Alter the Gut Microbiome and Increase Disease Incidence in a Spontaneous Mouse Model of Autoimmune Insulin-Dependent Diabetes. PLoS One. 2016;11(1):e0147888. doi:10.1371/journal.pone.0147888
20. Ho J, Ip IM. Antibiotic-Resistant Community-Acquired Bacterial Pneumonia. Infect Dis Clin North Am. 2019;33(4):1087–1103. doi:10.1016/j.idc.2019.07.002
21. Pultz NJ, Donskey CJ. Effect of antibiotic treatment on growth of and toxin production by Clostridium difficile in the cecal contents of mice. Antimicrob Agents Chemother. 2005;49(8):3529–3532. doi:10.1128/AAC.49.8.3529-3532.2005
22. Gajdács M, Spengler G, Urbán E. Identification and Antimicrobial Susceptibility Testing of Anaerobic Bacteria: rubik’s Cube of Clinical Microbiology? Antibiotics. 2017;6:4.
23. National Research Council Committee for the Update of the Guide for the C, Use of Laboratory A. The National Academies Collection: Reports Funded by National Institutes of Health. In: Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US) Copyright © 2011. National Academy of Sciences; 2011.
24. Gu S, Chen Y, Zhang X, et al. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. 2016;18(1):30–38. doi:10.1016/j.micinf.2015.09.008
25. Kc N. A brief history of antibiotics and select advances in their synthesis. J Antibiotics. 2018;71(2):153–184.
26. Benkő R, Gajdács M, Matuz M, et al. Prevalence and Antibiotic Resistance of ESKAPE Pathogens Isolated in the Emergency Department of a Tertiary Care Teaching Hospital in Hungary: A 5-Year Retrospective Survey. Antibiotics. 2020;9:9.
27. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–1270. doi:10.1016/j.cell.2012.01.035
28. Korpela K, Salonen A, Virta LJ, et al. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun. 2016;7(1):10410. doi:10.1038/ncomms10410
29. Tan SY. Alexander Fleming (1881–1955): discoverer of penicillin. Singapore Medical Journal. 2013;494(7435):463–466. doi:10.1016/j.explore.2016.08.015
30. Sanders ME. Probiotics: definition, sources, selection, and uses. Clin Infect Dis. 2008;46(Suppl 2):S58–S61.
31. Hammad AM, Shimamoto T. Towards a compatible probiotic-antibiotic combination therapy: assessment of antimicrobial resistance in the Japanese probiotics. J Appl Microbiol. 2010;109(4):1349–1360.
32. Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75(1):149–160.
33. Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun. 2009;77(6):2367–2375.
34. Semenitz E. [Mechanism of action of beta-lactam antibiotics and problems concerning the development of resistance in antibacterial chemotherapy]. Wien Klin Wochenschr Suppl. 1983;142:7–11.
35. Nakano V. Antimicrobial resistance and prevalence of resistance genes in intestinal Bacteroidales strains. Clinics. 2011;66(4):543–547.
36. Wrzosek L, Miquel S, Noordine ML, et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013;11:61.
37. Faber F, Tran L, Byndloss MX, et al. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature. 2016;534(7609):697–699.
38. Lu S, Huang Q, Wei B, Chen Y. Effects of β-Lactam Antibiotics on Gut Microbiota Colonization and Metabolites in Late Preterm Infants. Curr Microbiol. 2020.
39. Fiore E, Van Tyne D, Gilmore MS. Pathogenicity of Enterococci. Microbiology Spectrum. 2019;7:4.
40. Britton RA, Young VB. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology. 2014;146(6):1547–1553.
41. Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2(3):145–158.
42. Vardakas KZ, Trigkidis KK, Boukouvala E, Falagas ME. Clostridium difficile infection following systemic antibiotic administration in randomised controlled trials: a systematic review and meta-analysis. Int J Antimicrob Agents. 2016;48(1):1–10.
43. Elshaer D, Begun J. The role of barrier function, autophagy, and cytokines in maintaining intestinal homeostasis. Semin Cell Dev Biol. 2017;61:51–59.
44. Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci U S A. 2016;113(1):E7–15.
45. Ferrer M. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut Microbes. 2014;5(1):64–70.
46. Croom KF, Goa KL. Levofloxacin: a review of its use in the treatment of bacterial infections in the United States. Drugs. 2003;63(24):2769–2802.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.