")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Effect of the Lipoxin Receptor Agonist BML-111 on Cigarette Smoke Extract-Induced Macrophage Polarization and Inflammation in RAW264.7 Cells
Authors Cao E, Xu J, Gong Y, Yuan J, Chen A, Liu J, Fan Y, Fan X, Kuang X
Received 9 November 2022
Accepted for publication 29 April 2023
Published 19 May 2023 Volume 2023:18 Pages 919—932
DOI https://doi.org/10.2147/COPD.S395569
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
En Cao,1,* Jun Xu,1,* Yuanqi Gong,2,* Jingjing Yuan,3 Anbang Chen,1 Jiayi Liu,4 Yunfei Fan,4 Xiangyang Fan,4 Xiaodong Kuang1
1Department of Pathology, Basic Medical College of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Critical Care Medicine/ICU (Intensive Care Unit), Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 3Department of Physiology, School of Basic Medicine, Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 4The Basic Medical School of Nanchang University, Nanchang, Jiangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaodong Kuang, Department of Pathology, Basic Medical College of Nanchang University, Nanchang, Jiangxi, People’s Republic of China, Email [email protected]
Background: Macrophages are known to play a crucial role in the chronic inflammation associated with Chronic Obstructive Pulmonary Disease (COPD). BML-111, acting as a lipoxin A4 (LXA4) receptor agonist, has shown to be effective in protecting against COPD. However, the precise mechanism by which BML-111 exerts its protective effect remains unclear.
Methods: In order to establish a cell model of inflammation, cigarette smoke extract (CSE) was used on the RAW264.7 cell line. Afterwards, an Enzyme-linked immunosorbent assay (ELISA) kit was employed to measure concentrations of tumor necrosis factor-α (TNF-α), interleukin-1beta (IL-1β), interleukin-18 (IL-18), and interleukin-10 (IL-10) in the cell supernatants of the RAW264.7 cells.In this study, we examined the markers of macrophage polarization using two methods: quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot analysis. Additionally, we detected the expression of Notch-1 and Hes-1 through Western blotting.
Results: BML-111 effectively suppressed the expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-18, as well as inflammasome factors NLRP3 and Caspase-1, while simultaneously up-regulating the expression of the anti-inflammatory cytokine IL-10 induced by CSE. Moreover, BML-111 reduced the expression of iNOS, which is associated with M1 macrophage polarization, and increased the expression of Arg-1, which is associated with M2 phenotype. Additionally, BML-111 downregulated the expression of Hes-1 and the ratio of activated Notch-1 to Notch-1 induced by CSE. The effect of BML-111 on inflammation and macrophage polarization was reversed upon administration of the Notch-1 signaling pathway agonist Jagged1.
Conclusion: BML-111 has the potential to suppress inflammation and modulate M1/M2 macrophage polarization in RAW264.7 cells. The underlying mechanism may involve the Notch-1 signaling pathway.
Keywords: BML-111, macrophage polarization, Notch-1 signaling pathway, COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is defined as a common, preventable and treatable disease characterised by persistent respiratory symptoms and airflow limitation due to airway or alveolar abnormalities usually caused by significant exposure to noxious particles or gases.1 COPD is a leading cause of morbidity and mortality worldwide, ranked third among the leading causes of death globally in 2019.2 In 2016, the estimated global prevalence of COPD was 251 million cases; with a population of 1397 million in China, the estimated prevalence would suggest between 113 and 187 million of the global cases being in China.3 A number of 910,809 deaths due to COPD occurred in China in 2013, which accounted for about one-third of COPD-related deaths in the world.4 Even worse, it is projected to affect more people in the next decade due to the ageing population and increasing exposure to risk factors.5 Current treatments for COPD are mostly symptomatic and have limited efficacy. Research has revealed that COPD is a complex pulmonary inflammatory disease with multiple underlying causes, indicating that anti-inflammatory therapy should be a primary approach for managing COPD. Therefore, there is a need for more effective anti-inflammatory drugs to improve the treatment outcomes of COPD.6,7
Lipoxins (LXs) are known to possess potent anti-inflammatory properties by inhibiting the release of pro-inflammatory mediators.8 Among the various LXs, Lipoxin A4 (LXA4) has been found to bind with its receptor, a G protein-coupled receptor, also known as formyl peptide receptor 2 (FPR-2), to exert its anti-inflammatory effects.9–11 BML-111 is a stable and potent FPR-2 agonist that has demonstrated anti-inflammatory and pro-resolving effects in conditions such as acute lung injury,12 collagen-induced arthritis,13 and acute pancreatitis.14 In our previous study, we observed that BML-111 exhibited anti-inflammatory effects in COPD;15 however, the specific mechanisms underlying these effects remain unclear.
Studies have identified two major polarization states of macrophages:16–18 M1 and M2. M1 macrophages are pro-inflammatory, activated by lipopolysaccharide (LPS) with Th1 cytokines such as IFN-γ and GM-CSF, and produce cytokines such as IL-1β, IL-6, IL-12, IL-23, and TNF-α. In contrast, M2 macrophages are anti-inflammatory and immunoregulatory, polarized by Th2 cytokines such as IL-4 and IL-13, and produce cytokines including IL-10 and TGF-β. These macrophage types have distinct functions and transcriptional profiles.19,20
Recent data indicates that activated macrophages in COPD release pro-inflammatory factors, including TNF-α, TGF-β, IL-1β, and IL-18, which promote inflammation.11 However, BML-111 activation can also induce a strong anti-inflammatory response in macrophages.12,13 Macrophages exhibit bidirectional inflammatory regulation due to their plasticity, which allows for reversible polarization between the pro-inflammatory M1 and anti-inflammatory M2 phenotypes in response to various challenges. Differentiated M1 and M2 macrophages can be transformed into each other, with M2 transforming to M1 and vice versa, depending on the reversible expression levels of M1 and M2 subtype marker proteins.14–16 Long-term exposure to cigarette smoke can activate oxidants, inflammation, oxidative stress, and apoptosis, leading to the expansion of alveolar spaces and the development of chronic obstructive pulmonary disease. Cigarette smoke is considered to be the primary cause of chronic lung inflammation, protease/antiprotease imbalance, and oxidative stress. As a result, cigarette smoke extract (CSE) is commonly used to create COPD models.17,18
Given the anti-inflammatory potential of BML-111 and the role of macrophage polarization in regulating inflammation, we investigated the impact of BML-111 on COPD inflammation by examining the polarization state of macrophages using a CSE-induced inflammation model in RAW264.7 macrophages.
Materials and Methods
Cells and Culture
We obtained RAW264.7 mouse macrophages from the Shanghai Cell Bank, Chinese Academy of Sciences and cultured them in DMEM (Solarbio, Beijing, China) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% Penicillin-Streptomycin (Gibco, Grand Island, NY, USA) Solution. The cells were maintained at 37°C in a 5% CO2 incubator and subcultured when cell density reached 40–60% confluency.
CSE Preparation and Cell Treatment
For this study, according to the reference,21 we utilized two cigarettes (Jiangxi Jinsheng Tobacco Group, Nanchang, China; roasted tobacco type, with tar amount of 11mg; smoke nicotine amount of 1.0 mg; smoke carbon monoxide amount of 12 mg). We attached one end of a rubber tube to the filter and the other end to the mouth of a 50mL syringe. The cigarette was lit, and we slowly (8mL/s) pulled the 50mL syringe at a uniform speed to collect all the cigarette smoke in one tube.22 We then transferred the smoke into a glass bottle containing 50mL of culture medium. This process was repeated for the second cigarette, and the smoke from both cigarettes was combined in the glass bottle. After allowing the smoke to dissolve completely in the medium, which constituted 100% stock solution, we proportionally diluted it to 1% CSE and used it in the cell experiment within 30 minutes. The cells were incubated for 24 hours.
Experimental Grouping
Prior to each experiment, the cells were cultured in serum-free medium for 12 hours. BML-111 (5S, 6R, 7-trihydroxyheptanoic acid methyl ester; Cayman Chemicals, Michigan, USA) and Dex (Dexamethasone) (the positive control) (Sigma-Aldrich, München, Germany) were administered to different groups. In addition, we conducted pretreatment with either the lipoxin receptor antagonist Boc-2 or the Notch signaling pathway-specific agonist Jagged1 (CST, Boston, MA, USA). The CSE concentration chosen for the experiment was 1%.23
Part 1: Investigating the impact of BML-111 on CSE-induced inflammation in macrophages.
The RAW264.7 cells were divided into four groups:
- Control group: Cells were cultured in serum-free medium containing 1% penicillin-streptomycin.
- Model group: Cells were challenged with 1% CSE (2 mL) for 24 hours.
- BML-111 + Model group: Cells were pretreated with serum-free medium comprising different concentrations of BML-111 (2.5, 5, 10 μM) for 30 minutes before adding 1% CSE.
- Dex + BML-111 + Model group: Cells were pretreated with serum-free medium containing 100 nM Dex for 30 minutes before adding 1% CSE;24
Part 2: Investigating the effect of BML-111 on macrophage polarization and the Notch-1 signaling pathway in macrophages.
To interdict the lipoxin receptor, we pretreated Boc-2 (Butoxycarbonyl-Phe-Leu-Phe-Leu-Phe; Calbiochem, San Diego, CA, USA) at a final concentration of 10 μM for one hour.25 The remaining steps were performed according to the methods described in Part 1.
RAW264.7 cells were divided into four groups: (1) control group, (2) Model group, (3) BML-111 + Model group, and (4) Boc-2 + BML-111 + Model group.
Part 3: Investigating the intervention of the Notch signaling pathway-specific agonist Jagged1 on BML-111 in CSE-treated macrophages.
To promote the Notch signaling pathway, Jagged1 was pretreated with a final concentration of 8 μM for one hour. The remaining steps were performed according to the methods described in Part 1.
RAW264.7 cells were divided into five groups: (1) control group, (2) Model group, (3) Jagged1 + Model group, (4) BML-111 + Model group, and (5) Jagged1 + BML-111 + Model group.
CCK-8 Assay
To investigate the effects of BML-111 on RAW264.7 cells under normal or high glucose conditions, a CCK-8 (GLPBIO, Montclair, CA, USA) assay was performed. RAW264.7 cells were seeded into 96-well plates at a density of 2×103 cells/well and cultured for 24 hours. The cells were then treated with different concentrations of BML-111 (0, 2.5, 5, 10, 20 μM) in the presence of either 5.5 mM glucose or high glucose (30 mM) for 24 hours. After incubation, CCK-8 reagent was added to the cells, and the optical density (OD) value was measured using a microplate reader after 1.5 hours.
Enzyme-Linked Immunosorbent Assays (ELISAs)
The levels of IL-1β, IL-18, and TNF-α in the supernatant of RAW264.7 cells were quantified using an ELISA kit (R&D Systems, Minneapolis, MN, USA). The manufacturer’s protocol was followed, and the optical density values were measured at 450 nm. The control group was left untreated for 24 hours, while the remaining groups were treated with cigarettes for 24 hours, and the cell supernatant was collected for analysis.
Western Blotting
In this experiment, protein blotting was employed to detect the expression of M1 marker iNOS and M2 marker Arg-1, as well as NLRP3 (Ab263899, Abcam, San Francisco, CA, USA) inflammasome components including iNOS (Ab178945, Abcam, San Francisco, CA, USA), Arg-1 (Ab233548, Abcam, San Francisco, CA, USA), caspase-1 and cleaved caspase-1 (Ab893325, CST, Boston, MA, USA), Notch-1 (Ab52627, Abcam, San Francisco, CA, USA), activated Notch-1 (Ab52301, Abcam, San Francisco, CA, USA), and Hes-1 (Ab108937, Abcam, San Francisco, CA, USA). RAW264.7 cells were treated with a specific CSE concentration for 24 hours, and subsequently lysed in RIPA lysis buffer (Solarbio, Beijing, China).Protein concentrations were determined using a protein concentration analysis kit (GLPBIO, Montclair, CA, USA). The protein samples were denatured by heating and loaded onto SDS-PAGE gels prepared under specific conditions, followed by pre-electrophoresis, electrophoresis, and electrotransfer to polyvinylidene fluoride (PVDF) membranes. The membranes were then blocked with 5% skimmed milk powder in closure solution at room temperature for 1 hour on a shaker. Next, the membranes were washed once with TBST (Solarbio, Beijing, China) and incubated overnight at 4°C with primary antibodies against iNOS (1:1000), Arg-1 (1:5000), NLRP3 (1:1000), cleaved caspase-1 (1:1000), Notch-1 (1:2000), activated Notch-1 (1:1000), and Hes-1 (1:1000). After washing three times with TBST, the membranes were incubated for 2 hours in secondary antibody before being washed again three times with TBST. Finally, the membranes were submerged in ultrasensitive ECL luminescent solution (Yamei, shanghai, China), and the results were obtained using a gel imaging system (Bio-RAD, Hercules, CA, USA).
Quantitative Real-Time PCR (qRT-PCR)
Was extracted from RAW264.7 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The concentration of total RNA was measured with a Nanodrop-1000 spectrophotometer (Nanodrop Technologies, Waltham, MA, USA), and the total RNA was then subjected to qPCR using HiScript II Q RT Super-Mix with gDNA wiper (Vazyme Biotech Co, Nanjing, China). qRT-PCR was performed using the AceQ® qPCR SYBR® Green Master Mix kit (Vazyme Biotech Co., Ltd, Nanjing, China) with GAPDH (Proteintech, Wuhan, China) as the internal reference. The CT value obtained from real-time PCR was used to calculate relative changes in gene expression by the 2-ΔΔCT method.
Statistical Analyses
The data are presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used to compare multiple groups. All statistical analyses were performed using SPSS 22.0 software (SPSS Inc, Armonk, NY, USA), and figures were created using GraphPad Prism version 8.0 (GraphPad software, San Diego, CA, USA). A p-value less than 0.05 was considered statistically significant.
Results
The Effects of the Drugs on Cell Viability
To investigate the impact of BML-111 and Jagged-1 on RAW264.7 cells, different concentrations of BML-111 (0, 2.5, 5, 10, 20 μM) and Jagged-1 (0, 4, 8, 16, 32 μM) were added to RAW264.7 cells. The influence of these drugs on cell viability was assessed by conducting a CCK-8 assay, as shown in Figure 1A and B. The results indicated that after treatment with the drugs for 24 hours, the cell viability of RAW264.7 cells was affected differently at various concentrations. However, we observed that the selected drug concentrations and exposure times did not affect the viability of RAW264.7 cells. Hence, we chose BML-111 (10 μM) and Jagged-1 (8 μM) as the drug concentrations for subsequent experiments.
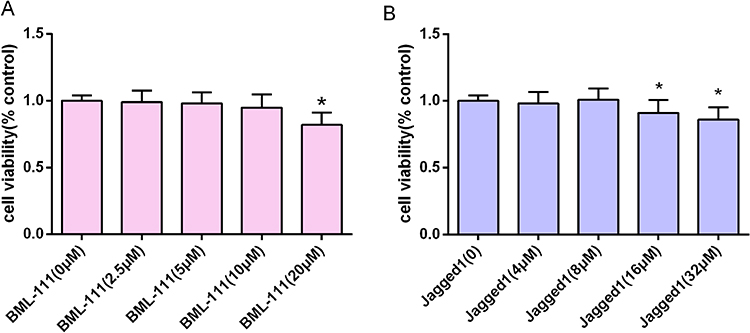 |
Figure 1 The effects of the drugs on RAW264.7 cell viability. (A and B) Cells were incubated with different concentrations of BML-111 and Jagged-1 for 24 h, and the effects of the drugs on the viability of RAW264.7 cells were detected by a CCK-8 assay. The values shown are the mean± SD; *p<0.05 vs the control group (0 μM). |
BML-111 Inhibited Inflammation in Macrophages
To investigate how BML-111 affects CSE-induced inflammation, we measured the release of inflammatory mediators TNF-α, IL-1β, IL-18, and IL-10 in cell supernatant. As depicted in Figure 2, CSE treatment significantly increased the levels of TNF-α (Figure 2A), IL-1β (Figure 2B), and IL-18 (Figure 2C) (p<0.01, respectively), while decreasing the level of IL-10 (Figure 2D) (p<0.01) as compared to the control group. However, treatment with BML-111 effectively suppressed the CSE-induced elevation of these proinflammatory cytokines (TNF-α, IL-1β, and IL-18) in a dose-dependent manner, and upregulated IL-10 level.
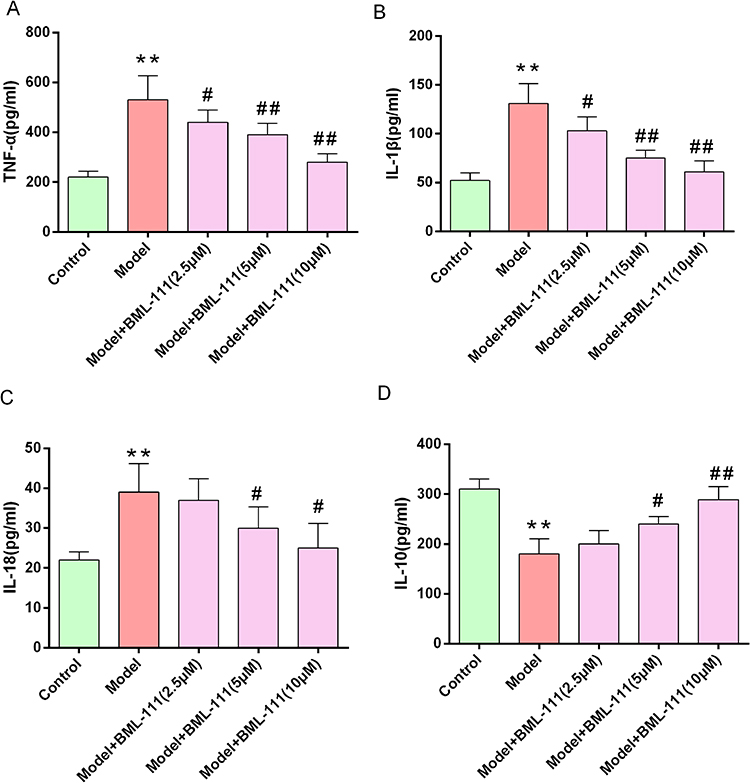 |
Figure 2 BML-111 reduced the expression of inflammatory mediators.(A–D) ELISAs illustrating the secretion levels of TNF-α, IL-1β, IL-18 and IL-10 protein at 24 h after macrophage exposure to CSE.Data are shown as the mean±SD. (n=6/**p<0.01 vs control group; #p<0.05, ##p<0.01 vs model group). |
BML-111 Compromised CSE-Induced NLRP3 Inflammasome Activation in Macrophages
To assess inflammasome activation in macrophages, we detected the expression of NLRP3, pro-caspase-1, and cleaved-caspase-1 (Figure 3A). As shown in Figure 3B and C, compared to the control group, NLRP3 and cleaved-caspase-1 were significantly upregulated in the CSE group, but not pro-caspase-1. However, treatment with BML-111 or Dex (Dexamethasone) alone or in combination noticeably decreased the expression of NLRP3 and cleaved-caspase-1 in macrophages as compared to the model group.
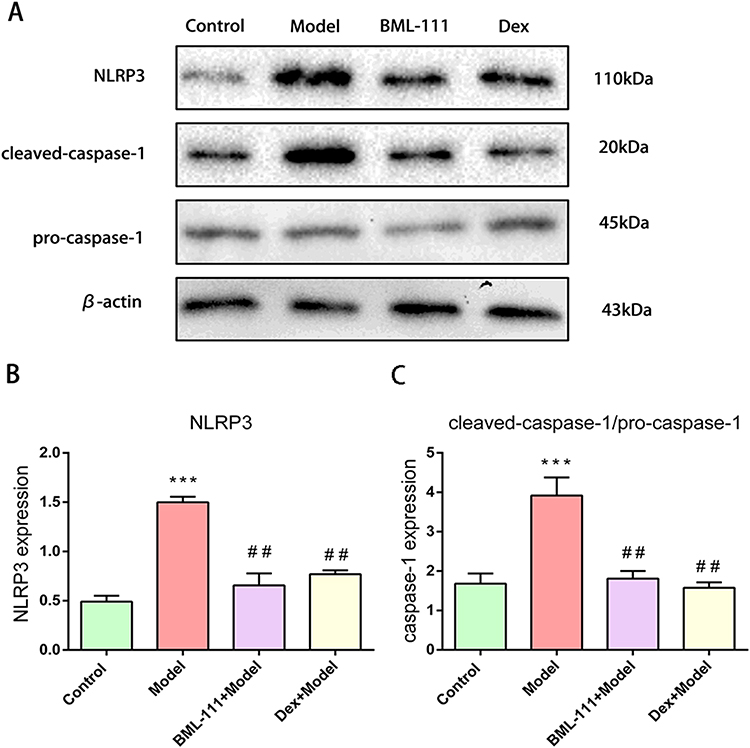 |
Figure 3 BML-111 inhibited CSE-induced NLRP3 inflammasome activation. (A) The protein expression levels of NLRP3, pro-caspase-1, and cleaved-caspase-1, observed using Western blot assay. (B and C) Semi-quantitative analysis of NLRP3 and cleaved-caspase-1/pro-caspase-1 ratio. The values shown are the mean±SD. (n=3/***p<0.01 vs Control group; ##p<0.01 vs model group). |
BML-111 Regulated CSE-Induced Macrophage Polarization
We utilized RT-PCR and Western blot to detect the expression of M1 macrophage polarization marker iNOS and M2 macrophage polarization marker Arg-1. As depicted in Figure 4A–E, compared to the control group, the model group exhibited high expression of iNOS (p<0.05, p<0.01), and low expression of Arg-1 at both transcription and protein levels (p<0.05). In contrast, BML-111 treatment resulted in decreased expression of iNOS (p<0.05, p<0.01) and increased expression of Arg-1 (p<0.05, p<0.01) as compared to the CSE group. Moreover, the lipoxin receptor antagonist, Boc-2, inhibited the effect of BML-111 on both iNOS (p<0.05) and Arg-1 (p<0.05) at transcription and protein levels.
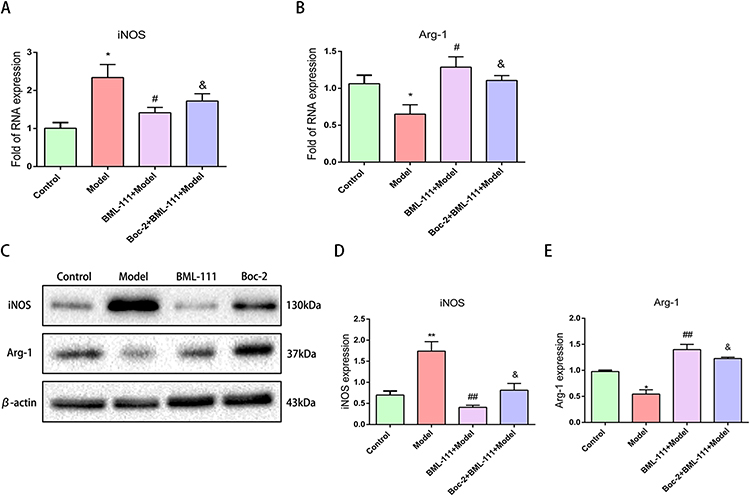 |
Figure 4 Effect of BML-111 on the expression of iNOS and Arg-1 after CSE stimulated mouse macrophages. (A and B) The transcription expression levels of iNOS and Arg-1, observed using RT-PCR assay by comparison with beta-actin; (C–E) The protein expression levels of iNOS and Arg-1, observed using Western blot assay by comparison with β-actin. Data are expressed as the mean±SD (n=3/*p<0.05, **p<0.01 vs control group; #p<0.05 ##p<0.01 vs model group; &p<0.05 vs BML-111 model group). |
The findings suggest that CSE-induced inflammation is associated with M1 macrophage polarization, while BML-111 treatment is linked to M2 macrophage polarization. Furthermore, the effect of macrophage polarization by BML-111 can be reversed by lipoxin receptor antagonists.
BML-111 Suppressed CSE-Induced Activation of Notch-1 Signaling Pathway in Macrophages
To explore the role of the Notch signaling pathway in CSE-induced inflammation, we measured the expression of Hes-1, Notch-1, and activated Notch-1 (Figure 5A). As shown in Figure 5B and C, exposure to CSE led to an increase in Hes-1 expression (p<0.01) and activated Notch-1/Notch-1 ratio (p<0.01) in macrophages. However, compared to the model group, the CSE+BML-111 group exhibited significantly decreased expression of Hes-1 (p<0.01) and a lower ratio of activated Notch-1 to Notch-1 (p<0.05).
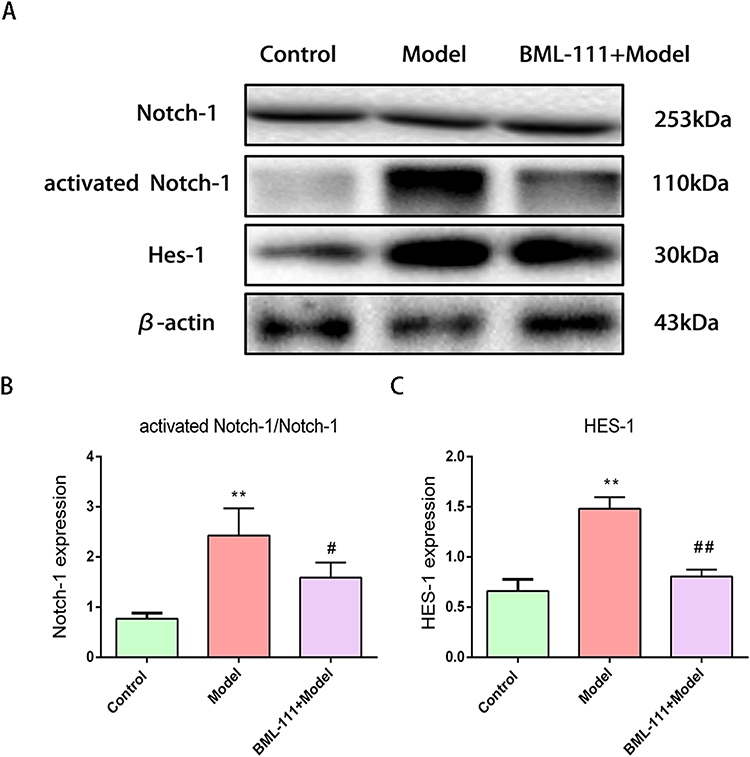 |
Figure 5 The effect of BML-111 on the expression of Notch-1, Hes-1 after CSE stimulated mouse macrophages. (A) The protein expression levels of Notch-1, activated Notch-1 and Hes-1, observed using Western blot assay by comparison with β-actin. (B and C) Semi-quantitative analysis of activated Notch-1/Notch-1, Hes-1 levels. Data are expressed as the mean±SD (n=3/ **p<0.01 vs control group; #p<0.05, ##p<0.01 vs model group). |
Jagged1 Weakened the Effect of BML-111 on Macrophage Polarization and Inflammation in CSE-Treated Macrophages
To investigate the mechanism by which BML-111 regulates CSE-induced macrophage polarization and inflammation, we used Jagged1, a specific agonist of the Notch signaling pathway. As depicted in Figure 6A and B, after 24 hours of CSE stimulation, the expression of Notch-1 significantly increased compared to the control group. However, pretreatment with BML-111 resulted in a significant decrease in Notch-1 expression (p<0.05) compared to the model group. Moreover, the combination of Jagged1 and BML-111 led to a further increase in Notch-1 expression as compared to BML-111 alone (p<0.05). Furthermore, compared to the model group, pretreatment with BML-111 significantly reduced iNOS expression (p<0.01) but increased Arg-1 expression (p<0.05).The co-treatment of Jagged1 and BML-111, however, resulted in increased expression of iNOS (p<0.05) but decreased expression of Arg-1 (p<0.05) compared to the BML-111 group, suggesting that Jagged1 reversed the effect of BML-111 on macrophage polarization as shown in Figure 6A, C and D. In addition, as depicted in Figure 7, we observed a significant decrease in TNF-α (Figure 7A), IL-1β (Figure 7B), and IL-18 (Figure 7C) secretion and an increase in IL-10 (Figure 7D) secretion following treatment with BML-111 compared to the model group (p<0.05, p<0.01). However, this effect was reversed by Jagged1 (p<0.05). These findings suggest that BML-111 exerts its strong anti-inflammatory effects through the Notch-1 signaling pathway in CSE-treated macrophages.
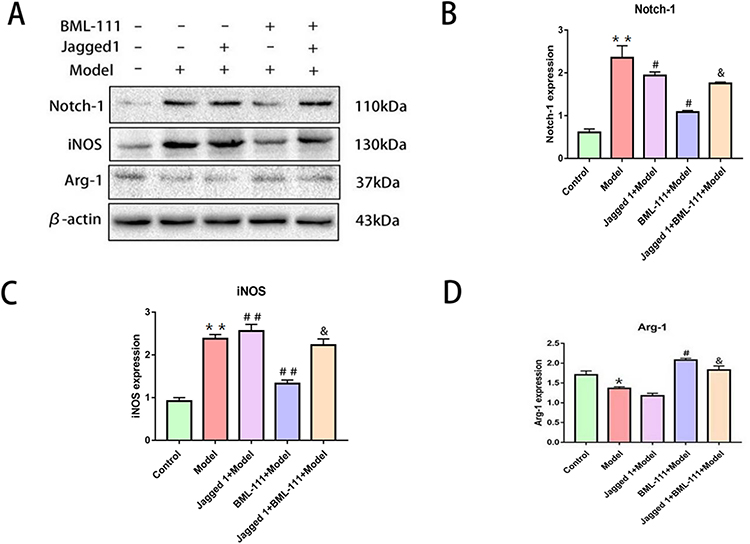 |
Figure 6 The expression of the Notch-1, iNOS, and Arg-1 after promoting the Notch signaling pathway with Jagged1 in macrophages following CSE treatment. (A) Western blot detection of Notch-1, iNOS, and Arg-1 in macrophages. (B–D) quantitative data on the expression of Notch signaling pathway related proteins Notch-1, the M1 marker iNOS and M2 marker Arg-1 in the cytosol and nucleus.The values shown are the mean±SD. (n = 3/*p<0.05, **p<0.01 vs control group; #p<0.05, ##p<0.01 vs model group; &p<0.05 vs the BML-111+Model group group). |
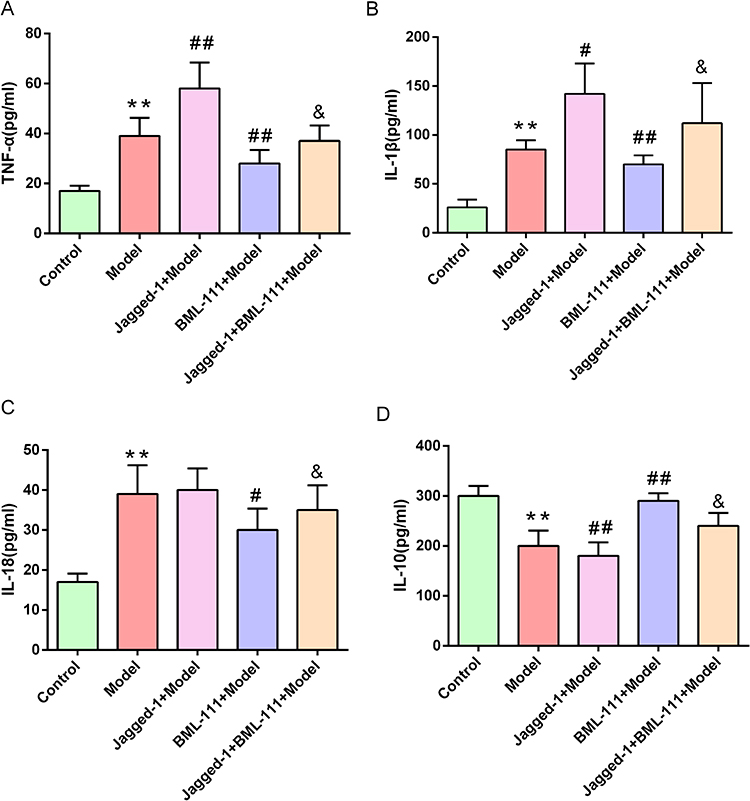 |
Figure 7 The expression of inflammatory cytokines after promoting the Notch signaling pathway with Jagged1 in macrophage supernatants following CSE treatment. (A–D) ELISA analysis of the secretion levels of inflammatory cytokines (TNF-α, IL-1β IL-18 and IL-10) in the supernatants. The values are shown as the mean±SD (n= 6/**p<0.01 vs control group; #p<0.05, ##p<0.01 vs model group; &p<0.05 vs the BML-111+Model group group). |
Discussion
Inflammation is a crucial factor in the pathogenesis of COPD, which primarily manifests as emphysema and airway remodeling. The development of COPD is closely associated with numerous inflammatory cells, among which macrophages play a vital role in the inflammatory response. Macrophages are widely distributed throughout the airways, lung parenchyma, and bronchoalveolar lavage (BAL) fluid of COPD patients,10 serving as the first line of defense against intracellular pathogens.9 These cells occupy a prominent position in orchestrating chronic inflammation in COPD, playing a significant role in the complex processes that underlie this condition.19,20
LXA4 is an endogenous anti-inflammatory and pro-resolving lipid mediator that regulates macrophages through enzymatic conversion of arachidonic acid by lipoxygenases (LOXs).26 This mediator is generated via dual lipoxygenation of arachidonic acid, involving either ALOX15/ALOX5 or ALOX5/ALOX12, and functions as a “stop signal” in inflammation.27 Previous studies have shown that LPS stimulation increases lipoxin receptor expression on monocyte-derived macrophages, suggesting the involvement of LXA4 in the inflammatory process of LPS-induced macrophages. In addition, LXA4 has been found to inhibit nuclear aggregation of NF-κB and AP-1, as well as downregulate IL-8 expression in LPS-induced macrophages, indicating its potential as an anti-inflammatory agent that can modulate the immune response of macrophages.Our goal was to determine whether LXA4 could effectively inhibit inflammation in COPD.28 However, due to its unstable methylesterified form, stable LX analogs or LXA4 receptor agonists have been designed. BML-111 is one such analog that is a synthetic LXA4 analogue and FPR2/ALX agonist with strong anti-inflammatory effects.29 BML-111 has been widely used as a lipoxin receptor agonist in experimental research because it retains the crucial functional group necessary for the lipoxin function and has stable characteristics consistent with lipoxin receptors. Multiple reports have shown that BML-111 can reduce inflammation and oxidative stress in rats based on various in vivo and in vitro experiments, and has multiple biological effects such as anti-inflammation and anti-apoptosis.30–32 In the present study, supplementation with BML-111 significantly restored the levels of inflammatory mediators TNF-α, IL-1β, and IL-18 in macrophages treated with CSE, indicating that BML-111 can effectively inhibit CSE-induced inflammation in macrophages. As CSE is commonly used to model COPD,17,18 these results suggest that BML-111 could be a potential drug for treating COPD.
Recent studies have suggested that the NLRP3 inflammasome is involved in the onset and development of COPD, as well as in CSE-induced inflammation, by promoting the release of inflammatory cytokines.33 The NLRP3 inflammasome is an important component of innate immunity, often located in macrophages. It is a polyprotein complex composed of NLR receptors, apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1 precursor.34,35 When stimulated, the NLRP3 protein acts as a scaffold to bind ASC, which then recruits and hydrolyzes pro-caspase-1 to form Caspase-1 with enzyme activity, promoting the processing, maturation, and release of inflammatory factors such as IL-1β and IL-18.36 Previous studies have shown that inhaling toxic and harmful particles or gases can directly activate pattern recognition receptors, primarily TLRs, in the lung. This activation leads to downstream activation of the NF-κB signaling pathway and promotes the transcription of NLRP3 and other inflammatory factors.37 Furthermore, exposure to these toxic particles or gases can cause extensive cell death in the lung, which releases a variety of endogenous danger molecules, such as high mobility group protein B1. These molecules further promote the activation of TLRs and downstream NLRP3.38–40 Increased expression of Caspase-1, IL-1β, and IL-18 in the lungs of patients with CSE-induced inflammation suggests that the NLRP3 inflammasome is activated during the occurrence and development of this condition.41 Dex, an α2 adrenergic receptor agonist with potent anti-inflammatory effects, has been shown to inhibit NLRP3 inflammasome activation in emerging literature.42–44 In our experiment, we found that the expression of NLRP3 and cleaved Caspase-1 significantly increased in the CSE-induced inflammatory model group, but decreased dramatically in the BML-111 and Dex groups. These results suggest that CSE-induced inflammation is associated with the NLRP3 inflammasome in macrophages, but BML-111 can effectively inhibit its activation. Therefore, regulation of the NLRP3 inflammasome and inflammatory mediators by BML-111 demonstrates its powerful anti-inflammatory properties.
The confusing pro-inflammatory or anti-inflammatory state of macrophages can be explained by their ability to polarize into two predominant phenotypes in response to different stimuli. Macrophages can be classified as classically activated M1 macrophages or alternatively activated M2 macrophages based on their activation states, functions, and secreted factors. Classically activated M1 macrophages, which are stimulated by granulocyte-macrophage colony-stimulating factor (GM-CSF), lipopolysaccharide (LPS), or interferon-gamma (IFN-γ), release IL-12, IL-18, IL-1β, and tumor necrosis factor-alpha (TNF-α). These cytokines have pro-inflammatory and cytotoxic effects that can lead to inflammatory diseases, including COPD.12 Recent evidence has also shown that NLRP3 inflammasome activation is associated with M1 macrophage polarization.41 In contrast to M1 macrophages, selectively activated M2 phenotypes are induced by stimuli such as alternating monocyte colony-stimulating factor (M-CSF), IL-4, IL-13, and glucocorticoids. M2 macrophages have been shown to have anti-inflammatory effects and promote tissue repair and wound healing during inflammation.11 In our experiment, we observed that the M1 marker protein iNOS was upregulated while the M2 marker protein Arg-1 was downregulated in the CSE-induced inflammation model group, indicating a strong association between CSE-induced inflammation and M1 macrophage polarization. However, treatment with BML-111 decreased iNOS levels and increased Arg-1 expression, suggesting that BML-111 has the potential to transform macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype.
To investigate the regulatory mechanism of macrophage polarization induced by BML-111, we examined the levels of Notch pathway-associated proteins in macrophages. The Notch signaling pathway is a highly conserved mechanism that plays a critical role in development, tissue homeostasis, and disease.45 The pathway consists of receptors, ligands expressed on adjacent cell membranes, intracellular transcription factors, regulatory molecules, and downstream effector molecules.46–49 Briefly, upon interaction between the Notch receptor and ligand, the intracellular domain (NICD) is released into the cytoplasm and transferred into the nucleus. This promotes the production of transcriptional activators and induces downstream target gene expression in the Notch pathway, including Hes1, Hes5, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and others.
The Notch signaling pathway plays an indispensable role in the process of macrophage polarization.50 Previous studies have shown that blocking the Notch signaling pathway can reduce inflammation and accelerate tissue repair.51,52 The upregulation of Notch-1 gene expression can promote macrophage activation and affect the function of mature macrophages by increasing the level of interferon-γ.53,54 Studies have also shown that the differentiation of macrophages to the M1 phenotype is closely related to increased levels of the Notch-1 receptor and downstream effector Hes-1.55,56 While few studies have shown that BML-111 can inhibit the Notch signaling pathway,57–59 the mechanism remains unclear. In our experiment, we observed that CSE upregulated the expression of Hes-1 and activated the Notch-1/Notch-1 ratio in macrophages.60 Nonetheless, BML-111 was found to decrease the expression of Hes-1 and the Notch-1/Notch-1 ratio. In subsequent experiments, we used the Notch-1 pathway activator Jagged-1 in combination with BML-111 in the RAW264.7 cell inflammation model to reactivate the Notch-1 pathway. Compared with the BML-111+CSE group, this led to a reversal of the expression trends of the M1 marker protein iNOS and the M2 marker protein Arg-1, as well as an abrogation of the effect of BML-111 on inflammatory mediators IL-1β, IL-18, and IL-10. Based on these results, we speculate that BML-111 may play an anti-inflammatory role in cigarette-induced macrophages through the Notch signaling pathway. Please see Figure 8 for more details on the mechanism of macrophage polarization and inflammation induced by BML-111 on RAW264.7 cells.
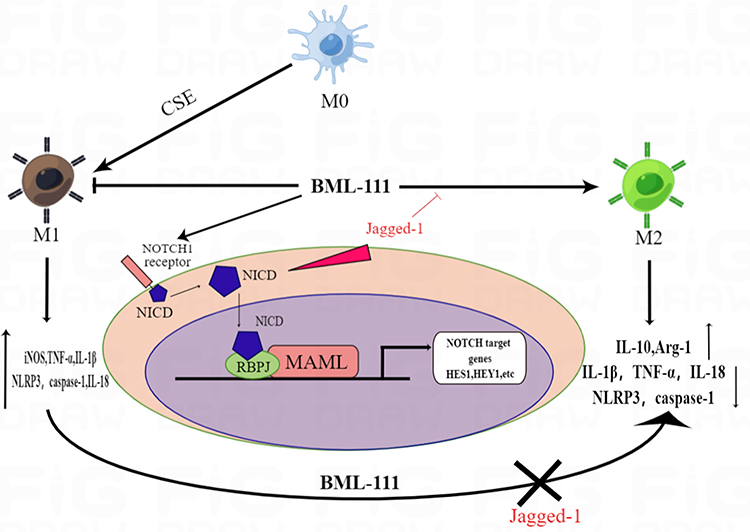 |
Figure 8 The mechanism of macrophage polarization and inflammation induced by BML-111 on RAW264.7 cells. |
To summarize, the lipoxin receptor agonist BML-111 inhibits CSE-induced inflammation and regulates macrophage polarization, possibly through the Notch-1 signaling pathway. However, it is regrettable that we were only able to inspect the effects of BML-111 on CSE-induced macrophage polarization and inflammation in vitro, and its action has not been confirmed in vivo. Therefore, further experimental studies are required to elucidate the specific anti-inflammatory mechanism of BML-111 in the CSE-induced mouse model. This experiment could provide a new possibility for drug development to treat CSE-induced inflammation, and establish a new theoretical basis for the drug treatment of CSE-induced inflammation.
Abbreviations
COPD, Chronic obstructive pulmonary disease; LXA4, Lipoxin A4; CSE, Cigarette smoke extract; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-18, interleukin 18; NLRP3, NOD-like receptor thermal protein domain associated protein 3; iNOS, inducible nitric oxide synthase; Arg-1, Arginase-1; Dex, Dexamethasone; LOXs, lipoxygenases; ASC, apoptosis-associated spot-like protein containing a card; GM-CSF, granulocyte-macrophage colony-stimulating factor; LPS, lipopolysaccharide; IFN-γ, interferon-γ.
Data Sharing Statement
The analyzed data sets generated during the present study are available from the corresponding author.
Ethics Approval
All experimental work was approved by Medical Ethics Committee of Second Affiliated Hospital of Nanchang University.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Project number: 81460002, 81960012, 81860349, 82260374), the Natural Science Foundation of Jiangxi province in China (Project number:20202BAB206038).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHLBI/WHO workshop report. 2021.
2. World Health Organization. The Top 10 Causes of Death. World Health Organization; 2020.
3. Fang L, Gao P, Bao H, et al. Chronic obstructive pulmonary disease in China: a nationwide prevalence study. Lancet Respir Med. 2018;6:421–430. doi:10.1016/S2213-2600(18)30103-6
4. Yin P, Wang H, Vos T, et al. A subnational analysis of mortality and prevalence of COPD in China from 1990 to 2013: findings from the global burden of disease study 2013. Chest. 2016;150:1269–1280. doi:10.1016/j.chest.2016.08.1474
5. Khakban A, Sin DD, FitzGerald JM, et al. The projected epidemic of chronic obstructive pulmonary disease hospitalizations over the next 15 years. A population-based perspective. Am J Respir Crit Care Med. 2017;195:287–291. doi:10.1164/rccm.201606-1162PP
6. Keogh E, Mark Williams E. Managing malnutrition in COPD: a review. Respir Med. 2021;176:106248. doi:10.1016/j.rmed.2020.106248
7. Shnoda M, Gajjar K, Ivanova V. COPD and cardiovascular disease: a review of association, interrelationship, and basic principles for integrated management. Crit Care Nurs Q. 2021;44:91–102. doi:10.1097/CNQ.0000000000000342
8. Yang JX, Li M, Chen XO, et al. Lipoxin A4 ameliorates lipopolysaccharide-induced lung injury through stimulating epithelial proliferation, reducing epithelial cell apoptosis and inhibits epithelial-mesenchymal transition. Respir Res. 2019;20:192. doi:10.1186/s12931-019-1158-z
9. Liu C, Guan H, Cai C, Li F, Xiao J. Lipoxin A4 suppresses osteoclastogenesis in RAW264.7 cells and prevents ovariectomy-induced bone loss. Exp Cell Res. 2017;352:293–303. doi:10.1016/j.yexcr.2017.02.018
10. Bozinovski S, Uddin M, Vlahos R, et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2012;109:935–940. doi:10.1073/pnas.1109382109
11. Cao Y, Zhou X, Yin Z, et al. The anti-inflammatory effect of BML-111 on COPD may be mediated by regulating NLRP3 inflammasome activation and ROS production. Prostaglandins Other Lipid Mediat. 2018;138:23–30. doi:10.1016/j.prostaglandins.2018.08.001
12. Zhao J, Geng W, Wan K, et al. Lipoxin A4 promotes autophagy and inhibits overactivation of macrophage inflammasome activity induced by Pg LPS. J Int Med Res. 2021;49:300060520981259. doi:10.1177/0300060520981259
13. Ali M, Kucko N, Jansen JA, Yang F, Walboomers XF. The effect of lipoxin A4 on E. coli LPS-induced osteoclastogenesis. Clin Oral Investig. 2021;25:957–969. doi:10.1007/s00784-020-03385-3
14. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. doi:10.1016/j.ejphar.2020.173090
15. Wu H, Zheng J, Xu S, et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflammation. 2021;18:2. doi:10.1186/s12974-020-02041-7
16. Zhang B, Yang Y, Yi J, Zhao Z, Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res. 2021;56:991–1005. doi:10.1111/jre.12912
17. Cho WK, Lee CG, Kim LK. COPD as a Disease of Immunosenescence. Yonsei Med J. 2019;60:407–413. doi:10.3349/ymj.2019.60.5.407
18. Son ES, Kim SH, Ryter SW, et al. Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy. Toxicol in Vitro. 2018;48:170–178. doi:10.1016/j.tiv.2018.01.011
19. Baker JM, Hammond M, Dungwa J, et al. Red blood cell-derived iron alters macrophage function in COPD. Biomedicines. 2021;17:1939. doi:10.3390/biomedicines9121939
20. Singh R, Belchamber KBR, Fenwick PS, et al.; COPDMAP consortium. Defective monocyte-derived macrophage phagocytosis is associated with exacerbation frequency in COPD. Respir Res. 2021;22:113. doi:10.1186/s12931-021-01718-8
21. Pezzuto A, Citarella F, Croghan I, Tonini G. The effects of cigarette smoking extracts on cell cycle and tumor spread: novel evidence. Future Sci OA. 2019;5:FSO394. doi:10.2144/fsoa-2019-0017
22. Wang M, Zhang Y, Xu M, et al. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic Biol Med. 2019;134:229–238. doi:10.1016/j.freeradbiomed.2019.01.004
23. Higashi T, Mai Y, Mazaki Y, Horinouchi T, Miwa S. A standardized method for the preparation of a gas phase extract of cigarette smoke. Biol Pharm Bull. 2016;39:898–902. doi:10.1248/bpb.b16-00062
24. Saraiya NV, Goldstein DA. Dexamethasone for ocular inflammation. Expert Opin Pharmacother. 2011;12:1127–1131. doi:10.1517/14656566.2011.571209
25. Bourne-Branchu Y, Gosmini C, Danoun G. N-Boc-amides in cross-coupling reactions. Chemistry. 2019;25:2663–2674. doi:10.1002/chem.201802635
26. Fu T, Mohan M, Brennan EP, et al. Therapeutic potential of lipoxin A4 in chronic inflammation: focus on cardiometabolic disease. ACS Pharmacol Transl Sci. 2020;3:43–55. doi:10.1021/acsptsci.9b00097
27. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi:10.1038/nri2294
28. Kotlyarov S, Kotlyarova A. Anti-inflammatory function of fatty acids and involvement of their metabolites in the resolution of inflammation in chronic obstructive pulmonary disease. Int J Mol Sci. 2021;22:12803. doi:10.3390/ijms222312803
29. Archambault AS, Poirier S, Lefebvre JS, et al. 20-Hydroxy- and 20-carboxy-leukotriene (LT) B(4) downregulate LTB(4) -mediated responses of human neutrophils and eosinophils. J Leukoc Biol. 2019;105(6):1131–1142. doi:10.1002/JLB.MA0718-306R
30. Liu J, Peng L, Li J. The lipoxin A4 receptor agonist BML-111 alleviates inflammatory injury and oxidative stress in spinal cord injury. Med Sci Monit. 2020;26:e919883. doi:10.12659/MSM.919883
31. Lin L, Wang Q, Xu F, et al. BML-111, the lipoxin A4 agonist, modulates VEGF or CoCl2-induced migration, angiogenesis and permeability in tumor-derived endothelial cells. Immunol Lett. 2021;230:27–35. doi:10.1016/j.imlet.2020.12.007
32. Pan S, Wu Y, Pei L, et al. BML-111 reduces neuroinflammation and cognitive impairment in mice with sepsis via the SIRT1/NF-κB signaling pathway. Front Cell Neurosci. 2018;12:267. doi:10.3389/fncel.2018.00267
33. Tian X, Xue Y, Xie G, et al. (-)-Epicatechin ameliorates cigarette smoke-induced lung inflammation via inhibiting ROS/NLRP3 inflammasome pathway in rats with COPD. Toxicol Appl Pharmacol. 2021;429:115674. doi:10.1016/j.taap.2021.115674
34. Kurita Y, Araya J, Minagawa S, et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir Res. 2017;18:114. doi:10.1186/s12931-017-0600-3
35. Gupta A, Anjomani-Virmouni S, Koundouros N, et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol Cell. 2017;65:999–1013.e7. doi:10.1016/j.molcel.2017.02.019
36. Xu F, Ji Q, Zhang J, Huang W, Cao Z, Li Y. AlCl3 inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production through suppressing NF-κB signaling pathway in murine peritoneal macrophages. Chemosphere. 2018;209:972–980. doi:10.1016/j.chemosphere.2018.06.171
37. Shen P, Jia S, Wang Y, et al. Mechanical stress protects against chondrocyte pyroptosis through lipoxin A4 via synovial macrophage M2 subtype polarization in an osteoarthritis model. Biomed Pharmacother. 2022;153:113361. doi:10.1016/j.biopha.2022.113361
38. Xu X, Zhang L, Ye X, et al. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflamm Res. 2018;67:57–65. doi:10.1007/s00011-017-1095-6
39. Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. 2017;79:517–539. doi:10.1146/annurev-physiol-022516-034314
40. An Y, Zhang H, Wang C, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019;33:12515–12527. doi:10.1096/fj.201802805RR
41. Benedikter BJ, Wouters EFM, Savelkoul PHM, Rohde GGU, Stassen FRM. Extracellular vesicles released in response to respiratory exposures: implications for chronic disease. J Toxicol Environ Health B Crit Rev. 2018;21:142–160. doi:10.1080/10937404.2018.1466380
42. Demiri M, Antunes T, Fletcher D, Martinez V. Perioperative adverse events attributed to α2-adrenoceptor agonists in patients not at risk of cardiovascular events: systematic review and meta-analysis. Br J Anaesth. 2019;123(6):795–807. doi:10.1016/j.bja.2019.07.029
43. Flanders CA, Rocke AS, Edwardson SA, Baillie JK, Walsh TS. The effect of dexmedetomidine and clonidine on the inflammatory response in critical illness: a systematic review of animal and human studies. Crit Care. 2019;23(1):402. doi:10.1186/s13054-019-2690-4
44. Zheng B, Zhang S, Ying Y, et al. Administration of dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci Rep. 2018;38(5). doi:10.1042/BSR20180892
45. Hasan SS, Tsaryk R, Lange M, et al. Endothelial notch signalling limits angiogenesis via control of artery formation. Nat Cell Biol. 2017;19(8):928–940. doi:10.1038/ncb3574
46. Tyagi A, Sharma AK, Damodaran C. A review on notch signaling and colorectal cancer. Cells. 2020;9(6). doi:10.3390/cells9061549
47. Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. 2003;23(4):543–553. doi:10.1161/01.ATV.0000060892.81529.8F
48. Oswald F, Liptay S, Adler G, Schmid RM. NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Mol Cell Biol. 1998;18(4):2077–2088. doi:10.1128/MCB.18.4.2077
49. Zhang YM, Chen SX, Dai QF, et al. Effect of acupuncture on the notch signaling pathway in rats with brain injury. Chin J Integr Med. 2018;24(7):537–544. doi:10.1007/s11655-015-1969-9
50. Huang YH, Cai K, Xu PP, et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-Notch-CCL2/CSF1 axis. Signal Transduct Target Ther. 2021;6:10. doi:10.1038/s41392-020-00437-8
51. De Nigris V, Prattichizzo F, Iijima H, Ceriello A. DPP-4 inhibitors have different effects on endothelial low-grade inflammation and on the M1-M2 macrophage polarization under hyperglycemic conditions. Diabetes Metab Syndr Obes. 2021;14:1519–1531. doi:10.2147/DMSO.S302621
52. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389:1931–1940. doi:10.1016/S0140-6736(17)31222-9
53. Kirkling ME, Cytlak U, Lau CM, et al. Notch signaling facilitates in vitro generation of cross-presenting classical dendritic cells. Cell Rep. 2018;23:3658–3672.e6. doi:10.1016/j.celrep.2018.05.068
54. Chen W, Liu Y, Chen J, et al. The Notch signaling pathway regulates macrophage polarization in liver diseases. Int Immunopharmacol. 2021;99:107938. doi:10.1016/j.intimp.2021.107938
55. Fan L, Xu C, Ge Q, et al. A. Muciniphila suppresses colorectal tumorigenesis by inducing TLR2/NLRP3-mediated M1-like TAMs. Cancer Immunol Res. 2021;9:1111–1124. doi:10.1158/2326-6066.CIR-20-1019
56. Du X, Xu Y, Chen S, Fang M. Inhibited CSF1R alleviates ischemia injury via inhibition of microglia M1 polarization and NLRP3 pathway. Neural Plast. 2020;2020:8825954. doi:10.1155/2020/8825954
57. Wu J, Ding DH, Li QQ, Wang XY, Sun YY, Li LJ. Lipoxin A4 regulates lipopolysaccharide-induced BV2 microglial activation and differentiation via the notch signaling pathway. Front Cell Neurosci. 2019;13:19. doi:10.3389/fncel.2019.00019
58. Li QQ, Ding DH, Wang XY, Sun YY, Wu J. Lipoxin A4 regulates microglial M1/M2 polarization after cerebral ischemia-reperfusion injury via the Notch signaling pathway. Exp Neurol. 2021;339:113645. doi:10.1016/j.expneurol.2021.113645
59. Liu X, Cong N, Cheng X, et al. The role of the notch signal pathway in mucosal cell metaplasia in mouse acute otitis media. Sci Rep. 2017;7:4588. doi:10.1038/s41598-017-04639-z
60. Xu X, Wu Y, Li H, Xie J, Cao D, Huang X. Notch pathway inhibitor Jagged1 accelerates in vitro proliferation and adipogenesis in infantile hemangioma stem cells. Oncol Lett. 2021;22:854. doi:10.3892/ol.2021.13115
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.