")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
Effect of Smoking and Its Cessation on the Transcript Profile of Peripheral Monocytes in COPD Patients
Authors Trivedi A, Bade G, Madan K, Bhat MA, Guleria R, Talwar A
Received 13 September 2021
Accepted for publication 13 December 2021
Published 5 January 2022 Volume 2022:17 Pages 65—77
DOI https://doi.org/10.2147/COPD.S337635
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Min Zhang
Anjali Trivedi, 1 Geetanjali Bade, 1 Karan Madan, 2 Muzaffer Ahmed Bhat, 1 Randeep Guleria, 2 Anjana Talwar 1
1Department of Physiology, All India Institute of Medical Sciences, New Delhi, India; 2Department of Pulmonary, Critical Care and Sleep Medicine, All India Institute of Medical Sciences, New Delhi, India
Correspondence: Anjana Talwar
Department of Physiology, All India Institute of Medical Sciences, Room No. 6003, 6th Floor, Convergence Block, New Delhi, India
Tel +9811505070
Email [email protected]
Rationale: Smoking is the primary cause of chronic obstructive pulmonary disease (COPD); however, only 10– 20% of smokers develop the disease suggesting possible genomic association in the causation of the disease. In the present study, we aimed to explore the whole genome transcriptomics of blood monocytes from COPD smokers (COPD-S), COPD Ex-smokers (COPD-ExS), Control smokers (CS), and Control Never-smokers (CNS) to understand the differential effects of smoking, COPD and that of smoking cessation.
Methods: Exploratory analyses in form of principal component analysis (PCA) and hierarchical component analysis (uHCA) were performed to evaluate the similarity in gene expression patterns, while differential expression analyses of different supervised groups of smokers and never smokers were performed to study the differential effect of smoking, COPD and smoking cessation. Differentially expressed genes among groups were subjected to post-hoc enrichment analysis. Candidate genes were subjected to external validation by quantitative RT-PCR experiments.
Results: CNS made a cluster completely segregated from the other three subgroups (CS, COPDS and COPD-ExS). About 550, 8 and 5 genes showed differential expression, respectively, between CNS and CS, between CS and COPD-S, and between COPD-S and COPD-ExS. Apoptosis, immune response, cell adhesion, and inflammation were the top process networks identified in enrichment analysis. Two candidate genes (CASP9 and TNFRSF1A) found to be integral to several pathways in enrichment analysis were validated in an external validation experiment.
Conclusion: Control never smokers had formed a cluster distinctively separated from all smokers (COPDS, COPD-ExS, and CS), while amongst all smokers, control smokers had aggregated in a separate cluster. Smoking cessation appeared beneficial if started at an early stage as many genes altered due to smoking started reverting towards the baseline, whereas only a few COPD-related genes showed reversal after smoking cessation.
Keywords: chronic obstructive pulmonary disease, apoptosis, immune response, cell adhesion, smoking
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is characterized by persistent respiratory symptoms and airflow limitation due to airway and/or alveolar abnormalities, usually caused by significant exposure to noxious particles and gases.1 COPD has become the third most common cause of death and was predicted to become the fifth most common cause of chronic disability globally by 2020.2 Tobacco smoking is one of the major risk factors for COPD, however, not all smokers develop COPD, putatively due to the differential display of expression of candidate genes in various target tissues in smokers.3,4 It is however generally believed that whole-genome expression studies, rather than candidate-based genomic expression data, may provide meaningful and discovery-level molecular understanding of any complex disease including COPD.5–7 Macrophages are the key players as these cells appear to play a pivotal role in the pathophysiology of COPD.8 These cells are activated by cigarette smoke to release chemokines and cytokines involved in many aspects of the disease processes in COPD including recruitment of the inflammatory cells.9 Monocytes are the precursors of macrophages and blood is a relatively more assessable sample for isolating monocytes.10,11 COPD being a systemic disease shows inflammatory changes in the peripheral circulation as well.12,13 Peripheral monocytes also carry a COPD-related gene expression pattern. An overlap of gene expression signatures of COPD patients between alveolar macrophages and monocytes have been reported.14 While the role of monocytes is evident in the pathogenesis of COPD,15,16 there is only one reported study by Poliska and co-workers 2011,14 in which global gene expression profiles in alveolar macrophages and circulating monocytes obtained from COPD patients, as well as, from control subjects were examined. However, pooled samples of RNA from ex-smoker as well as smoker COPD patients were employed in that study, thus making it difficult to attribute the observed changes in the gene expression specific to the pathology of COPD, vis-à-vis, smoking effect and the effect of cessation of smoking. To address this hiatus, global gene transcript profiles in the peripheral blood monocytes isolated from COPD smokers, COPD Ex-smokers, Control smokers, and Control never smokers were examined in the present study. It was assumed that the application of exploratory and differential analyses of whole-genome transcriptomics obtained from the above-mentioned four groups would reveal the effect of smoking, COPD and smoking cessation on the genomic expression in the peripheral monocytes in COPD patients. Figure 1 depicts the schema of the study design.
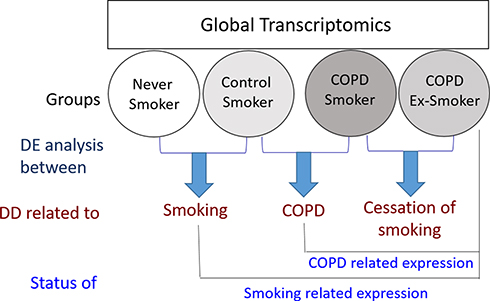 |
Figure 1 Global transcript profiling of the peripheral blood monocytes isolated from Control (Never Smokers and Control Smokers) and COPD (COPD Smokers and COPD Ex-Smokers) groups was performed. To identify the effect of smoking, differentially expressed genes between Never-smokers and Control Smokers were studied, while the effect of COPD was studied by performing differential display between control smokers and COPD smokers. To obtain information about the effect of smoking cessation on expression of genes associated with COPD, genes differentially expressed in COPD smokers and COPD Ex-smokers were studied. To study the status of COPD related expression, genes which were found to be differentially regulated in the control smokers and COPD smokers were compared with the Ex-smoker COPD group. The genes which were found to be differentially regulated in the Never-smokers and Control smokers were compared with that in the Ex-smoker COPD group to study the status of smoking related genes. It was assumed that the effects of target variables on the whole-genome transcript expression were liable to exhibit simple summative effect. Abbreviations: DD, differential display; DE, differentially expressed. |
Methods
The study was conducted at All India Institute of Medical Sciences, New Delhi. The ethical clearance for the proposed study was obtained from the Institute Ethics Committee (Ref. No. IESC/T-148/04.04.2014, OT-2/29.08.2016).
Patient Selection and Sample Collection
Clinically diagnosed COPD patients were recruited from the OPD of the Department of Pulmonary, Critical Care and Sleep Medicine in accordance with the norms laid down by the Institute Ethics Committee (IEC) for Post Graduate Research, All India Institute of Medical Sciences (AIIMS), New Delhi and the Helsinki Declaration amendment 2013. Group 1 comprised of male subjects aged between 30 and 70 years with stable, mild to severe COPD (stages I–III according to GOLD guidelines) and either smoker having a smoking history of at least 10 pack-years (group 1a, COPD-S; n = 4), or former smokers who had relinquished smoking for at least past one year (group 1b, COPD-ExS; n = 4). Group 2 comprised of control male subjects aged between 30 and 70 years with normal spirometry values and either non-smoker with no smoking history of <10 cigarettes in a lifetime (group 2a, CNS; n = 4) or smokers having a smoking history of at least 10 pack-years with no airflow obstruction or COPD (group 2b; CS; n = 4). All subjects voluntarily participated in the proposed study and provided duly signed informed consent forms prior to the commencement of the study. Subjects with any lung diseases apart from COPD, having a history of any chronic respiratory disorder or any active inflammatory disorder were excluded from the study. Subjects who had lung volume reduction surgery or lung transplantation were also excluded from the present study. Subjects who had diabetes and those with the intake of oral steroids past one month were not included in the present study. Figure 2 demonstrates the flow diagram of the experimental proceeding undertaken in the present study.
 |
Figure 2 Whole genome human transcriptomics of peripheral blood monocytes isolated from Never smokers, Control smokers, COPD smokers and COPD Ex-smokers was performed. COPD Patients (GOLD 1–3) were recruited from hospital OPD and controls from hospital staff following written consent. Anthropometric parameters were collected, spirometry was performed followed by blood sample collection. Peripheral blood mononuclear cells (PBMCs) were isolated by using Histopaque followed by isolation of monocytes using CD14 microbeads. Total RNA was extracted from CD14+ monocytes followed by quality check and quantification of RNA using Bioanalyzer and Nanodrop. Whole genome expression microarray was performed using Agilent one colour (Cy3) microarray protocol. Out of 41 samples, 25 samples did not pass the quality check and were not used further for microarray experiment. Samples (n = 16) with RIN score ≥8 were used further. Alongside exploratory analyses, viz. PCA and unsupervised HCA, differential display from the expression microarray data between different subgroups were done. Orthogonal validation of expression for two genes (TNFRSF1A and CASP9) was done using different samples (n = 5) by RT-PCR according to MIQE guidelines. |
Isolation of Peripheral Blood Monocytes
Peripheral blood samples were collected in EDTA vials (Cat# 455036, Greiner Bio-one GmbH, Kremsmunster, Austria) from subjects of both groups, which were processed immediately. Peripheral blood mononuclear cells (PBMCs) were separated by Histopaque (Cat# 79382, Sigma Aldrich Inc., St. Louis, Mo, USA) density gradient centrifugation using a standardized protocol.17 PBMC pellet was re-suspended in phosphate-buffered saline (PBS, pH 7.4), which was subjected to enrichment for monocytes by positive selection using CD14 microbeads (Cat# 130–050-201, MiltenyiBiotec GmbH, Friedrich, Bergisch Gladbach, Germany) in MACS separator (MiltenyiBiotec, Bergisch Gladbach, Germany) as previously described.18
RNA Extraction and Quality Check
Total RNA was extracted using a Direct-zol RNA MicroPrep kit (Cat# R2060, Zymo Research, Irvine, California) according to the manufacturer’s instructions. RNA was eluted in 15–20 µL of nuclease-free water, and purity was evaluated using Nanodrop 2000/2000c spectrophotometer (Thermo Fisher Scientific, MA, USA). Absorbance at 260 nm, 280 nm, and 230 nm was determined by the Nanodrop, and ratios of 260/280 and 260/230 were also noted to determine the purity of nucleic acid loaded. RIN scores of individual samples were assessed using Agilent 2100 Bioanalyzer, RNA 6000 Nano kit, and Agilent 2100 Expert software (Agilent Technologies, Santa Clara, CA, USA) employing optimized protocol as described elsewhere.19,20
Gene Expression Microarray and Array Analysis
Individual RNA samples isolated from peripheral blood monocytes having RIN score >8.0 and obtained from group 1a (n = 4) and 1b (n = 4) and group 2a (n = 4) and 2b (n = 4) were used for genome-wide microarray experiments on 4 × 44 K array glass chip format with 44,000 human oligo probes (Cat# 014850) using microarray system obtained from Agilent Technologies, Santa Clara, CA, USA. Sample preparation and hybridization of Poly-A RNA extracted from monocytes was done as per previously validated protocols.19,20 Gene expression was measured and the information from the probe features was extracted from microarray scan data. Scanned images of all the COPD patients and control subjects were used for data extraction using feature extraction software 10.7.3.1 (Agilent Technologies) and image processing was performed by using Agilent one-color (Cy3) microarray protocol. Hybridization signals were quantified after logarithmic transformation. After averaging, the signal spots that were lower than average background plus 2SD values were removed. Normalization was performed on log-transformed data using GeneSpringv11.5.1 (Agilent Technologies, Santa Clara, CA, USA). Thus, individual hybridization intensities are adjusted and balanced to make meaningful biological comparisons. The signal value of the 75th percentile of all the probes on the microarray was used to normalize the expression values,21 which were further used for exploratory analysis and differential expression analysis as described below.
Exploratory Analysis
In the present study, two kinds of exploratory analysis, namely Principal Component Analysis (PCA) and unsupervised Hierarchical Clustering Analysis (uHCA) were applied using GeneSpring software v.11.5.1 (Agilent Technologies, Santa Clara, CA, USA). PCA was applied to all expressed genes in the annotated samples to evaluate the similarity in gene expression patterns based on underlying variability and cluster structures and the clustered groups were displayed in a three-dimensional (3D) graph.22 uHCA was applied to unravel the patterns in the gene expression profiles from the data obtained in the present study. Pair-wise comparisons among samples were performed by using a standardized Pearson’s uncentered correlation vector with average linkage for measuring the distance.23 Heat maps and dendrograms were generated to visualize the data. The measured intensity values of genes were graphically represented by heat maps and the relationships between the samples were displayed by the dendrograms.
Differential Expression (DE) Analysis
Differential gene expression analysis was performed between the subgroups of COPD and controls to study the effect of smoking, COPD, and cessation of smoking. Normalized data were used for overall differential expression analysis between different sub-groups. Briefly, Venn analysis was done to obtain the genes commonly expressed in all the four subgroups: Control Never-smokers (CNS), Control Smokers (CS), COPD Ex-smokers (COPD-ExS,) and COPD Smokers (COPD-S). Similarly, Venn analysis was done to obtain the genes expressed in all smoking subgroups, which included COPD Ex-smokers, COPD Smokers, and Control Smokers. All statistical analyses and presentations were performed using GeneSpring software v.11.5.1 (Agilent Technologies, Santa Clara, CA, USA). List of genes showing statistically significant (p < 0.05) differences (>3-fold) in expression is given in Supplementary Table 1.
Post-Hoc Analysis
Post-hoc Enrichment Analysis was performed based on the gene list obtained from Venn analysis based on COPD and controls. Similarly, another list was obtained based on Venn analysis comparing gene expression obtained from Smoking versus Nonsmoking analysis. Metacore platform (GeneGo, St. Joseph, MI, USA) was used for the analysis and it was based on the a-priory setting of a cut-off threshold of pFDR(p)=0.05.
Orthogonal Validation
Since the quantity of residual RNA samples after microarray was not adequate for internal validation, two (2) candidate genes (CASP9 and TNFRSF1A) which were integral to several pathways in enrichment analysis were selected for orthogonal validation using different sets of five (5) samples from four subgroups and real-time RT-PCR analysis based on SYBR-Green chemistry and real-time PCR iCycler machine (BioRad CFX 96 Real-Time PCR system, BioRad Laboratories, Hercules, CA, USA). It was done as per MIQE guidelines and protocols as described elsewhere.20,24 Transcripts abundance of three housekeeping (GAPDH, ACTB, and B2M) normalization genes were also validated. Forward and reverse primers for two selected genes were designed using Beacon designer v12.1 (Premier Biosoft, California, USA) as provided in Supplementary Table 2 and were obtained from Integrated DNA Technologies (Skokie, Illinois, USA). The changes in gene expression were calculated from the threshold cycle (Ct) and then normalized with housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the transcripts of the other two genes (ACTB and B2M) yielded inconsistent profiles.25
Results
COPD patients had a mean age of 59.6 ± 8.3 years, while control subjects had a mean age of 51.4 ± 7.3 years. Demographic and spirometric details of the subjects in both groups are shown in Table 1. Statistical differences were analyzed among the groups using an unpaired t-test. P <0.0001 was obtained in COPD as compared to controls for weight, BMI, 6MWD, FEV1 (% predicted), FVC (% predicted), and FEV1/FVC (see Table 1). Supplementary Table 3 provides the summary of the patient profiles. Out of the total of 41 volunteers, RNA samples extracted from CD14+ monocytes isolated from blood cells of 16 (n = 8/each group) subjects were selected for microarray analysis since those samples could qualify pre-fixed requirements of quality (RIN score >8) and quantity (>500 ng) for undertaking whole-genome expression array of an individual sample. Expression microarray data are archived at NCBI GEO with accession number GSE146560. Supplementary Figure 1 shows Giemsa stained typical CD14+ monocytes isolated from blood cells.
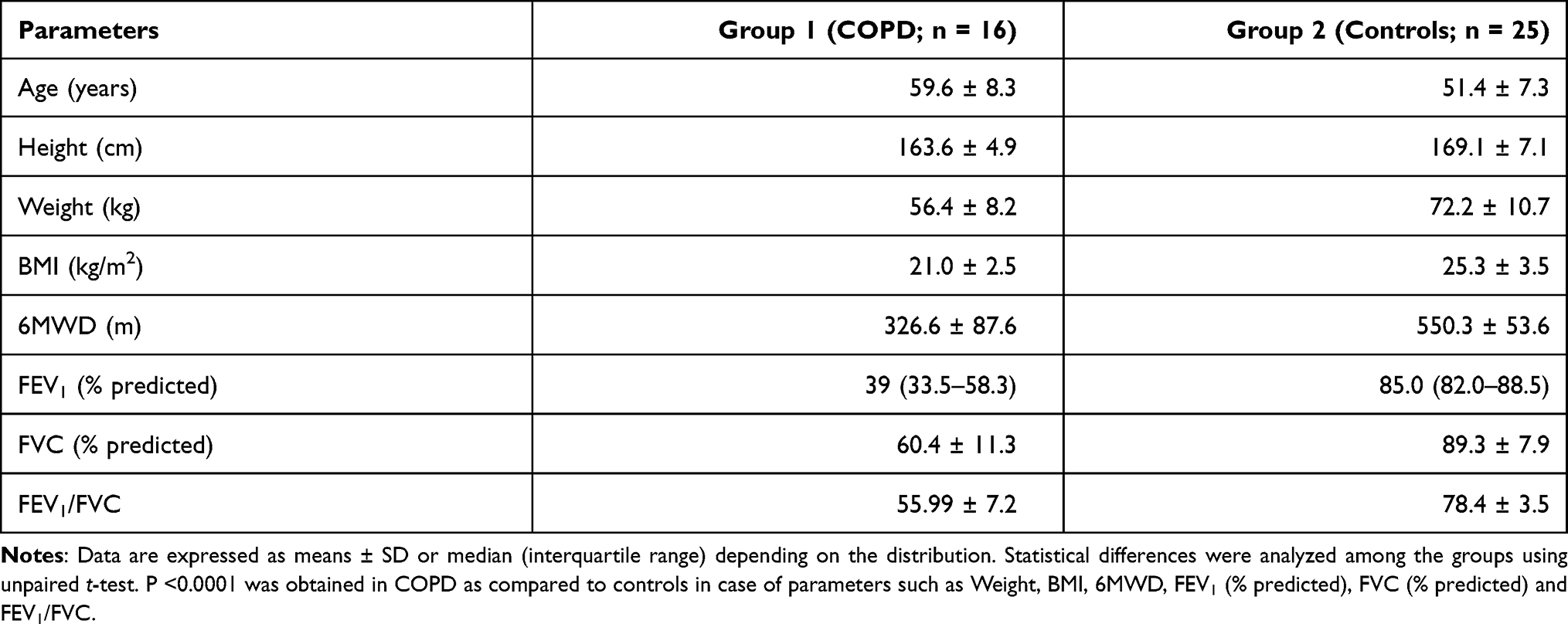 |
Table 1 Demographic and Spirometric Data of the Subjects in Two Groups |
Exploratory Analysis
Exploratory analysis using PCA (see Supplementary Figure 2 for details) and uHCA (see Supplementary Figure 3 for details) revealed three noteworthy points: (i) data of CNS (group 2a) made a cluster completely segregated from other groups (r = 0.09), (ii) data of CS (group 2b) clustered together (r = 0.68) but not much segregated from COPD-S (group 1a) samples (r = 0.85) and (iii) data from COPD-ExS (group 1b) did not show any significant clustering (r = 0.25).
Differential Expression (DE) Analysis
Differential expression (DE) analysis was performed between the subgroups of COPD and controls to analyze the effect of smoking, COPD, and cessation of smoking on the transcript profiles of CD14+ monocytes, the results of which are detailed in the following sections.
Effect of Smoking
To analyze the effect of smoking, differential expression analysis was performed between CNS (group 2a) and CS (group 2b). As seen in Figure 3A, a large number of genes (~11k) were detected in a mutually exclusive manner in these two subgroups of controls. Of 8170 genes found to be expressed in both groups, 550 genes showed differential expression (>3-fold at p < 0.05) between CNS and CS (Figure 3B). The expression of genes, eg, TUBB2B, MLLT4, and PCDHB10 showed greater than 90-fold down-regulation in Control smokers as compared to Control Never smokers.
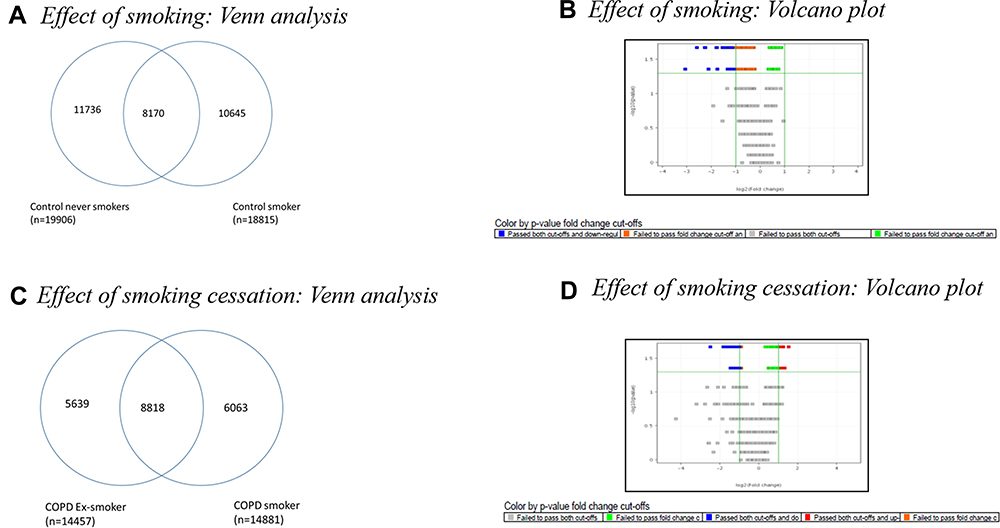 |
Figure 3 (A) Venn diagram showing commonly regulated genes between the subgroups of Control: Control Never Smokers (CNS) and Control Smokers (CS). Out of 19,906 genes expressed in CNS and 18,815 genes expressed in CS, 8170 common genes were expressed in the two subgroups of controls, while 11,736 genes were expressed only in Control Never smokers and 10645 genes were expressed only in Control smokers. (B) Volcano plot of Control Smoker Vs Control Never Smoker analysed using Mann–Whitney U-test at >2-fold, p value<0.05. (C) Venn diagram showing common genes expressed in the subgroups of COPD: COPD Ex-Smokers and COPD Smokers. Out of 14,457 genes expressed in COPD Ex-Smokers and 14,881 genes expressed in COPD S, 8818 genes were expressed in both subgroups of COPD, while 5639 genes were expressed only in COPD Ex-Smokers and 6063 genes were expressed only in COPD Smokers. (D)Volcano plot of COPD Smoker Vs COPD Ex-Smoker analysed using Mann Whitney U-test at>2-fold; p value<0.05. |
Effect of Smoking Cessation
Differential expression analysis was performed between COPDS (group 1a) and COPD-ExS (group 1b) to study the effect of smoking cessation. As seen in Figure 3C, a large number of genes (~6k) were detected in a mutually exclusive manner in the subgroups of COPD. Of 8818 genes found to be expressed in both groups, 5 genes showed differential expression (>3-fold at p < 0.05) between COPDS and COPD-ExS (Figure 3D). Upon cessation of cigarette smoking, genes such as MLLT4, MAGI2-AS3, PHLPP1 showed a reversal in gene expression in COPD Ex-smokers. These genes showed greater than 100-fold down-regulation in Control smokers when compared with Control Never smokers. The expression of genes related to forming the major component of microtubules and cell adhesion molecules namely TUBB2B, MLLT4, and PCDHB10 started reverting showing down-regulation of greater than 35–40 fold in COPD Ex-smokers as compared to Control Never smokers upon cessation of smoking.
TRIM60 gene showed up-regulation of 3.6-folds whereas ABHD12 gene showed about 3-fold down-regulation in COPD Ex-smokers as compared to COPD smokers. The expression of genes encoding for mitochondrial proteins involved in oxidative phosphorylation, eg, GFM1 and MRPL43 showed greater than 5-folds up-regulation whereas PEX6 gene showed downregulation of 3-folds in COPD Ex-smokers as compared to COPD smokers. FERMT3 gene was found to be up-regulated by more than 2-fold in COPD smokers when compared with control smokers and it started showing downregulation by more than 2-fold in COPD Ex-smokers upon cessation of cigarette smoking. GABRG2 gene showed up-regulation by 4-fold in COPD smokers as compared to control smokers. However, upon cessation of cigarette smoking, this gene showed up-regulation by 2-fold in COPD Ex-smokers.
Effect of COPD
Differential expression analysis was performed between CS (group 2b) and COPDS (group 1a) to study the effect of COPD. As seen in Figure 4A, a large number of genes (~9k) were detected in a mutually exclusive manner in the subgroups of smokers. Of 8943 genes found to be expressed in both groups, 8 genes showed differential expression (>3-fold at p < 0.05) between CS and COPDS (Figure 4B). Oncogenes such as ARM6 and PLOD3 were found to be up-regulated by more than 6-fold and 3-fold, respectively in COPD smokers as compared to control smokers and the TMEM30B gene which plays role in lipid metabolism was also found to be up-regulated in COPD smokers.
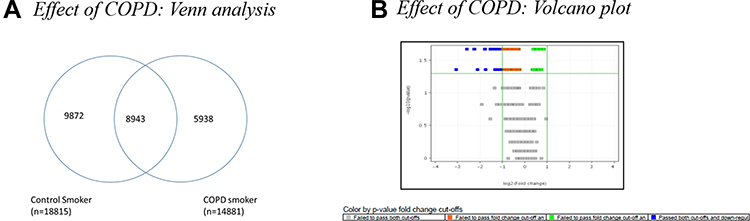 |
Figure 4 (A) Venn diagram showing common genes expressed in the subgroups of Control and COPD: Control Smokers (CS) and COPD Smokers (COPD (S). Out of 18,815 genes expressed in CS and 14,881 genes expressed in COPD S, 8943 genes were expressed in both subgroups of controls and COPD: CS and COPD S, while 9872 genes were expressed only in Control smokers and 5938 genes were expressed only in COPD smokers. (B) Volcano plot of Control Smoker Vs COPD Smoker analysed using Mann Whitney U-test at>2-fold; p value<0.05. |
Post-Hoc Enrichment Analysis
Differentially expressed genes among groups were subjected to enrichment analysis based on a curated database to obtain processes and pathways putatively associated with COPD and smoking. Flowchart of steps adopted for post hoc analysis among the groups is shown in Figure 5.
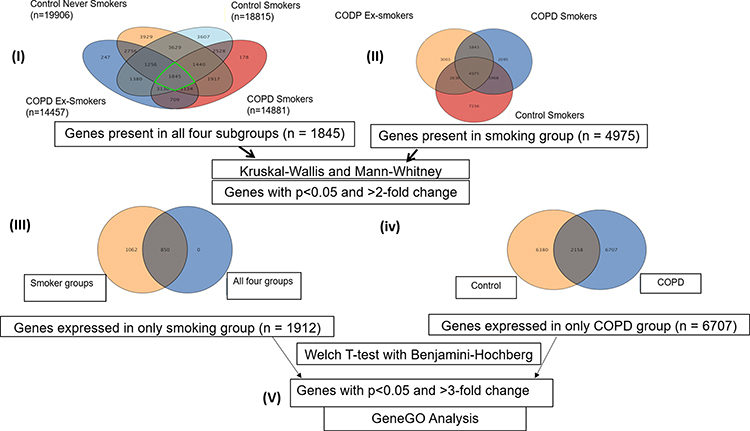 |
Figure 5 Work flow of analysis. Number in parenthesis shows the number of (I) genes expressed in that particular subgroups: CNS (n=19,906), CS (n=18,815), COPD Ex-S (n=14,457) and COPD S (n=14,481), (II) genes (n =1845) expressed in all 4 subgroups, (III) n=1912 genes were found to be commonly expressed in smoking group (p<0.05 and FC>2). (IV) n= 6707 genes were found to be uniquely expressed in the COPD group when compared to controls. (V) Genes uniquely expressed in smoking group and COPD group were subjected to GeneGOMetacore analysis. |
As seen in Figure 6, the top ten (10) process networks were obtained when the genes which were expressed only in the smoking group and COPD group, were subjected to enrichment analysis. Apoptosis (p = 0.0005), Immune response (p = 0.0011), Cell adhesion (p = 0.0017) and Inflammation (p = 0.0019) processes were selected from the obtained pathways based on information curated from database. Multiple genes were involved in each of these processes. Out of those, genes that were involved in more than one of these processes were selected and expressions of such two genes (CASP9 and TNFRSF1A) were subjected to real-time qPCR validation using different sets of samples from all four sub-groups. As seen in Figure 7, the differential expression profiles of these genes were comparable between two sets of samples in microarray and real-time qPCR experiments, respectively.
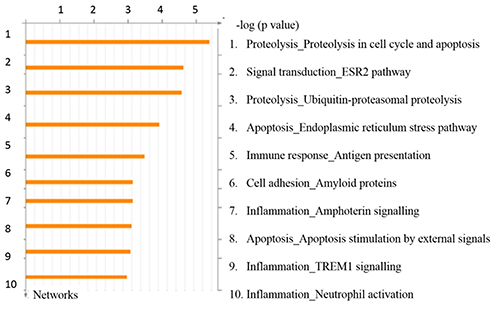 |
Figure 6 Top scored process networks enriched by GeneGO analysis. The details are provided in Methods section. |
 |
Figure 7 Fold changes in microarray and real time RT-PCR experiment. Microarray data (shown in blue) were validated (p< 0.01) by estimating transcript copy numbers by performing real time RT-PCR (shown in Orange) of two (2) candidate genes selected on the basis of post-hoc enrichment analysis using different sets of five (5) samples from four subgroups (COPD-S, COPD-ExS, CNS and CS) as explained in the Methods section. |
Discussion
COPD is a major and growing cause of morbidity and mortality across the world.2 Smoking is recognized as the most important causative factor of COPD, however, only 10–20% of smokers develop the disease.26 The available therapeutic interventions and our understanding of COPD are still very limited. In the present study, we examined the differential genomic expression in circulatory CD14+ monocytes obtained from the patients of subgroups of COPD and controls to study the effect of smoking (Control Never-Smokers, CNS, ie, group 2a versus Control Smokers, CS, ie, group 2b), the effect of COPD (Control Smokers, CS, ie, group 2b versus COPD Smokers, COPD-S, ie, group 1a) and the effect of smoking cessation (COPD Smokers, COPD-S, ie, group 1a versus COPD Ex-smokers, COPD-ExS, ie, group 1b). To the best of our knowledge, this is the first study to explore the whole genome transcriptomics of blood monocytes from COPD smokers, COPD Ex-smokers, Control smokers, and Control Never-smokers.
Exploratory analyses in form of Principal Component Analysis (PCA) and unsupervised Hierarchical Component Analysis (uHCA) of whole-genome expression results revealed that Control never smokers had formed a cluster distinctively separated from all smokers (COPD smoker, COPD ex-smoker, and control smoker), while amongst all smokers, control smokers had aggregated in a separate cluster. This gave us the lead to perform differential expression analysis of different supervised groups of smokers and never smokers, so that the putative genes associated with smoking and that associated with COPD and cessation of smoking, respectively may be identified as explained in Figure 1.
As summarized in Tables 2,27–45 differential display between COPD Smokers and Control Smokers identified different categories of genes which include oncogenes, mucin overproduction genes, inflammatory genes, and those involved in acid-base disorders. In this study, differential expression analysis between COPD Smokers and Control Smokers revealed that oncogenes such as ARM6 and PLOD3 which played role in promoting the metastasis via STAT3 pathways were found to be up-regulated by more than 6-fold and 3-fold, respectively in COPD Smokers as compared to Control Smokers in the present study. Such differential regulation may have a role in modulating the transcription of several genes involved in the regulation of apoptosis, angiogenesis, metastasis, immune responses, cell differentiation, and proliferation thus contributing to the pathophysiology of COPD.46 Smoking appears to cause dysfunctional lipid metabolism within the lung macrophages that may contribute to inflammation.47 TMEM30B gene was found to be up-regulated in COPD Smokers. Thus, up regulation of this gene in COPD smokers suggests its role in augmented inflammatory responses in COPD which may be caused due to the abnormalities of lipid metabolism.48
 |
Table 2 Categorical List of Genes Showing Differential Expression in the Monocytes Isolated from COPD (COPD Smokers and COPD Ex-Smokers) and Control (Control Smokers and Control Never Smokers) Subjects |
The genes, expressions of which are perturbed by smoking play a major role in COPD development. Though smoking cessation is the only treatment towards an effective slowing down of the progression of COPD as seen by the reduced decline in FEV1, our knowledge regarding the effects of cessation of smoking on genomic expression is rather thin. Tumor suppressor genes such as MLLT4, MAGI2-AS3, PHLPP1 which showed greater than 100-fold downregulation in control smokers when compared with Control never smokers, showed a reversal in gene expression after smoking cessation in COPD Ex-smokers. This observation corroborates well with previous reports.49,50 Besides this, other processes and pathways associated with smoking and its cessation included cellular signaling, triglyceride metabolism, and receptor-mediated endocytosis. Cigarette smoking putatively affected cell adhesion and components of microtubules as genes involved in these processes namely TUBB2B, MLLT4 and PCDHB10 showed greater than 90-fold down-regulation in Control smokers as compared to Control Never smokers.
Improved prognosis is reported if smoking cessation is started at an early stage of the disease and there is evidence that it is more effective if done at an early stage of COPD than in the later stages.51–53 However, Turato et al (1995) reported that the ongoing inflammation in airway mucosa of ex-smokers who had cough and sputum production even after 13-years of smoking cessation.54 Both the current and Ex-smokers tend to show an increased number of macrophages in the bronchial biopsies.54 Airway inflammation is not resolved in COPD patients even after 1-year of smoking abstinence and even found to be increased in some aspects. In the present study, the beneficial effects of smoking cessation were seen by the change in expression of genes involved in mediating protein–protein interactions (TRIM60 gene) and inflammation (ABHD12 gene). In the present study, the TRIM60 gene showed up-regulation of 3.6-folds in COPD Ex-smokers as compared to COPD smokers55 and ABHD12 gene showed about 3-fold down-regulation in COPD Ex-smokers as compared to COPD smokers. Thus, our results suggested that the TRIM60 gene might help in improving protein–protein interactions after smoking cessation in COPD Ex-smokers. As the ABHD12 gene mediates the hydrolysis of 2-arachidonoyl glycerol (2-AG), which is the main endocannabinoid lipid-transmitter; this cannabinoid system is known to be involved in inflammation.56,57 Thus, downregulation of the ABHD12 gene might be responsible for the decreased inflammatory process in COPD Ex-smokers, thus indicating the beneficial effects of smoking cessation.
Interestingly, genes differentially expressed in COPD smokers and COPD Ex-smokers also included genes that are known to be involved in maintaining the structure and function of mitochondria. In the present study, GFM1 and MRPL43 showed greater than 5-folds up-regulation in COPD Ex-smokers as compared to COPD smokers. These genes encode mitochondrial proteins involved in oxidative phosphorylation.40,41 Hence, our results suggest that after cessation of cigarette smoking, oxidative phosphorylation might be improved in COPD Ex-smokers. Higher levels of mtROS and pro-inflammatory cytokines were seen in COPD smokers as compared to healthy smokers.42 The exact role of smoking cessation on the airway inflammation process in COPD is still not known.58 In general, the inflammatory and structural changes in the airways increase with disease severity and persist despite smoking cessation.59 PEX6 gene mediates peroxisomal protein import and is required for PTS1 receptor activity.60 Thus, it plays a role in peroxisome assembly, where degradation of fatty acids takes place thereby contributing to the reduction of ROS.61 As the PEX6 gene showed downregulation of 3-folds in COPD Ex-smokers as compared to COPD smokers, thus our results indicated the possibility that despite smoking cessation, peroxisomal assembly was decreased in COPD Ex-smokers, thereby increasing the load of ROS and leading to various damages caused due to ROS.
It is indeed intriguing to note from the results of the present study that, upon cessation of cigarette smoking, Ex-smokers showed reversibility of a large number of genes that were found to be associated with smoking. However, only two COPD-related genes (FERMT3 and GABGR2) showed reversibility in COPD Ex-smokers when compared with COPD smokers. This implicates that smoking cessation is beneficial if started at an early age and that once COPD is developed, the reversal of genes is declined. Increased expression of the FERMT3 gene has been associated with primary tumor growth, angiogenesis, and metastasis.29 In our study, the FERMT3 gene was found to be up-regulated by more than 2-fold in COPD smokers when compared with control smokers, started reverting to baseline values in COPD Ex-smokers upon cessation of cigarette smoking. GABRG2 gene, on the other hand, has been associated with mucin overproduction along with differential regulation in COPD Ex-smokers.33 GABRG2 gene showed up-regulation by 4-fold in COPD smokers as compared to control smokers. However, upon cessation of cigarette smoking, this gene showed up-regulation by 2-fold in COPD Ex-smokers in the present study. Hence, our results suggest that with smoking cessation, this gene started reverting to baseline values in COPD Ex-smokers, thus reducing the mucin overproduction in COPD Ex-smokers. Our study thus collectively supports the hypothesis that smoking cessation is beneficial if started at an early stage.
Apoptosis, immune response, cell adhesion, and inflammation were among the top-scored enriched process pathways revealed in the present study (Figure 6). Apoptosis plays an important role in amplifying the inflammatory processes in COPD.62 Apoptosis of interstitial cells, alveolar epithelial cells, inflammatory cells, and endothelial cells in lung tissue samples is associated with COPD.63–65 Mathanraj et al (2017) reported elevated levels of serum TNFA levels associated with increased severity of COPD.66 Production of TNFA from peripheral blood monocytes is also increased in COPD patients and has been implicated in skeletal muscle apoptosis found in some COPD patients.67 Interactions of TNFA and TNFR1 leads to clustering of TRADD (TNF (Tumor Necrosis Factor) R1 (Receptor Type 1)-Associated Death Domain) results in activation of caspases leading to “Caspase Cascade” and apoptosis.68,69 It is notable that the differential display in the expression of two (2) candidate genes, CASP9 and TNFRSF1A that were integral to these pathways were externally validated by quantitative RT-PCR experiments in the present study.
It appears from the present study that the expression of genes perturbed by smoking included majorly oncogenes and tumor suppressor genes. Further, genes related to mitochondrial structure and function as well as a transcriptional regulator were also perturbed due to cigarette smoking. The expression of genes related to cell adhesion and ion transport, triglyceride metabolism, and those related to receptor-mediated endocytosis were also altered by smoking. Apoptosis, immune response, cell adhesion, and inflammation processes were identified as major pathophysiological process modules for COPD in the bioinformatics analysis of the results of the present study. In the present study, however, the group of Control Ex-smokers subjects was not included. This would have provided additional information about the expression of the genes showing a return to baseline after smoking cessation when compared with COPD Ex-smokers.
Conclusions
In conclusion, the profile of genomic expression revealed that control never smokers had formed a cluster distinctively separate from all the smokers, while all the smokers form a cluster irrespective of disease status. Smoking cessation is beneficial, if started at an early stage as many genes altered due to smoking started reverting towards baseline whereas only two COPD-related genes showed reversal after smoking cessation. Two (2) candidate genes, namely, TNFRSF1A and CASP9, that were validated by quantitative RTPCR experiments were integral to four critical pathways: Apoptosis, Immune response, Cell adhesion, and Inflammation. Functional validation by animal models is valuable to measure the downstream effects, for further investigation of mechanisms involved in COPD, and for the translation of the findings from bench to bedside. Further studies are required to substantiate the observed changes in gene expression in different groups and subgroups of COPD patients and controls and the physiological significance of the same.
Acknowledgment
We would like to acknowledge Professor Dr. Ghosh (Department of Physiology, All India Institute of Medical Sciences, New Delhi) for his immense support and contribution in bringing the concept and overall study design, data analysis, editing, proofreading, and finalizing the manuscript. The authors also wish to thank the patients and personnel of the hospital unit for their cooperation during this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study was funded by the Department of Science and Technology (DST), India.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Roversi S, Corbetta L, Clini E. GOLD 2017 recommendations for COPD patients: toward a more personalized approach. COPD Res Pract. 2017;3(1):5. doi:10.1186/s40749-017-0024-y
2. Quaderi SA, Hurst JR. The unmet global burden of COPD. Glob Health Epidemiol Genom. 2018;3:4. doi:10.1017/gheg.2018.1
3. Siafakas NM, Tzortzaki EG. Few smokers develop COPD. Why? Respir Med. 2002;96(8):615–624. doi:10.1053/rmed.2002.1318
4. Matsumura K, Shigeaki I. Novel biomarker genes which distinguish between smokers and chronic obstructive pulmonary disease patients with machine learning approach. BMC Pulm Med. 2020;20:29. doi:10.1186/s12890-020-1062-9
5. Kim V, Criner GJ. Chronic Bronchitis and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2013;187(3):228–237. doi:10.1164/rccm.201210-1843CI
6. Morrow JD, Qiu W, Chhabra D, Rennard SI, Belloni P, Belousov A. Identifying a gene expression signature of frequent COPD exacerbations in peripheral blood using network methods. BMC Med Genomics. 2015;8(1). doi:10.1186/s12920-014-0072-y
7. Ham S, Oh YM, Roh TY. Evaluation and interpretation of transcriptome data underlying heterogeneous chronic obstructive pulmonary disease. Genomics Inform. 2019;17(1):e2. doi:10.5808/GI.2019.17.1.e2
8. Rovina N, Koutsoukou A, Koulouris NG. Inflammation and Immune Response in COPD: where Do We Stand? Mediators Inflamm. 2013;2:413735.
9. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. doi:10.1038/nri2254
10. Varol C, Yona S, Jung S. Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol. 2009;87(1):30–38. doi:10.1038/icb.2008.90
11. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi:10.1126/science.1178331
12. Gan WQ, Man SFP, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59(7):574–580. doi:10.1136/thx.2003.019588
13. Karadag F, Karul AB, Cildag O, Yilmaz M, Ozcan H. Biomarkers of systemic inflammation in stable and exacerbation phases of COPD. Lung. 2008;186(6):403–409. doi:10.1007/s00408-008-9106-6
14. Poliska S, Csanky E, Szanto A, et al. Chronic Obstructive Pulmonary Disease-Specific Gene Expression Signatures of Alveolar Macrophages as well as Peripheral Blood Monocytes Overlap and Correlate with Lung Function. Respiration. 2011;81(6):499–510. doi:10.1159/000324297
15. Aldonyte R, Jansson L, Piitulainen E, Janciauskiene S. Circulating monocytes from healthy individuals and COPD patients. Respir Res. 2003;4:11. doi:10.1186/1465-9921-4-11
16. Cornwell WD, Kim V, Fan X, et al. Activation and polarization of circulating monocytes in severe chronic obstructive pulmonary disease. BMC Pulm Med. 2018;18(1):101. doi:10.1186/s12890-018-0664-y
17. Panda SK, Ravindran B. Isolation of Human PBMCs. Bio-Protocol. 2013;3(3):e323. doi:10.21769/BioProtoc.323
18. Trivedi A, Ghosh D, Guleria R, Mehta N, Talwar A. CD14 positive selection displays an edge in the isolation of macrophages from induced sputum of COPD patients using immunobead technology. Indian J Pharmacol. 2018;62(1):32–40.
19. Khan MA, Sengupta J, Mittal S, et al. Genome-wide expressions in autologous eutopic and ectopic endometrium of fertile women with endometriosis. Reprod Biol Endocrinol. 2012;10(1):84. doi:10.1186/1477-7827-10-84
20. Bhat MA, Sharma JB, Roy KK, et al. Genomic evidence of Y chromosome microchimerism in the endometrium during endometriosis and in cases of infertility. Reprod Biol Endocrinol. 2019;17(1):22. doi:10.1186/s12958-019-0465-z
21. Casares FM, Simple A. Method for Optimization of Reference Gene Identification and Normalization in DNA Microarray Analysis. Med Sci Monit Basic Res. 2016;22:45–52. doi:10.12659/MSMBR.897644
22. Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17(9):763–774. doi:10.1093/bioinformatics/17.9.763
23. Gil-Garcia RJ, Badia-Contelles JM. A General Framework for Agglomerative Hierarchical Clustering Algorithms A Pons-Porrata Pattern Recognition, 2006. ICPR. 2006;1:569–572.
24. Bustin SA, Benes V, Garson JA, et al. The MIQE Guidelines: minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin Chem. 2009;55(4):611–622. doi:10.1373/clinchem.2008.112797
25. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acid Res. 2002;30:e36. doi:10.1093/nar/30.9.e36
26. Terzikhan N, Verhamme KMC, Hofman A, et al. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur J Epidemiol. 2016;31(8):785–792. doi:10.1007/s10654-016-0132-z
27. Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA. 2004;101(27):10143–10148. doi:10.1073/pnas.0401422101
28. Bhattacharya S, Tyagi S, Srisuma S, DeMeo DL, Shapiro SD, Bueno R. Peripheral blood gene expression profiles in COPD subjects. J Clin Bioinform. 2011;1(1):12. doi:10.1186/2043-9113-1-12
29. Sossey-Alaoui K, Pluskota E, Davuluri G, et al. Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis. FASEB J. 2014;28(5):2260–2271. doi:10.1096/fj.13-244004
30. Hackett NR, Heguy A, Harvey B-G, O’Connor TP, Luettich K, Flieder DB. Variability of Antioxidant-Related Gene Expression in the Airway Epithelium of Cigarette Smokers. Am J Respir Cell Mol Biol. 2003;29(3):331–343.
31. Singh D, Fox SM, Tal-Singer R, Plumb J, Bates S, Broad P. Induced sputum genes associated with spirometric and radiological disease severity in COPD ex-smokers. Thorax. 2011;66(6):489–495. doi:10.1136/thx.2010.153767
32. Pierrou S, Broberg P, O’Donnell RA, Pawłowski K, Virtala R, Lindqvist E. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(6):577–586. doi:10.1164/rccm.200607-931OC
33. Fu XW, Wood K, Spindel ER. Prenatal Nicotine Exposure Increases GABA Signaling and Mucin Expression in Airway Epithelium. Am J Respir Cell Mol Biol. 2011;44(2):222–229. doi:10.1165/rcmb.2010-0109OC
34. Golpon HA, Coldren CD, Zamora MR, Cosgrove GP, Moore MD, Tuder RM.Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol. 2004;31(6):595–600. doi:10.1165/rcmb.2004-0008OC
35. Almansa R, Socias L, Garcia M, et al. Critical COPD respiratory illness is linked to increased transcriptomic activity of neutrophil proteases genes. BMC Res Notes. 2012;5:401. doi:10.1186/1756-0500-5-401
36. Wang M, Stepaniants S, Boie Y, Mortimer JR, Kennedy B, Elliott M. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med. 2008;177(4):402–411. doi:10.1164/rccm.200703-390OC
37. Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. 2004;101(41):14895–14900. doi:10.1073/pnas.0401168101
38. Bahr TM, Hughes GJ, Armstrong M, Reisdorph R, Coldren CD, Edwards MG. Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol. 2013;49(2):316–323. doi:10.1165/rcmb.2012-0230OC
39. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. doi:10.1378/chest.12-2664
40. Coenen MJH, Antonicka H, Ugalde C, et al. Mutant Mitochondrial Elongation Factor G1 and Combined Oxidative Phosphorylation Deficiency. New Engl J Med. 2004;351(20):2080–2086. doi:10.1056/NEJMoa041878
41. Kenmochi N, Suzuki T, Uechi T, et al. The Human Mitochondrial Ribosomal Protein Genes: mapping of 54 Genes to the Chromosomes and Implications for Human Disorders. Genomics. 2001;77(1–2):65–70. doi:10.1006/geno.2001.6622
42. Wiegman CH, Michaeloudes C, Haji G, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769–780. doi:10.1016/j.jaci.2015.01.046
43. Agustí AGN, Sauleda J, Miralles C, et al. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):485–489. doi:10.1164/rccm.2108013
44. Picard M, Godin R, Sinnreich M, et al. The mitochondrial phenotype of peripheral muscle in chronic obstructive pulmonary disease: disuse or dysfunction? Am J Respir Crit Care Med. 2008;178(10):1040–1047.
45. Guo Y, Gosker HR, Schols AMWJ, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(11):1313–1320. doi:10.1164/rccm.201304-0732OC
46. Johnston PA, Grandis JR. STAT3 SIGNALING: anticancer Strategies and Challenges. Mol Interv. 2011;11(1):18–26. doi:10.1124/mi.11.1.4
47. Katoh Y, Katoh M. Identification and characterization of CDC50A, CDC50B and CDC50C genes in silico. Oncol Rep. 2004;12(4):939–943.
48. Higham A, Lea S, Simpson K, et al. Lipids in the lung: respiratory inflammation in COPD. Eur Respir J. 2012;40(56):385.
49. Chen M, Pratt CP, Zeeman ME, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20(2):173–186. doi:10.1016/j.ccr.2011.07.013
50. Lai Y, Xu P, Liu J, et al. Decreased expression of the long non-coding RNA MLLT4 antisense RNA 1 is a potential biomarker and an indicator of a poor prognosis for gastric cancer. Oncol Lett. 2017;14(3):2629–2634. doi:10.3892/ol.2017.6478
51. Xu X, Dockery DW, Ware JH, et al. Effects of cigarette smoking on rate of loss of pulmonary function in adults: a longitudinal assessment. Am Rev Respir Dis. 1992;146(51):1345–1348. doi:10.1164/ajrccm/146.5_Pt_1.1345
52. Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. JAMA. 1994;272(19):1497–1505. doi:10.1001/jama.1994.03520190043033
53. Wagena EJ, van der Meer RM, Ostelo RJWG, Jacobs JE, van Schayck CP. The efficacy of smoking cessation strategies in people with chronic obstructive pulmonary disease: results from a systematic review. Respir Med. 2004;98(9):805–815. doi:10.1016/j.rmed.2004.06.001
54. Turato G, Di Stefano A, Maestrelli P, et al. Effect of smoking cessation on airway inflammation in chronic bronchitis. Am J Respir Crit Care Med. 1995;152(41):1262–1267. doi:10.1164/ajrccm.152.4.7551380
55. Jiang M-X, Hong X, Liao -B-B, et al. Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci Rep. 2017;7(1). doi:10.1038/srep42781
56. Fiskerstrand T, H’mida-Ben Brahim D, Johansson S, et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: an Inborn Error of Endocannabinoid Metabolism. Am J Hum Genet. 2010;87(3):410–417. doi:10.1016/j.ajhg.2010.08.002
57. Barrie N, Manolios N. The endocannabinoid system in pain and inflammation: its relevance to rheumatic disease. Eur J Rheumatol. 2017;4(3):210–218. doi:10.5152/eurjrheum.2017.17025
58. Willemse BWM. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26(5):835–845. doi:10.1183/09031936.05.00108904
59. Rutgers SR. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax. 2000;55(1):12–18. doi:10.1136/thorax.55.1.12
60. Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi:10.1146/annurev.biochem.74.082803.133329
61. Steinberg SJ, Dodt G, Raymond GV, et al. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763(12):1733–1748. doi:10.1016/j.bbamcr.2006.09.010
62. Schmidt EP, Tuder RM. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J Cell Death. 2010;3:
63. Kasahara Y, Tuder RM, Cool CD, et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–744. doi:10.1164/ajrccm.163.3.2002117
64. Plataki M, Tzortzaki E, Rytila P, et al. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):161–171. doi:10.2147/copd.2006.1.2.161
65. Song Q, Chen P, Liu XM. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir Res. 2021;22:39. doi:10.1186/s12931-021-01630-1
66. Mathanraj S, Vysak K, Yuvarajan S, Vikram R. Correlation of serum TNF alpha level with severity of chronic obstructive pulmonary disease. Int J Res Med Sci. 2017;5(8):3309–3316. doi:10.18203/2320-6012.ijrms20173020
67. Montes MM, Torres SH, De SJ, Mata A, Hernández N, Tálamo C. Skeletal muscle inflammation and nitric oxide in patients with COPD. Eur Respir J. 2005;26(3):390–397. doi:10.1183/09031936.05.00107404
68. Pobezinskaya YL, Liu Z. The role of TRADD in death receptor signaling. Cell Cycle. 2012;11(871):10. doi:10.4161/cc.11.5.19300
69. Yu X, James S, Felce JH. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun Biol. 2021;4:772. doi:10.1038/s42003-021-02309-5
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.