")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Effect of honokiol on the induction of drug-metabolizing enzymes in human hepatocytes
Authors Cho Y, Jeong H, Kim J, Lee HS
Received 5 August 2014
Accepted for publication 29 August 2014
Published 3 November 2014 Volume 2014:8 Pages 2137—2145
DOI https://doi.org/10.2147/DDDT.S72305
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Yong-Yeon Cho,1 Hyeon-Uk Jeong,1 Jeong-Han Kim,2 Hye Suk Lee1
1College of Pharmacy, The Catholic University of Korea, Bucheon, Korea; 2Department of Agricultural Biotechnology, Seoul National University, Seoul, Korea
Abstract: Honokiol, 2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol, an active component of Magnolia officinalis and Magnolia grandiflora, exerts various pharmacological activities such as antitumorigenic, antioxidative, anti-inflammatory, neurotrophic, and antithrombotic effects. To investigate whether honokiol acts as a perpetrator in drug interactions, messenger ribonucleic acid (mRNA) levels of phase I and II drug-metabolizing enzymes, including cytochrome P450 (CYP), UDP-glucuronosyltransferase (UGT), and sulfotransferase 2A1 (SULT2A1), were analyzed by real-time reverse transcription polymerase chain reaction following 48-hour honokiol exposure in three independent cryopreserved human hepatocyte cultures. Honokiol treatment at the highest concentration tested (50 µM) increased the CYP2B6 mRNA level and CYP2B6-catalyzed bupropion hydroxylase activity more than two-fold in three different hepatocyte cultures, indicating that honokiol induces CYP2B6 at higher concentrations. However, honokiol treatment (0.5–50 µM) did not significantly alter the mRNA levels of phase I enzymes (CYP1A2, CYP3A4, CYP2C8, CYP2C9, and CYP2C19) or phase II enzymes (UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1) in cryopreserved human hepatocyte cultures. CYP1A2-catalyzed phenacetin O-deethylase and CYP3A4-catalyzed midazolam 1'-hydroxylase activities were not affected by 48-hour honokiol treatment in cryopreserved human hepatocytes. These results indicate that honokiol is a weak CYP2B6 inducer and is unlikely to increase the metabolism of concomitant CYP2B6 substrates and cause pharmacokinetic-based drug interactions in humans.
Keywords: honokiol, human hepatocytes, drug interactions, cytochrome P450, UDP-glucuronosyltransferases
Introduction
Herb–drug interactions resulting from concurrent use of botanical drugs with prescription and over-the-counter drugs may cause adverse reactions such as toxicity and treatment failure. The mechanisms underlying herb–drug interactions involve inhibition or induction of drug-metabolizing enzymes such as cytochrome P450 (CYP), UDP-glucuronosyltransferase (UGT), and sulfotransferase (SULT) enzymes, and drug transporters.1–7 Herbal drugs such as ginkgo (Ginkgo biloba), ginseng (Panax ginseng), garlic, danshen (Salvia miltiorrhiza), licorice (Glycyrrhiza glabra), resveratol, milk thistle (Silybum marianum), and St John’s wort (Hypericum perforatum) reportedly cause drug interactions with anticoagulant, antiretroviral, anticancer, immunosuppressant, and antidepressant drugs.8–18 Therefore, it is necessary to evaluate herb–drug interactions at an early stage to prevent potentially dangerous clinical outcomes.
Cultured fresh and cryopreserved human hepatocytes enable the integrated evaluation of phase I drug-metabolizing enzymes (eg, CYPs) and phase II drug-metabolizing enzymes (eg, UGTs and SULTs).19–21 The function of drug-metabolizing enzymes and transporters is regulated by their expression at the transcriptional and posttranscriptional levels.22,23 The primary hepatocyte culture system contains nuclear receptors such as aromatic hydrocarbon receptor, pregnane X-receptor, and constitutive androstane receptor.20,24 Quantification of CYP messenger ribonucleic acid (mRNA) and protein levels and enzyme activities is recommended to evaluate drug interactions.25–27 Moreover, drug interaction knowledge might increase efficacy of a drug and reduce its undesirable side effects and toxicity. According to the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for drug–drug interactions, the effect of a drug on CYP mRNA levels in fresh or cryopreserved human hepatocytes is critical for CYP-induced drug interactions.28,29 Numerous drug–drug interaction studies using primary or cryopreserved human hepatocyte culture systems have been reported.30–35
Honokiol, 2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol, is a pharmacologically active component isolated from Magnolia officinalis, Magnolia grandiflora, and other plants. Honokiol exerts antitumor,36–39 antioxidative,40,41 anti-inflammatory,42–44 antithrombotic,45 neuroprotective,46 antiseptic,47 antinociceptive,48–50 and antidepressant activities.51 Honokiol is extensively metabolized to 18 metabolites via hydroxylation, methylation, sulfation, and glucuronidation in rats.52–54 Honokiol potently inhibits CYP1A2, CYP2C8, CYP2C9, CYP2C19, and UGT1A9 enzyme activities with Ki values of 1.2, 4.9, 0.54, 0.57, and 0.3 μM, respectively, but weakly inhibits those of CYP2B6, CYP2D6, CYP3A4, UGT1A1, and UGT2B7 in human liver microsomes.55 Thus, honokiol should be examined for potential pharmacokinetic drug interactions in vivo due to its inhibition of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and UGT1A9 enzyme activities.
There are no reports describing the effects of honokiol on the induction of various phase I or II drug-metabolizing enzymes in human hepatocytes after chronic treatment. In this study, we investigated the effect of honokiol on the expression of various drug-metabolizing enzymes using attachable cryopreserved human hepatocytes to evaluate the potential of honokiol as a perpetrator in drug interactions.
Materials and methods
Materials and reagents
Honokiol (>98% by high-performance liquid chromatography), acetaminophen, phenacetin, lansoprazole, phenobarbital, rifampicin, l-glutamine, William’s medium E, dimethyl sulfoxide (DMSO), and midazolam were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 13C2,15N-acetaminophen, bupropion, 1′-hydroxymidazolam, hydroxybupropion, cryopreserved human hepatocytes (HMC520, HFC443, and HF382), high-viability cryohepatocyte recovery kit, Biocoat™ Hepatocyte Culture Medium, and Biocoat™ Collagen I 24-Well Plates were obtained from Corning Life Sciences (Woburn, MA, USA). TaqMan® RNA-to-CT™ 1-Step Kit, TaqMan® Gene Expression Assays, and gene-specific probes and primers (Table 1) for real-time reverse transcription polymerase chain reaction (RT-PCR) were obtained from Applied Biosystems (Foster city, CA, USA). Fetal bovine serum and TRIzol® were obtained from Invitrogen (Carlsbad, CA, USA). Acetonitrile and methanol (liquid chromatography mass spectrometry [LC-MS] grade) were purchased from Burdick & Jackson Inc. (Ulsan, Korea).
 | Table 1 Probe and primer sets used in this study |
Cell culture
Cryopreserved human hepatocyte lines (HF382, HFC443, and HMC520) were thawed in hepatocyte recovery medium, and viability was determined using Trypan Blue according to the manufacturer’s suggested protocols (Corning Life Sciences). Viable cells (4×105) were seeded in collagen type I precoated 24-well plates in 400 μL of hepatocyte plating medium, maintained at 37°C with 5% CO2 in an incubator for 4 hours. Plating medium was then removed and the cells were supplied with matrigel medium containing 0.25 mg/mL of MatrigelTM matrix and cultured for 24 hours. Honokiol stock solutions (0.5, 5, or 50 mM in DMSO), 10 mM lansoprazole, 10 mM rifampicin, and 1 M phenobarbital in DMSO were diluted 1,000 times with hepatocyte culture medium. The cells were treated with 0.5, 5, or 50 μM honokiol, vehicle (0.1% DMSO), or prototypical inducers, including lansoprazole (10 μM), phenobarbital (1 mM), or rifampicin (10 μM), in triplicate. The cells were then incubated at 37°C with 5% CO2 for 48 hours, and the medium was exchanged with fresh medium containing drugs or vehicle (400 μL) every 24 hours.
CYP1A2, CYP2B6, and CYP3A4 activity measurement
A cocktail (200 μL) containing 20 μM phenacetin, 10 μM bupropion, and 5 μM midazolam in William’s E buffer solution was added to each well treated with vehicle (0.1% DMSO), rifampicin, lansoprazole, phenobarbital, or honokiol, and the plate was returned to the CO2 incubator. After incubation for 1 hour, 200 μL of the medium was removed from each well and stored in a −70°C deep freezer until sample analysis. 13C2,15N-acetaminophen (50 μL; 0.15 μg/mL, internal standard for acetaminophen) and d9-hydroxybufuralol (0.01 μg/mL, internal standard for hydroxybupropion and 1′-hydroxymidazolam) in methanol were added to 50 μL of the medium obtained from each well. The mixtures were vortex-mixed for 2 minutes and then centrifuged at 13,000 rpm for 4 minutes at 4°C. The supernatant (40 μL) was diluted with 60 μL of deionized water and then mixed for 2 minutes by vortexing. An aliquot (5 μL) was analyzed by LC-MS/MS.56 The LC-MS/MS system consisted of a tandem mass spectrometer (TSQ Quantum Access, Thermo Scientific, San Jose, CA, USA) coupled with a Nanospace SI-2 LC system (Shiseido, Tokyo, Japan). Separation was performed on an Atlantis dC18 (5 μm, 2.1 mm internal diameter ×100 mm; Waters Co., MA, USA) using a gradient elution of 5% methanol in 0.1% formic acid (mobile phase A) and 95% methanol in 0.1% formic acid (mobile phase B) at a flow rate of 0.4 mL/min: 15% mobile phase B for 1.5 minutes, 15%–95% mobile phase B for 0.5 minute, and 95% mobile phase B for 5 minutes. The column and autosampler temperatures were 50°C and 6°C, respectively. After 1.5 minutes, the LC eluent was diverted from waste to a mass spectrometer fitted with an electrospray ionization source and operated in the positive ion mode. Electrospray ionization source settings for ionization of the metabolites were as follows: electrospray voltage, 4,000 V; vaporizer temperature, 350°C; capillary temperature, 330°C; sheath gas pressure, 35 psi; and auxiliary gas pressure, 15 psi. Quantification of each metabolite was performed by selected reaction monitoring mode: acetaminophen, m/z 152.0>110.0; 13C2,15N-acetaminophen (internal standard for acetaminophen), m/z 155.0>111.0; hydroxybupropion, m/z 256.2>238.2; 1′-hydroxymidazolam, m/z 342.1>203.0; and d9-1-hydroxybufuralol, 287.1>187.0 (internal standard). CYP1A2, CYP2B6, and CYP3A4 enzyme activities are expressed as formation rates (pmol/million cells/h).
Total RNA isolation
At the end point of the experiment, William’s E buffer solutions containing substrate cocktail were recovered and total RNA was immediately isolated using TRIzol. Briefly, TRIzol (500 μL) was added to each well of 24-well culture plates and transferred into 1.5 mL microcentrifuge tubes. Cell lysates were combined with 100 μL of chloroform, mixed by vortexing, and the RNA aqueous layer was separated by centrifugation. Total RNA was obtained by ethanol precipitation and dissolved in diethylpyrocarbonate-treated water. The RNA concentration and purity were determined by absorbance test at OD260/280 nm using a NanoVue Plus spectrophotometer (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and stored at −70°C until RT-PCR analysis.
RT-PCR
RT-PCR was conducted using an RT-PCR detection system (Bio-Rad, Foster City, CA, USA) with a TaqMan® RNA-to-CT™ 1-Step Kit and TaqMan® Gene Expression Assays Kit (Table 1), as follows. Total RNA (25 ng) in each reaction sample was used for RT-PCR: 48°C for 25 minutes for reverse transcription, 95°C for 15 minutes for enzyme activation, 44 cycles of denaturation at 95°C for 15 seconds, and annealing/extension at 60°C for 1 minute. The relative (ΔCt) values of all samples were normalized to the glyceraldehyde 3-phosphate dehydrogenase Ct value, and then the relative Ct values (ΔΔCt) of each sample, including CYP3A4, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1, were obtained by comparison (ΔCt) of vehicle and rifampicin, lansoprazole, phenobarbital, or honokiol (0.5, 5, and 50 μM). The relative gene expression rate was calculated using the (ΔΔCt) increase/decrease compared with the vehicle control.
Data analysis
The data are presented throughout as means ± standard deviation for three organ donors. On the basis of EMA and FDA guidelines, the induction results were evaluated separately for each donor. The treatment mRNA level is compared with those of the vehicle incubations. Enzyme induction in an in vitro study was considered to be demonstrated if drug treatment resulted in a more than two-fold increase in mRNA level in a concentration-dependent manner. A 50% decrease in mRNA that was not attributable to cytotoxicity was taken to indicate downregulation of the enzyme in question.
Results
Effect of honokiol on CYP mRNA levels in human hepatocytes
The effect of honokiol on the mRNA levels of CYP1A2, CYP3A4, CYP2B6, CYP2C8, CYP2C9, and CYP2C19 enzymes was evaluated by RT-PCR. Honokiol and prototypical inducers did not exert toxicity at the indicated concentrations (0.5–50 μM) using 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium calorimetric assay. Rifampicin, a pregnane X-receptor inducer, and phenobarbital, a constitutive androstane receptor inducer, increased the mRNA levels of CYP3A4, CYP2B6, CYP2C8, CYP2C9, and CYP2C19 in three human hepatocyte lines (Table 2). Lansoprazole, an aromatic hydrocarbon receptor inducer, significantly increased the CYP1A2 mRNA level by 6.93- to 70.0-fold (Table 2). The fold increase was 6.61–21.2 for CYP2B6 mRNA in phenobarbital-treated hepatocytes and 36.6–113 for CYP3A4 mRNA in rifampicin-treated hepatocytes (Table 2). These results demonstrated that the cryopreserved human hepatocytes significantly responded to prototypical inducers and that these test systems were adequate to address FDA and EMA guidelines.
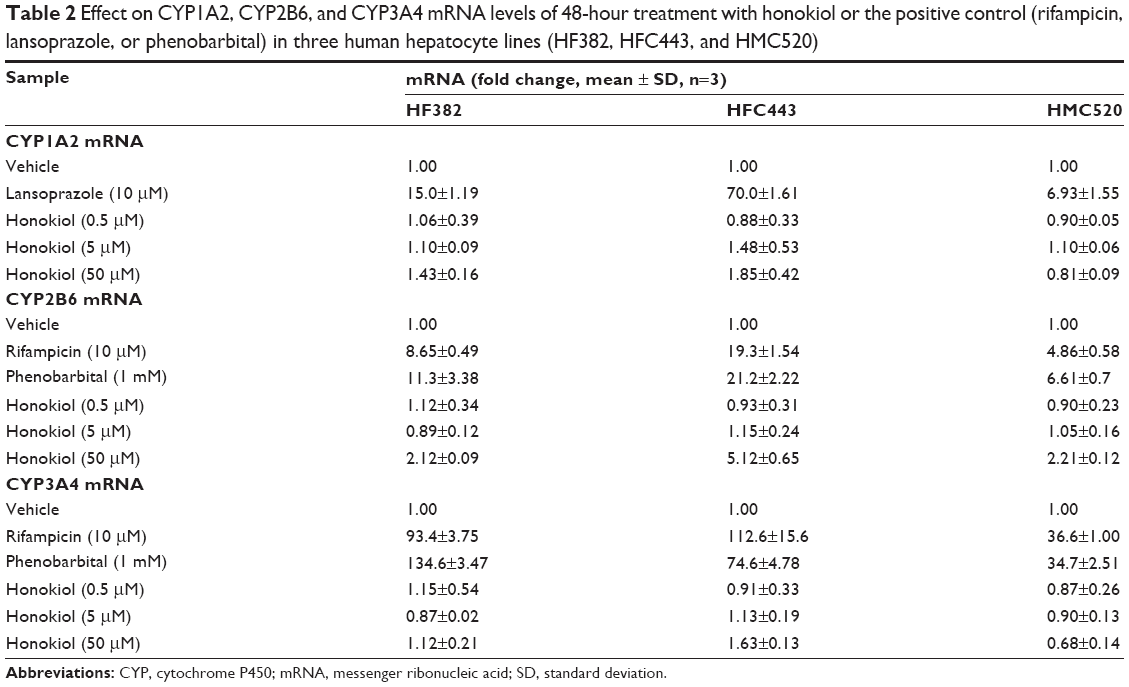 | Table 2 Effect on CYP1A2, CYP2B6, and CYP3A4 mRNA levels of 48-hour treatment with honokiol or the positive control (rifampicin, lansoprazole, or phenobarbital) in three human hepatocyte lines (HF382, HFC443, and HMC520) |
Honokiol did not affect the mRNA levels of CYP1A2, CYP3A4, CYP2C8, CYP2C9, or CYP2C19 at the three concentrations tested (0.5, 5, and 50 μM) in three human hepatocyte lines (Table 2, Figure 1). Honokiol (50 μM) induced a more than two-fold increase in the CYP2B6 mRNA level in three human hepatocyte lines (HF382, HFC443, and HMC520; Table 2), indicating that honokiol at higher concentrations may cause drug interactions in vivo due to CYP2B6 induction, based on FDA and EMA guidelines.
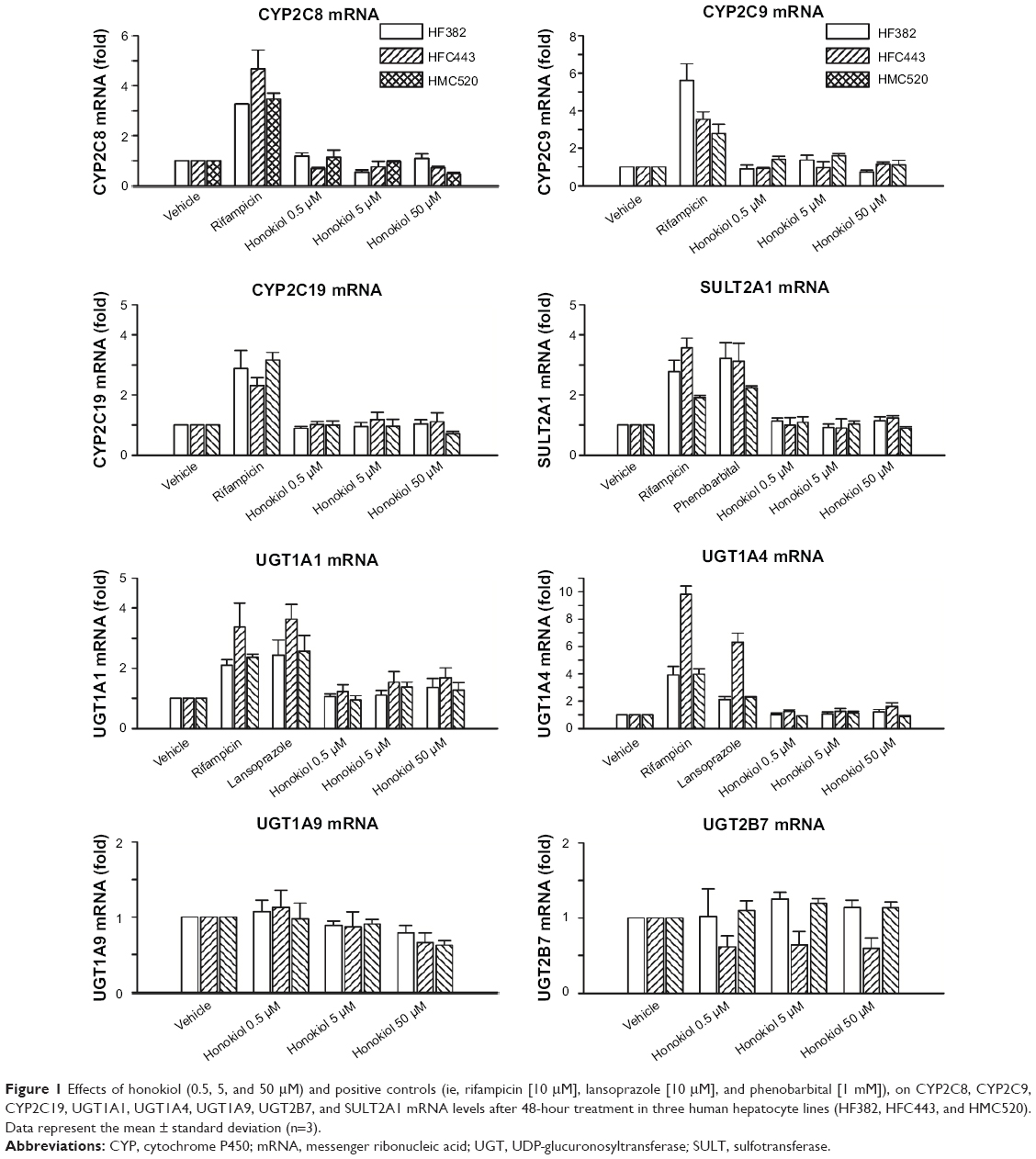 | Figure 1 Effects of honokiol (0.5, 5, and 50 μM) and positive controls (ie, rifampicin [10 μM], lansoprazole [10 μM], and phenobarbital [1 mM]), on CYP2C8, CYP2C9, CYP2C19, UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1 mRNA levels after 48-hour treatment in three human hepatocyte lines (HF382, HFC443, and HMC520). Data represent the mean ± standard deviation (n=3). |
Effects of honokiol on CYP1A2, CYP2B6, and CYP3A4 enzyme activities in three human hepatocyte lines
The effects of honokiol and prototypical inducers on CYP1A2, CYP2B6, and CYP3A4 enzyme activities were evaluated in three human hepatocyte lines (Table 3). CYP1A2-catalyzed phenacetin O-deethylase activity was significantly increased by 12.1- to 47.8-fold by lansoprazole treatment in three human hepatocyte lines (Table 3). Rifampicin and phenobarbital treatment significantly increased CYP2B6-catalyzed bupropion hydroxylase activities by 4.14- to 12.7-fold, and CYP3A4-catalyzed midazolam 1′-hydroxylase activities by 2.04- to 4.40-fold, compared with the vehicle control (Table 3). CYP1A2-catalyzed phenacetin O-deethylase and CYP3A4-catalyzed midazolam 1′-hydroxylase activities were not significantly altered by treatment with 0.5–50 μM honokiol in three human hepatocyte lines, indicating that honokiol did not affect CYP1A2 and CYP3A4 enzyme activities (Table 3). However, chronic 50 μM honokiol treatment resulted in 1.94- to 4.66-fold increases in CYP2B6-catalyzed bupropion hydroxylase activity in three human hepatocyte lines (Table 3).
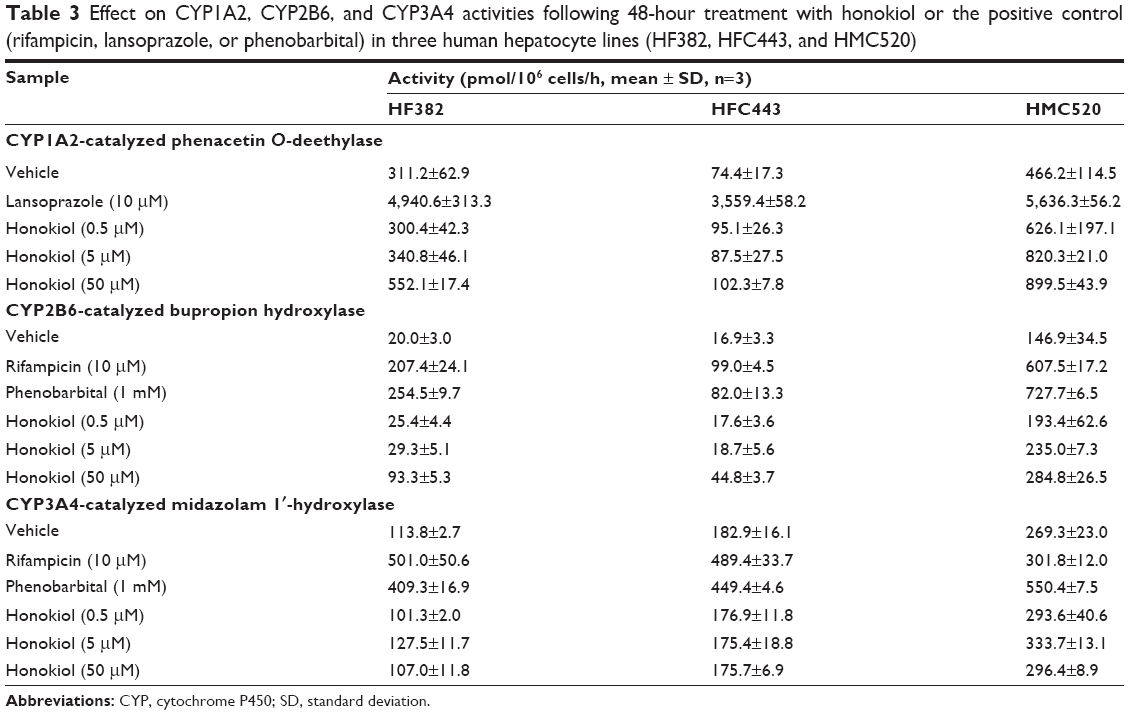 | Table 3 Effect on CYP1A2, CYP2B6, and CYP3A4 activities following 48-hour treatment with honokiol or the positive control (rifampicin, lansoprazole, or phenobarbital) in three human hepatocyte lines (HF382, HFC443, and HMC520) |
Effect of honokiol on SULT and UGT mRNA levels in human hepatocytes
Phase II drug metabolic enzymes such as UGT and SULT play key roles in drug metabolism to increase the hydrophilicity of xenobiotics by conjugation of chemical moieties such as glucuronide or sulfate. The mRNA levels of human UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1 were determined and compared against the fold induction levels of vehicle-treated hepatocytes. The UGT1A1 mRNA level was increased 2.10- to 3.36-fold by rifampicin and 2.42- to 3.63-fold by lansoprazole compared with vehicle in three human hepatocyte lines (Figure 1). UGT1A4 mRNA levels were increased 3.89- to 9.81-fold by rifampicin and 2.10- to 6.29-fold by lansoprazole compared with vehicle in three human hepatocyte lines (Figure 1). SULT2A1 mRNA levels were induced 1.92- to 3.56-fold by rifampicin and 2.23- to 3.22-fold by phenobarbital in three human hepatocyte lines (Figure 1). We found that honokiol did not affect the UGT1A1, UGT1A4, UGT1A9, UGT2B7, or SULT2A1 mRNA levels in three human hepatocyte lines (Figure 1). Therefore, honokiol likely does not induce drug–drug interactions by up- or downregulating the expression of phase II drug-metabolizing enzymes such as UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1.
Discussion
Drug-metabolizing enzymes play major roles in the metabolism of xenobiotics or endogenous compounds, and are up- or downregulated following xenobiotic exposure.57 Hyperforin, an active component of St John’s wort, potently inhibited CYP2C9 (Ki, 1.8 μM) and CYP3A4 (Ki, 0.49 μM) activity in human complementary deoxyribonucleic acid-expressing CYP microsomes,58 but chronic 48-hour hyperforin treatment (1 μM) in human hepatocytes resulted in significant increases in the mRNA, activity, and protein levels of CYP3A4 and CYP2C9.31 These results suggest that cotreatment with hyperforin in the clinic significantly decreases the plasma concentrations of CYP3A4 substrates, including cyclosporine, digoxin, simvastatin, and oral contraceptives.59
In our previous study,55 30-minute honokiol treatment resulted in marked inhibition of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and UGT1A9 activities in pooled human liver microsomes, with Ki values of 1.2, 4.9, 0.54, 0.57, and 0.3 μM, respectively. However, chronic treatment with a higher concentration (50 μM) of honokiol increased CYP2B6 mRNA and activity levels without affecting the expression of CYP1A2, CYP3A4, CYP2C8, CYP2C9, CYP2C19, UGT1A1, UGT1A4, UGT1A9, UGT2B7, or SULT2A1 in three human hepatocyte lines (Tables 2 and 3, Figure 1). CYP1A1-catalyzed phenacetin O-deethylase and CYP3A4-catalyzed midazolam 1′-hydroxylase activities were unaffected in honokiol-treated hepatocytes compared with vehicle-treated hepatocytes (Table 3), suggesting that honokiol had no effect on CYP1A2 and CYP3A4 mRNA levels in three human hepatocyte lines. These results indicate that honokiol coadministration may not result in any clinically significant drug interactions with CYP1A2, CYP3A4, CYP2C8, CYP2C9, CYP2C19, UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1 substrates in vivo.
Treatment with a higher concentration (50 μM) of honokiol increased the CYP2B6 mRNA level and CYP2B6-catalyzed bupropion hydroxylase activity by more than two-fold in three human hepatocyte lines, indicating that a higher concentration of honokiol induces CYP2B6 expression in vivo according to FDA and EMA drug interaction guidelines. High doses of honokiol should be used cautiously with CYP2B6-metabolized drugs such as cyclophosphamide, ifosfamide, tamoxifen, bupropion, valproic acid, and efavirenz to avoid drug interactions.60
Honokiol has poor oral absorption and is extensively metabolized in liver with an elimination half-life of 49.05 minutes and 56.24 minutes for 5 mg/kg and 10 mg/kg intravenous doses, respectively, in rats.36 After intravenous administration of honokiol in rats, 18 metabolites via hydroxylation, sulfation, glucuronidation, and methylation were identified in plasma.53 Incubation of human hepatocytes with honokiol (5 μM) for 1 hour resulted in the detection of metabolites such as honokiol glucuronide, honokiol sulfates, hydroxyhonokiol, hydroxyhonokiol glucuronide, and hydroxyhonokiol sulfate, but not honokiol, by LC high-resolution orbitrap mass spectrometry. At present, no data on honokiol pharmacokinetics in humans are available; such data are indispensable for prediction of honokiol–drug interactions.
Conclusion
In conclusion, the present study demonstrated the potential of honokiol to increase CYP2B6 mRNA and activity levels following chronic exposure to a high concentration (50 μM). However, honokiol had negligible effects on the CYP1A2, CYP3A4, CYP2C8, CYP2C9, CYP2C19, UGT1A1, UGT1A4, UGT1A9, UGT2B7, and SULT2A1 mRNA levels in human hepatocytes. Our results suggest that honokiol at the usual dosage may not cause pharmacokinetic-based drug interactions with the substrates of these drug-metabolizing enzymes in humans via enzyme induction.
Acknowledgments
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI12C1852), and The Catholic University of Korea, 2012 (M-2012-B0002-00024).
Disclosure
The authors report no conflicts of interest in this work.
References
Goey AK, Mooiman KD, Beijnen JH, Schellens JH, Meijerman I. Relevance of in vitro and clinical data for predicting CYP3A4-mediated herb-drug interactions in cancer patients. Cancer Treat Rev. 2013;39(7):773–783. | ||
Gouws C, Steyn D, Du Plessis L, Steenekamp J, Hamman JH. Combination therapy of Western drugs and herbal medicines: recent advances in understanding interactions involving metabolism and efflux. Expert Opin Drug Metab Toxicol. 2012;8(8):973–984. | ||
Mukherjee PK, Ponnusankar S, Pandit S, Hazam PK, Ahmmed M, Mukherjee K. Botanicals as medicinal food and their effects on drug metabolizing enzymes. Food Chem Toxicol. 2011;49(12):3142–3153. | ||
Na DH, Ji HY, Park EJ, Kim MS, Liu KH, Lee HS. Evaluation of metabolism-mediated herb-drug interactions. Arch Pharm Res. 2011; 34(11):1829–1842. | ||
Mohamed ME, Frye RF. Effects of herbal supplements on drug glucuronidation. Review of clinical, animal, and in vitro studies. Planta Med. 2011;77(4):311–321. | ||
Zhou SF, Zhou ZW, Li CG, et al. Identification of drugs that interact with herbs in drug development. Drug Discov Today. 2007; 12(15–16):664–673. | ||
Chen XW, Serag ES, Sneed KB, et al. Clinical herbal interactions with conventional drugs: from molecules to maladies. Curr Med Chem. 2011; 18(31):4836–4850. | ||
Rahimi R, Abdollahi M. An update on the ability of St John’s wort to affect the metabolism of other drugs. Expert Opin Drug Metab Toxicol. 2012;8(6):691–708. | ||
Wang X, Zhang H, Chen L, Shan L, Fan G, Gao X. Liquorice, a unique “guide drug” of traditional Chinese medicine: a review of its role in drug interactions. J Ethnopharmacol. 2013;150(3):781–790. | ||
Zadoyan G, Rokitta D, Klement S, et al. Effect of Ginkgo biloba special extract EGb 761(R) on human cytochrome P450 activity: a cocktail interaction study in healthy volunteers. Eur J Clin Pharmacol. 2012;68(5):553–560. | ||
Russo E, Scicchitano F, Whalley BJ, et al. Hypericum perforatum: pharmacokinetic, mechanism of action, tolerability, and clinical drug-drug interactions. Phytother Res. 2014;28(5):643–655. | ||
Zhang Z, Ge B, Zhou L, Lam TN, Zuo Z. Induction of liver cytochrome P450s by Danshen-Gegen formula is the leading cause for its pharmacokinetic interactions with warfarin. J Ethnopharmacol. 2014;154(3):672–686. | ||
Muller AC, Kanfer I. Potential pharmacokinetic interactions between antiretrovirals and medicinal plants used as complementary and African traditional medicines. Biopharm Drug Dispos. 2011;32(8):458–470. | ||
Zhou X, Chan K, Yeung JH. Herb-drug interactions with Danshen (Salvia miltiorrhiza): a review on the role of cytochrome P450 enzymes. Drug Metabol Drug Interact. 2012;27(1):9–18. | ||
Chow HH, Garland LL, Hsu CH, et al. Resveratrol modulates drug- and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev Res. 2010;3(9):1168–1175. | ||
Wu JW, Lin LC, Tsai TH. Drug-drug interactions of silymarin on the perspective of pharmacokinetics. J Ethnopharmacol. 2009;121(2): 185–193. | ||
Haefeli WE, Carls A. Drug interactions with phytotherapeutics in oncology. Expert Opin Drug Metab Toxicol. 2014;10(3):359–377. | ||
Unger M. Pharmacokinetic drug interactions involving Ginkgo biloba. Drug Metab Rev. 2013;45(3):353–385. | ||
Grime K, Ferguson DD, Riley RJ. The use of HepaRG and human hepatocyte data in predicting CYP induction drug-drug interactions via static equation and dynamic mechanistic modelling approaches. Curr Drug Metab. 2010;11(10):870–885. | ||
Honma M, Kozawa M, Suzuki H. Methods for the quantitative evaluation and prediction of CYP enzyme induction using human in vitro systems. Expert Opin Drug Discov. 2010;5(5):491–511. | ||
Klieber S, Torreilles F, Guillou F, Fabre G. The use of human hepatocytes to investigate drug metabolism and CYP enzyme induction. Methods Mol Biol. 2010;640:295–308. | ||
Zhu BT. On the general mechanism of selective induction of cytochrome P450 enzymes by chemicals: some theoretical considerations. Expert Opin Drug Metab Toxicol. 2010;6(4):483–494. | ||
Hukkanen J. Induction of cytochrome P450 enzymes: a view on human in vivo findings. Expert Rev Clin Pharmacol. 2012;5(5):569–585. | ||
Mattinen L, Kublbeck J, Rechardt O, Honkakoski P, Rautio J. Direct and rapid transcript analysis assay for CYP mRNA expression and inducibility in human primary hepatocytes. Drug Metab Lett. Epub 2014 Jun 15. | ||
Gerin B, Dell’Aiera S, Richert L, Smith S, Chanteux H. Assessment of cytochrome P450 (1A2, 2B6, 2C9 and 3A4) induction in cryopreserved human hepatocytes cultured in 48-well plates using the cocktail strategy. Xenobiotica. 2013;43(4):320–335. | ||
Einolf HJ, Chen L, Fahmi OA, et al. Evaluation of various static and dynamic modeling methods to predict clinical CYP3A induction using in vitro CYP3A4 mRNA induction data. Clin Pharmacol Ther. 2014;95(2):179–188. | ||
Halladay JS, Wong S, Khojasteh SC, Grepper S. An ‘all-inclusive’ 96-well cytochrome P450 induction method: measuring enzyme activity, mRNA levels, protein levels, and cytotoxicity from one well using cryopreserved human hepatocytes. J Pharmacol Toxicol Meth. 2012;66(3):270–275. | ||
Zhang JG, Ho T, Callendrello AL, et al. Evaluation of calibration curve-based approaches to predict clinical inducers and non-inducers of CYP3A4 using plated human hepatocytes. Drug Metab Dispos. 2014;42(9):1379–1391. | ||
Shen C, Meng Q. Prediction of cytochrome 450 mediated drug-drug interactions by three-dimensional cultured hepatocytes. Mini Rev Med Chem. 2012;12(10):1028–1036. | ||
Koe XF, Lim EL, Seah TC, et al. Evaluation of in vitro cytochrome P450 induction and inhibition activity of deoxyelephantopin, a sesquiterpene lactone from Elephantopus scaber L. Food Chem Toxicol. 2013;60:98–108. | ||
Komoroski BJ, Zhang S, Cai H, et al. Induction and inhibition of cytochromes P450 by the St John’s wort constituent hyperforin in human hepatocyte cultures. Drug Metab Dispos. 2004;32(5):512–518. | ||
Komoroski BJ, Parise RA, Egorin MJ, Strom SC, Venkataramanan R. Effect of the St John’s wort constituent hyperforin on docetaxel metabolism by human hepatocyte cultures. Clin Cancer Res. 2005;11(19 Pt 1):6972–6979. | ||
Srovnalova A, Svecarova M, Kopecna Zapletalova M, et al. Effects of anthocyanidins and anthocyanins on the expression and catalytic activities of CYP2A6, CYP2B6, CYP2C9, and CYP3A4 in primary human hepatocytes and human liver microsomes. J Agric Food Chem. 2014;62(3):789–797. | ||
Williamson B, Dooley KE, Zhang Y, Back DJ, Owen A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob Agents Chemother. 2013;57(12):6366–6369. | ||
Takusagawa S, Miyashita A, Iwatsubo T, Usui T. In vitro inhibition and induction of human cytochrome P450 enzymes by mirabegron, a potent and selective beta3-adrenoceptor agonist. Xenobiotica. 2012;42(12):1187–1196. | ||
Arora S, Singh S, Piazza GA, Contreras CM, Panyam J, Singh AP. Honokiol: a novel natural agent for cancer prevention and therapy. Curr Mol Med. 2012;12(10):1244–1252. | ||
Chilampalli C, Zhang X, Kaushik RS, et al. Chemopreventive effects of combination of honokiol and magnolol with alpha-santalol on skin cancer developments. Drug Discov Ther. 2013;7(3):109–115. | ||
Tian W, Xu D, Deng YC. Honokiol, a multifunctional tumor cell death inducer. Pharmazie. 2012;67(10):811–816. | ||
Kumar A, Kumar Singh U, Chaudhary A. Honokiol analogs: a novel class of anticancer agents targeting cell signaling pathways and other bioactivities. Future Med Chem. 2013;5(7):809–829. | ||
Guerra-Araiza C, Alvarez-Mejia AL, Sanchez-Torres S, et al. Effect of natural exogenous antioxidants on aging and on neurodegenerative diseases. Free Radic Res. 2013;47(6–7):451–462. | ||
Shen JL, Man KM, Huang PH, et al. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules. 2010;15(9):6452–6465. | ||
Chen YJ, Tsai KS, Chan DC, et al. Honokiol, a low molecular weight natural product, prevents inflammatory response and cartilage matrix degradation in human osteoarthritis chondrocytes. J Orthop Res. 2014;32(4):573–580. | ||
Kim BH, Cho JY. Anti-inflammatory effect of honokiol is mediated by PI3K/Akt pathway suppression. Acta Pharmacol Sin. 2008;29(1):113–122. | ||
Zhang P, Liu X, Zhu Y, Chen S, Zhou D, Wang Y. Honokiol inhibits the inflammatory reaction during cerebral ischemia reperfusion by suppressing NF-kappaB activation and cytokine production of glial cells. Neurosci Lett. 2013;534:123–127. | ||
Hu H, Zhang XX, Wang YY, Chen SZ. Honokiol inhibits arterial thrombosis through endothelial cell protection and stimulation of prostacyclin. Acta Pharmacol Sin. 2005;26(9):1063–1068. | ||
Woodbury A, Yu SP, Wei L, Garcia P. Neuro-modulating effects of honokiol: a review. Front Neurol. 2013;4:130. | ||
Li N, Xie H, Li L, et al. Effects of honokiol on sepsis-induced acute kidney injury in an experimental model of sepsis in rats. Inflammation. 2014;37(4):1191–1199. | ||
Lin YR, Chen HH, Lin YC, Ko CH, Chan MH. Antinociceptive actions of honokiol and magnolol on glutamatergic and inflammatory pain. J Biomed Sci. 2009;16:94. | ||
Matsui N, Takahashi K, Takeichi M, et al. Magnolol and honokiol prevent learning and memory impairment and cholinergic deficit in SAMP8 mice. Brain Res. 2009;1305:108–117. | ||
Lee YJ, Lee YM, Lee CK, Jung JK, Han SB, Hong JT. Therapeutic applications of compounds in the Magnolia family. Pharmacol Ther. 2011;130(2):157–176. | ||
Xu Q, Yi LT, Pan Y, et al. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(3):715–725. | ||
Dong Y, Tang M, Song H, et al. Characterization of metabolic profile of honokiol in rat feces using liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry and (13)C stable isotope labeling. J Chromatogr B. 2014;953–954:20–29. | ||
Lai H, Tang M, Liu J, et al. Identification of honokiol metabolites in rats by the method of stable isotope cluster technique and ultra-high performance liquid chromatography/quadrupole-time-of-flight mass spectrometry. J Chromatogr B. 2013;931:157–163. | ||
Liu J, Tang M, Lai H, et al. Identification of metabolites of honokiol in rat urine using 13C stable isotope labeling and liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry. J Chromatogr A. 2013;1295:48–56. | ||
Jeong HU, Kong TY, Kwon SS, et al. Effect of honokiol on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules. 2013;18(9):10681–10693. | ||
Ji HY, Liu KH, Jeong JH, et al. Effect of a new prokinetic agent DA-9701 formulated with corydalis tuber and pharbitidis semen on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Evid Based Complement Alternat Med. 2012;2012:650718. | ||
Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28(3):249–268. | ||
Obach RS. Inhibition of human cytochrome P450 enzymes by constituents of St John’s Wort, an herbal preparation used in the treatment of depression. J Pharmacol Exp Ther. 2000;294(1):88–95. | ||
Madabushi R, Frank B, Drewelow B, Derendorf H, Butterweck V. Hyperforin in St John’s wort drug interactions. Eur J Clin Pharmacol. 2006;62(3):225–233. | ||
Turpeinen M, Zanger UM. Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metab Drug Interact. 2012;27(4):185–197. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.