")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 11
Effect of glutaminase inhibition on cancer-induced bone pain
Received 14 May 2019
Accepted for publication 23 July 2019
Published 11 September 2019 Volume 2019:11 Pages 273—282
DOI https://doi.org/10.2147/BCTT.S215655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Jennifer Fazzari, Gurmit Singh
Department of Pathology and Molecular Medicine, Mcmaster University, Hamilton, ON, Canada
Correspondence: Gurmit Singh
McMaster University, 1280 Main St. W, 2102 Michael G DeGroote Centre for Learning and Discovery, Hamilton, Ontario L8S 4L8, Canada
Tel +1 905 525 9140 x28144
Email [email protected]
Purpose: The complex nature of cancer-induced bone pain (CIBP) has led to investigation into cancer-targeted therapies. This has involved targeting glutamate release from the tumor, secreted as a byproduct of antioxidant responses and metabolic disruption. Cancer cells undergo many metabolic changes that result in increased glutamine metabolism and subsequently the production of glutamate. Glutaminase (GLS) is the enzyme that mediates the conversion of glutamine to glutamate and has been shown to be upregulated in many cancer types including malignancies of the breast. This enzyme, therefore, represents another potential therapeutic target for CIBP, one that lies upstream of glutamate secretion.
Methods: A recently developed inhibitor of GLS, CB-839, was tested in an animal model of CIBP induced by intrafemoral MDA-MB-231 xenografts. CIBP behaviors were assessed using Dynamic Weight Bearing and Dynamic Plantar Aesthesiometer readings of mechanical hyperalgesia and allodynia.
Results: CB-839 failed to modulate any of the associated nociceptive behaviors induced by intrafemoral MDA-MB-231 tumor growth. Further investigation in vitro revealed the sensitivity of the drug is dependent on the metabolic flexibility of the cell line being tested which can be modulated by cell culture environment.
Conclusion: Adaptation to metabolic disturbances may explain the failure of CB-839 to exhibit any significant effects in vivo and the metabolic flexibility of the cell line tested should be considered for future investigations studying the metabolic effects of glutaminase inhibition.
Keywords: Cancer-induced bone pain, breast cancer, glutaminase, anaplerosis
Introduction
Circulating glutamine is the most abundant amino acid in the plasma.1,2 Glutamine is readily consumed by rapidly dividing cells, including cancer, for both energy synthesis and carbon/nitrogen source. Rapidly proliferating cancer cells have a modified metabolism increasing their dependence on glutamine to sustain uncontrolled proliferation at rates that exceed normal intracellular glutamine production, making this otherwise non-essential amino acid become conditionally essential.1,2 The nitrogen from glutamine supports the levels of many amino acids in the cell via aminotransferase activity,3 where at least half of the non-essential amino acids used for protein synthesis in cancer are derived from glutamine.4,5
Cancer-induced bone pain (CIBP) is a multifaceted pain state that poses a clinical challenge for effective treatment and control in oncology patients with advanced, metastatic disease. In particular, colonization of cancer in the bone most commonly results in the development of CIBP6 with cancer-secreted factors playing a major role in modulating the bone microenvironment and eliciting nociceptive stimuli.7,8 Previously, we have targeted the cystine/glutamate antiporter system xc- (xCT) to modulate CIBP induced by intrafemoral inoculation of an aggressive breast carcinoma, MD-MB-231.9,10 Targeting the secretion of glutamate from malignant cells via xCT inhibition has contributed to the delay in severity and/or onset of CIBP in animal models bearing an intrafemoral tumor.9,10 Although effective in pre-clinical models, existing inhibitors of xCT are limited for clinical use. We, therefore, investigated other targets that contribute to the production of cancer-derived glutamate. Glutaminase (GLS) is the enzyme responsible for the generation of glutamate from glutamine and therefore offered a new therapeutic target to investigate the role of cancer-derived glutamate in CIBP.
Not only have breast cancer cells been shown to derive a large proportion of intracellular glutamate from glutamine,11 but GLS is transcriptionally upregulated in triple-negative breast cancer (TNBC) cells increasing their reliance on exogenous glutamine.12,13 Increased glutamine oxidation results in increased reactive oxygen species (ROS) production via the mitochondrial electron transport chain14 but is also required for ROS detoxification via glutathione (GSH).15 Glutamate production is essential for GSH synthesis with glutamate itself as a component of the antioxidant but also as a driver of xCT activity to acquire cystine, the rate-limiting substrate in GSH synthesis. Amino acid utilization by cancer cells is therefore essential for both cellular metabolism and redox homeostasis.16–18
CB-839 is a potent allosteric inhibitor of GLS in Phase I clinical trials with TNBC cells reported to show great sensitivity to this compound. CB839 exhibits antiproliferative effects, depletion of key metabolic intermediates and anti-tumor activity in TNBCs exploiting a metabolic alteration in these cells for therapeutic benefit.19 Based on this work, GLS appeared to be an attractive target for CIBP based on the glutamate-induced nociception hypothesis as GLS represents a glutamate-modulating target upstream of its release through the activity of xCT.
Despite the results reported by Gross et al 2014, we did not observe any significant delay in the development of CIBP in our MDA-MB-231 xenograft animal model after chronic CB-839 treatment. Subsequent in vitro analysis suggests its susceptibility to CB-839 may be nutrient-dependent. Therefore, the metabolic flexibility of this cell line and its ability to adapt to different nutrient conditions which may provide variable responses in vivo versus in vitro.
Materials and methods
Cell culture
MDA-MB-231 human breast adenocarcinoma (American Type Culture Collection) were maintained at sub-confluent densities with 5% CO2 at 37°C in DMEM supplemented with 10% fetal bovine serum and 1X antibiotic/antimycotic (Life Technologies).
Animals
Female athymic Balb/c nu/nu homozygous nude mice (Jackson Laboratories) at 4–6 weeks of age were used. The mice were housed in a sterile environment with room temperature and lighting maintained at 24°C with a 12 hrs light/dark cycle, respectively. Autoclaved food and water were provided ad libitum. All procedures involving animals were approved by the McMaster University Animal Research Ethics Board in accordance with the Canadian Council on Animal Care’s Guidelines to the Care and Use of Experimental Animals (Volume 1, 2nd edition).
Quantification of extracellular glutamate
Media samples are collected after incubation with CB-839 for 72 hrs and the concentration of glutamate is quantified using the Amplex Red Glutamic Acid Kit (Life Technologies). Cells are fixed with 10% formalin and stained with crystal violet to quantify cell number. Final glutamate concentrations are normalized to cell number.
Quantification of intracellular ROS production
Intracellular ROS was quantified using a choloromethyl 2ʹ,7ʹ–dichlorofluorescein diacetate derivative (CM-H2DCFDA). The DCFDA reagent is loaded onto the cells prior to drug treatment in phenol-red-free DMEM supplemented with 10% FBS, sodium pyruvate (1 mM) and L-glutamine (4 mM). Fluorescence was quantified after either 24 or 72 hrs of CB-839 treatment. Fluorescence was then read at 529 nm following the indicated time points.
Assessing xCT activity by monitoring uptake of radiolabeled cystine (14C-cystine)
The uptake of radiolabelled [14C]-cystine was measured as previously described.9 MDA-MB-231 cells incubated with CB-839 or DMSO for 72 hrs were washed and incubated in the uptake buffer (HBSS +0.45uCi 14C-cystine) for 5 mins and then subjected to washes with ice-cold HBSS and lysed in lysis buffer consisting of 0.1 N NaOH and 0.1% Triton-X for 15 mins. One hundred microliters of lysate is then added to 1 mL of scintillation fluid for quantification of radioactivity. Protein concentration for each sample is quantified using the Bradford reagent and used to normalize scintillation counts.
Development of subcutaneous MDA-MB-231 xenograft
Slow release (0.25 mg, 21-day release) 17β-estradiol pellets (Innovative Research of America, Sarasota, FL, USA) were implanted subcutaneously 3 days prior to tumor cell inoculation. Three million MDA-MB-231 cells were injected subcutaneously in the right flank of Balb/c nude mice. Tumors were allowed to develop until they reached on average approximately 75 mm3 before compound administration. CB-839 (n=4) or vehicle (n=4) was administered 2x per day at a dose of 200 mg/kg by oral gavage due to the high clearance of the drug (Calithera Biosciences). The treatment period extended for 17 days with daily tumor volume measurements.
Development of intrafemoral MDA-MB-231 xenograft
The intrafemoral xenograft model is used to simulate the effects of bone metastases including pain, a common experience of cancer patients with bone metastases. As mentioned above, estrogen pellet implantation preceded tumor cell inoculation. Animals were subject to isofluorane anesthesia and subcutaneous buprenorphine (0.05 mg/kg) prior to intrafemoral injection. Animals receiving MDA-MB-231 cells were injected with 500,000 cells suspended in 25 uL PBS into the right, distal epiphysis of the femur as previously reported.9,10 CB-839 (n=8) or vehicle (n=11) was administered as indicated above.
Behavioral testing and radiographic lesion assessment
Dynamic weight bearing (DWB) and dynamic plantar aesthesiometer (DPA) testing was used to assess weight distribution and mechanical withdrawal thresholds, respectively, as previously described.9,10 Briefly, at least 3 baseline measurements were collected before tumor inoculation. After tumor development, behavioral testing was done at least two times per week for continuous assessment of CIBP progression. Experimenter was blinded as to what treatment group each animal belonged. As animals had to be administered vehicle or CB-839 twice per day by oral gavage which is a potentially stressful procedure, animals were given ample time following their first gavage on testing day. For DPA testing, animals were allowed to equilibrate for 20 mins in the testing chamber prior to testing. No equilibration time was allotted for DWB testing as exploratory behavior (including rearing) is important to these measurements.20
Assessment of intrafemoral lesions was conducted using radiographic analysis at endpoint and blinded scoring of radiographs on a scale of 0 (no lesion) to 3 (extensive osteolysis) as described previously.10
Serum glutamate quantification by HPLC-MS
To quantify the concentration of glutamate in serum, a standard glutamate curve was made by spiking 10 μL serum with 0.2 μg/mL L-glutamate acid-2,3,3,4,4-d5 as an internal standard (Sigma 616,281). Injection volumes of 0.1, 0.2, 0.5 and 1 μL corresponded to 0.1, 0.2, 0.5 and 1 ng/μL of the L-glutamate d5 in the standard curve. Glutamine and glutamate elute in close proximity to each other and therefore, pH of mobile Phase A is essential to prevent overlap of peaks. Although not quantified, the ratio of glutamine to glutamate was determined by comparing peak areas of both analytes.
Serum samples were subject to an ice-cold methanol (MeOH) extraction at a ratio of 1:10 (serum:MeOH) for 1 hr on ice. Liquid chromatography was performed on an Agilent 1290 Infinity liquid chromatography system in isocratic mode equipped with a SeQuant ZIC-HILIC PEEK column (3.5 μm, 100 A; 150×2.1 mm; Millipore). Mobile phase consisted of (A) 10 mM ammonium formate (pH 6.5) and (B) acetonitrile. Injection volume was 5 μL with a flow rate of 0.2 mL/minute. LC/MS analysis was performed on an Agilent 6550 iFunnel Q-TOF in negative ion mode with a mass range of 100–200 m/z. Additional parameters are as follows: gas temperature at 275°C, nebulizer pressure at 30 psig, sheath gas temp at 320°C, sheath gas flow at 11 L/min, capillary voltage (VCap) at 3500 V and nozzle voltage at 1500 V. Data acquisition was performed using Agilent Mass Hunter (version B.07.00). The pH of mobile phase (A) is very important to maximize separation of glutamate from glutamine in the stationary phase to prevent co-elution of these molecules. Although not quantified, the ratio of glutamine to glutamate was determined by comparing peak areas of both analytes.
Statistics
Data are presented as means ± SEM. One-way ANOVA and two-way repeated measures ANOVA were used for statistical comparisons. P-values <0.05 were considered statistically significant.
Results
CB-839 does not prevent growth of MDA-MB-231 xenografts
Before testing CB-839 in the CIBP model, the effect of the drug on the growth of MDA-MB-231 xenografts was tested in a subcutaneous tumor-growth model. Differing from results observed by Gross et al 2014 with another TNBC cell line (HCC1806), CB-839 treatment did not affect the growth of MDA-MB-231 cells relative to the vehicle-treated control (Figure 1). Serum glutamate concentrations also did not differ significantly between groups despite the trend illustrating a decrease in serum glutamate concentrations from CB-839-treated animals (Figure 2). Furthermore, the ratio of glutamine to glutamate in the serum was significantly elevated in drug-treated animals, indicative of an accumulation of glutamine due to inhibition of GLS which is in agreement with Gross et al 2014 who report elevated glutamine in plasma within 4 hrs of CB-839 administration.
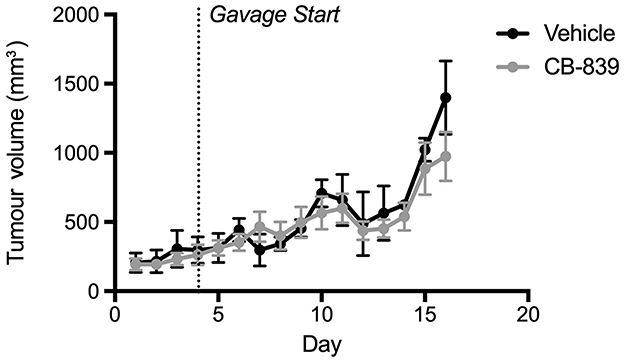 |
Figure 1 Growth of subcutaneous MDA-MB-231 tumors in Balb/c nude mice treated with CB-839 or vehicle. Tumor volume was measured daily. Treatment began once they reached approximately 75 mm3. CB-839 treatment (n=4) does not affect result in a reduction in tumor volume relative vehicle-treated controls (n=4). |
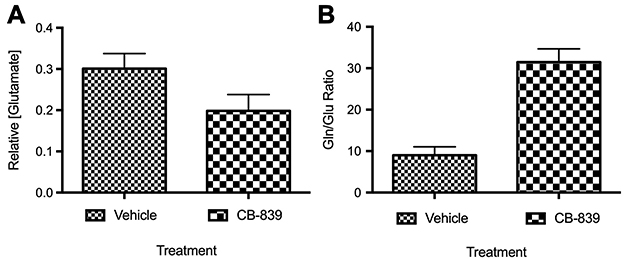 |
Figure 2 Concentration of glutamate in serum from animals with subcutaneous MDA-MB-231 tumors treated with CB-839 (n=4) or vehicle (n=4). (A) Concentration of glutamate in serum relative to vehicle-treated animals (P=0.0781 by unpaired t-test). (B) The ratio of glutamine to glutamate in serum indicating that glutamine levels significantly increase in the serum of mice-treated with CB-839 (P<0.001 by unpaired t-test). |
CB-839 does not prevent development of CIBP behaviors
CB-839-treated animals did not show any delay in the development of pain behaviors as measured by dynamic weight bearing (Figure 3A) or dynamic plantar aesthesiometer measurements (Figure 3B) relative to the vehicle-treated controls. Treatment with this compound was, therefore, not effective in preventing CIBP-related mechanical allodynia. The degree of osteolysis also did not differ significantly between vehicle and CB-839-treated groups (Figure 4) suggesting CB-839 did not affect tumor development consistent with our preliminary subcutaneous tumor growth analysis.
 |
Figure 3 Assessment of pain behaviors – dynamic weight bearing (A) dynamic plantar aesthesiometer (B) behavioral testing was done twice per week to monitor development of CIBP behaviors until endpoint. No significant changes between CB-839 (n=8) and vehicle-treated (n=11) groups were observed in both tests. |
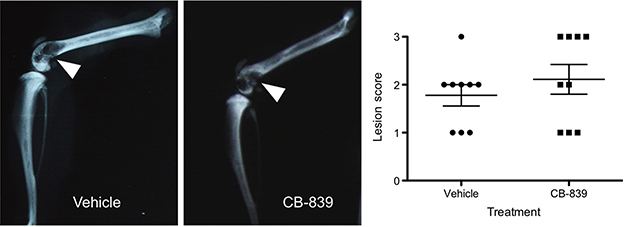 |
Figure 4 Radiographic lesion scoring of MDA-MB-231 tumors at endpoint. Representative images of vehicle and CB-839-treated groups are presented and osteolysis from tumor development is indicated by white arrows. A graphical representation of radiographic lesion scoring of all animals in each group is shown. |
CB-839 reduces glutamate release from MDA-MB-231 cells
After seeing negative results in our in vivo model, we wanted to confirm CB-839 reduces extracellular glutamate release from MDA-MB-231 cells as reported. CB-839’s ability to reduce extracellular glutamate is sensitive to the status of sodium pyruvate in the culture media. Extracellular glutamate concentrations decrease significantly in the absence of sodium pyruvate in the media (Figure 5A). Furthermore, only in the absence of sodium pyruvate is any significant impact on cell density observed suggesting that the presence of that metabolite in the media maintains cellular proliferation during GLS inhibition (Figure 5B).
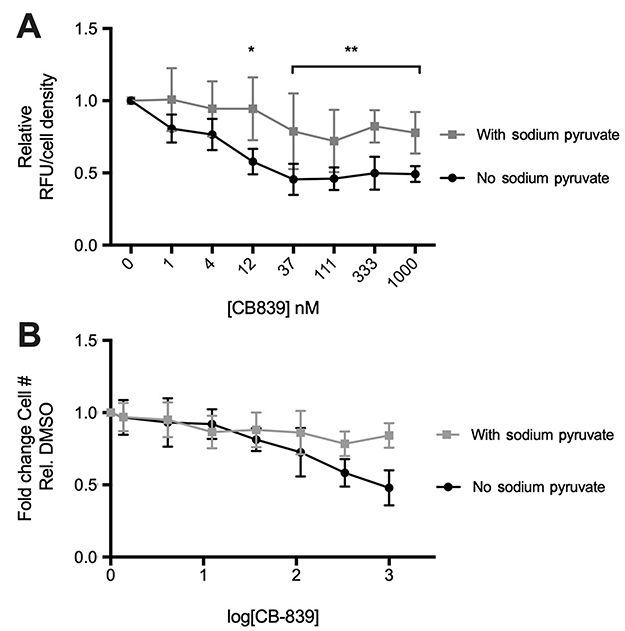 |
Figure 5 Effect of sodium pyruvate on CB-839 induced changes in glutamate release and cell survival in vitro CB-839 reduces extracellular glutamate (A) relative to the vehicle (DMSO) control at concentrations above 12 uM only in the absence of sodium pyruvate in culture media (P<0.005). No significant changes observed in the presence of sodium pyruvate. CB-839-induced decrease in cell number after treatment for 72 hrs observed only in the absence of sodium pyruvate in culture media (B). Non-linear regression of (B) results in a shift in EC50 values between “no sodium pyruvate” (EC50=4.997 nM) and “with sodium pyruvate“ (EC50=11.07) indicating that cellular proliferation and/or survival is affected by CB-839 treatment only when sodium pyruvate is absent from culture media. |
ROS production and uptake of radiolabeled cystine
In the absence of sodium pyruvate, there is an approximate 2.5 fold increase in ROS production by 72 hrs relative to the DMSO control (Figure 6A). Interestingly, this is not accompanied by a significant influx of cystine uptake unless there is sodium pyruvate present in the media (Figure 6B).
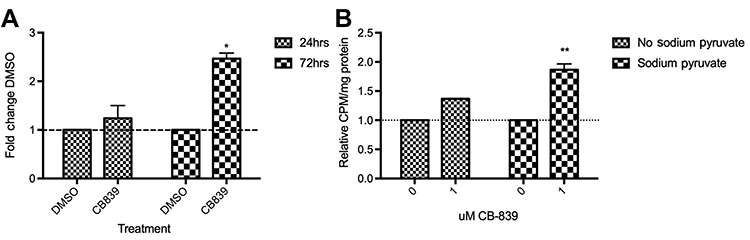 |
Figure 6 Production of reactive oxygen species over the course of 24 and 72 hrs (A) and cystine uptake after 72 hrs treatment with 1 μM CB-839 (B). A significant increase in ROS (A) is observed after a 72 hrs incubation with 1000 nM CB-839 (P<0.05; 2-way repeated measures ANOVA). Similarly, [14C]-cystine uptake (B) increases significantly by 72 hrs but only in the absence of sodium pyruvate (P<0.01; 2-way repeated measures ANOVA). |
Discussion
In an effort to target glutamate release from cancer cells associated with clinical CIBP characteristics, we targeted glutamate production at a major metabolic hub that is often associated with the reprogrammed metabolism of aggressive cancers. In such cells, GLS activity is a key mediator of anaplerosis fueling the Tricarboxylic Acid (TCA) cycle, as a result of a shunted glycolytic pathway (Warburg effect). GLS expression and activity is, therefore, upregulated in many cancers including MDA-MB-231 cells. In addition to generation of TCA metabolites, glutamine-derived glutamate is essential for production of the major antioxidant tripeptide, glutathione (GSH) composed of glutamate, cystine and glycine where glutamate is not only a component of the molecule itself, but is also necessary for the acquisition cystine, the rate-limiting substrate of GSH synthesis. This glutamate/cystine exchange occurs through the xCT antiporter. Expression of xCT has been identified as a marker for GLS inhibitor sensitivity21 as high xCT activity decreases intracellular glutamate pools, thus increasing the cell’s reliance on glutamine and GLS to replenish this pool. This allows the cell to maintain antioxidant production at the expense of regenerating TCA cycle intermediates from glutamine.22
It has become clear that the metabolic flexibility of the cancer must be considered when evaluating the efficacy of a potential therapeutic that targets a major metabolic pathway in the cell. To promote survival, the cell must balance fueling energy production and biomass accumulation with ROS clearance. This involves diverting potential metabolic intermediates, such as glutamate, toward antioxidant production rather than anabolism, exporting a large amount of glutamate in order to acquire cystine for GSH synthesis. Therefore, glutamine-derived glutamate can be sacrificed to promote survival over growth when ROS levels are high.23
After treatment with CB-839, in vitro, we observed an increase in ROS levels possibly due to impaired mitochondrial function and limited GSH production in the absence of GLS-derived glutamate. It was hypothesized that glutamate flux via xCT would be limited as GLS inhibition was expected to limit glutamate available to drive antiporter activity and as a result reduce cystine acquisition. Unexpectedly, however, a decrease in extracellular glutamate was not accompanied by significant changes in cystine uptake when sodium pyruvate was absent from the media. Under these culture conditions, decreased cell proliferation was also observed. When sodium pyruvate is present in the media, extracellular glutamate was diminished slightly and cystine uptake increased, as expected, in response to rising ROS levels (Figure 6). Therefore, ROS production was not dependent on the presence of sodium pyruvate in the media potentially due to mitochondrial dysfunction that accompanies CB-839 treatment,19 however, cystine uptake was. The cell must regenerate this depleted intracellular glutamate pool that accompanies xCT activity via glutamine uptake and GLS activity.24 However, with GLS activity inhibited, TCA cycle intermediates and GSH are depleted and in the absence of sodium pyruvate there no alternatives to restore glutamate levels; therefore, the growth of TNBCs is halted (Figure 5B).19
CB-839 sensitivity is associated with an increased basal ratio of glutamate to glutamine which is common of TNBCs including MDA-MB-231 cells and is indicative of GLS activity.19,25,26 Despite this, sensitivity of MDA-MB-231 xenografts to CB-839 treatment was limited (Figure 1). This has also been observed by Lampa et al27 where the same cell lines resistant to CB-839 treatment in vivo exhibited increased sensitivity to GLS in vitro, suggesting the metabolic profile of these cells differs under in vitro and in vivo conditions. This differential response to CB-839 in vitro and in vivo has also been observed with pancreatic carcinoma and attributed to compensatory metabolic networks.28
Flexibility to overcome perturbations in glutamine metabolism is important for survival of cells conventionally labeled as glutamine addicted. Under conditions of limited glutamine, pyruvate carboxylase (PC) has been found to be a compensatory anaplerotic mechanism allowing the cells to use glucose-derived pyruvate for anaplerosis over glutamine.29 In many cancer cells, glucose and glutamine are compensatory under different metabolic conditions in order to maintain the TCA cycle.30 Cells deemed glutamine-addicted can therefore become glutamine independent,29 using glucose to produce oxaloacetate (OAA) via pyruvate metabolism. When pyruvate is limited or not present, there is a shift to glutamine-dependent acetyl-CoA formation, which is suppressed when pyruvate is present.30 This may explain why there is an amplified impact of CB-839 on proliferation and glutamate release when pyruvate is removed from tissue culture media as under these conditions the cell is relying more heavily on glutaminolytic machinery including GLS. Pyruvate therefore aids in the cell’s resistance to this metabolic stress. In glioblastoma cells, GLS suppression is accompanied by glucose-dependent anaplerosis via PC which produces OAA from pyruvate (glucose-derived).29
Furthermore, MDA-MB-231 cells have been shown to have reduced TCA cycle activity due to oncogenic kRas activity.31 As a result, glycolysis is enhanced and these cells consume greater quantities of glucose relative to glutamine. This redundancy in glutamate production could be an explanation for why MDA-MB-231 cells show varied survival after CB-839 treatment in the presence or absence of sodium pyruvate and also show persistence of intrafemoral tumor development and CIBP in vivo where nutrient conditions are less controlled than those in vitro. This redundancy is further illustrated by glutamate production from alpha-ketoglutarate (α-KG). Pyruvate can feed the TCA cycle in the absence of GLS activity generating α-KG which can be diverted to make more glutamate and fuel system xc- exchange for antioxidant production. Glutamate dehydrogenase (GDH) can catalyze the conversion of α-KG to glutamate when there are sufficient levels of TCA cycle intermediates.32 This can explain the role of sodium pyruvate supplementation and the ability for MDA-MB-231 cells to survive GLS inhibition by CB-839. When pyruvate is absent in the media, the pull remains on GLS-derived glutamate to fuel the TCA cycle, increasing the cells' reliance on GLS activity and, as a result, making it more sensitive to GLS inhibition. However, when pyruvate is present, the TCA cycle pool can be restored in the absence of GLS activity making the cell less susceptible to GLS inhibition. Therefore, under these conditions, it is possible that sufficient levels of glutamate are being produced from the α-KG pool via transamination32 to drive system xc- activity under the pressure of increasing ROS as cystine uptake increases after 72 hrs when ROS is also increased. This compensatory mechanism is summarized in Figure 7.
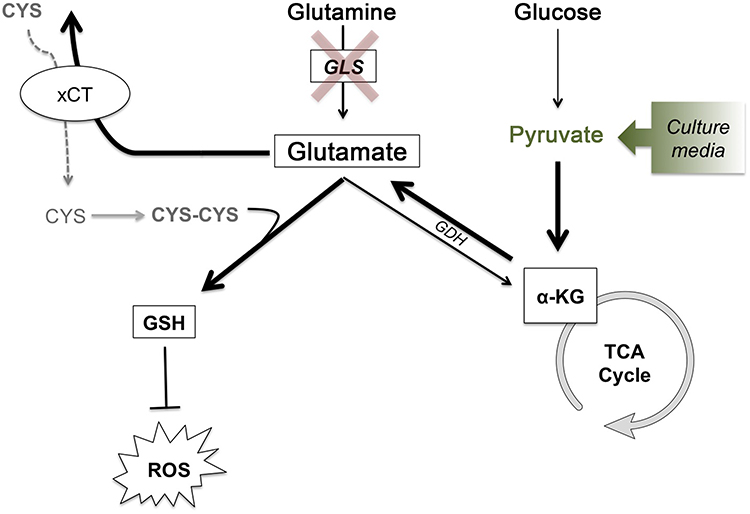 |
Figure 7 Glutathione synthesis for ROS detoxification drives metabolic adaptations to maintain glutamate production and cystine acquisition in the absence of GLS activity. Glutathione synthesis for ROS detoxification drives metabolic adaptations to maintain glutamate production and cystine acquisition in the absence of GLS activity. In the absence of GLS activity, glucose-derived pyruvate can feed the TCA cycle generating α-KG, which can then be used to generate glutamate via glutamate dehydrogenase. With the intracellular pool of glutamate maintained, GSH production can continue, both with glutamate itself as a component for GSH and as an exchange factor for cystine, the rate-limiting substrate of GSH synthesis. Bolded arrows represent the pathway that is hypothesized to compensate for loss of glutamine-derived glutamate under conditions of GLS inhibition or low glutamine conditions. Under in vitro conditions, the addition of sodium pyruvate to cell culture media drives this process.Abbreviations: CYS, cystine; CYS-CYS, cysteine; GSH, glutathione; ROS, reactive oxygen species; GLS, glutaminase; GDH, glutamate dehydrogenase; α-KG, alpha-ketoglutarate; TCA, tricarboxylic acid. |
Despite the fact that the majority of extracellular glutamate released from TNBCs is derived from glutamine, it is clear that this can vary between cell types and under different nutrient conditions. For example, where one TNBC line, HCC1806 reported by Gross et al 2014 showed limited metabolic flexibility when it comes to GLS inhibition, we have observed that another TNBC line, MDA-MB-231, potentially has greater nutrient flexibility and ability to overcome CB-839 treatment and ultimately survive perturbations in glutamine metabolism. The potential nutrient flexibility of MDA-MB-231 cells is presented as a hypothesis for the persistence of CIBP in our model despite GLS inhibition and highlights future investigations that can be made into determining the metabolic mechanisms behind cancer cell metabolism and CIBP.
Conclusion
Sensitivity of MDA-MB-231 cells to GLS inhibition with CB-839 is variable and dependent on culture conditions. CB-839’s effect on cell survival and glutamate release in these cells is dependent on the presence of sodium pyruvate in the culture media. Furthermore, the reported effect on tumor growth following CB-839 therapy was not observed with both the subcutaneous growth of MDA-MB-231 tumors and the development of intrafemoral lesions not differing significantly from vehicle-treated controls. Of most significance, CB-839 treatment also failed to modulate CIBP behaviors in vivo. It is possible the same metabolic adaptations observed in vitro are occurring in vivo and maintaining glutamate production in the absence of GLS activity. Balancing cellular proliferation with ROS homeostasis places different metabolic demands on the cell. When these demands can be met, cell survival persists even in the presence of metabolic perturbations.
By identifying what adaptations are taking place in MDA-MB-231 tumors following CB-839 administration will give more insight into the role of glutamine metabolism and intratumoural glutamate production in CIBP and reveal novel combinatorial treatment approaches to overcome the metabolic plasticity of these cells.
Abbreviation list
GLS, Glutaminase; CIBP, cancer-induced bone pain; DWM, dynamic weight bearing; DPA, dynamic plantar aesthesiometer; ROS, reactive oxygen species; TNBC, triple negative breast cancer; xCT, functional unit of system xc-; DCFDA, choloromethyl 2ʹ,7ʹ–dichlorofluorescein diacetate; TCA, tricarboxylic acid; α-KG, alpha ketoglutarate; GDH, glutamate dehydrogenase.
Acknowledgment
We would like to thank Susan Demo and Calithera Biosciences for generously providing us with their compound CB-839 for all of our experimentation. We would also like to acknowledge the Canadian Breast Cancer Foundation for funding this project.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
2. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168(4):657–669. doi:10.1016/j.cell.2016.12.039
3. Hosios AM, Hecht VC, Danai LV, et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. 2016;36(5):540–549. doi:10.1016/j.devcel.2016.02.012
4. Alberghina L, Gaglio D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014;5:e1561. doi:10.1038/cddis.2014.513
5. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–433. doi:10.1016/j.tibs.2010.05.003
6. Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69(1–2):1–18.
7. Skerry TM. The role of glutamate in the regulation of bone mass and architecture. J Musculoskelet Neuronal Interact. 2008;8(2):166–173.
8. Seidlitz EP, Sharma MK, Singh G. Extracellular glutamate alters mature osteoclast and osteoblast functions. Can J Physiol Pharmacol. 2010;88(9):929–936. doi:10.1139/y10-070
9. Fazzari J, Balenko MD, Zacal N, Singh G. Identification of capsazepine as a novel inhibitor of system xc- and cancer-induced bone pain. J Pain Res. 2017;10:915–925. doi:10.2147/JPR.S125045
10. Ungard RG, Seidlitz EP, Singh G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. Pain. 2014;155(1):28–36. doi:10.1016/j.pain.2013.08.030
11. Carrascosa JM, Martínez P, de Castro IN. Nitrogen movement between host and tumor in mice inoculated with ehrlich ascitic tumor cells. Cancer Res. 1984;44(9):3831–3835.
12. Kung H-N, Marks JR, Chi J-T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;7(8):e1002229. doi:10.1371/journal.pgen.1002229
13. Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–465. doi:10.1016/j.ccr.2013.08.020
14. Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107(19):8788–8793. doi:10.1073/pnas.1003428107
15. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi:10.1038/nature12040
16. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. doi:10.1038/nrc.2016.71
17. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16(10):650–662. doi:10.1038/nrc.2016.81
18. Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75(9):1782–1788. doi:10.1158/0008-5472.CAN-14-3745
19. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor cb-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi:10.1158/1535-7163.MCT-13-0870
20. Quadros AU, Pinto LG, Fonseca MM, Kusuda R, Cunha FQ, Cunha TM. Dynamic weight bearing is an efficient and predictable method for evaluation of arthritic nociception and its pathophysiological mechanisms in mice. Sci Rep. 2015;5:14648. doi:10.1038/srep14648
21. Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. 2017;6. doi:10.7554/eLife.27713
22. Shin C-S, Mishra P, Watrous JD, et al. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun. 2017;8:15074. doi:10.1038/ncomms15074
23. Gu Y, Albuquerque CP, Braas D, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell. 2017;67(1):128–138.e7. doi:10.1016/j.molcel.2017.05.030
24. Sayin VI, LeBoeuf SE, Singh SX, et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife Sciences. 2017;6:e28083. doi:10.7554/eLife.28083
25. Budczies J, Brockmöller SF, Müller BM, et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. J Proteomics. 2013;94:279–288. doi:10.1016/j.jprot.2013.10.002
26. Budczies J, Pfitzner BM, Györffy B, et al. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer. 2015;136(7):1619–1628. doi:10.1002/ijc.29152
27. Lampa M, Arlt H, He T, et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS One. 2017;12(9):e0185092. doi:10.1371/journal.pone.0185092
28. Biancur DE, Paulo JA, Małachowska B, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. 2017;8:15965. doi:10.1038/ncomms15965
29. Cheng T, Sudderth J, Yang C, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. PNAS. 2011;108(21):8674–8679.
30. Yang C, Ko B, Hensley CT, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56(3):414–424. doi:10.1016/j.molcel.2014.09.025
31. Gaglio D, Metallo CM, Gameiro PA, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi:10.1038/msb.2011.56
32. Whillier S, Garcia B, Chapman BE, Kuchel PW, Raftos JE. Glutamine and α-ketoglutarate as glutamate sources for glutathione synthesis in human erythrocytes. Febs J. 2011;278(17):3152–3163. doi:10.1111/j.1742-4658.2011.08241.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.