")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Effect of epigallocatechin-3-gallate, major ingredient of green tea, on the pharmacokinetics of rosuvastatin in healthy volunteers
Authors Kim TE, Ha N, Kim Y, Kim H, Lee JW, Jeon JY, Kim MG
Received 12 December 2016
Accepted for publication 3 April 2017
Published 9 May 2017 Volume 2017:11 Pages 1409—1416
DOI https://doi.org/10.2147/DDDT.S130050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Tae-Eun Kim,1 Na Ha,2 Yunjeong Kim,2 Hyunsook Kim,1 Jae Wook Lee,3 Ji-Young Jeon,2 Min-Gul Kim2,4
1Department of Clinical Pharmacology, Konkuk University Medical Center, Seoul, 2Center for Clinical Pharmacology, Biomedical Research Institute, Chonbuk National University Hospital, Jeonju, Jeonbuk, 3Nephrology Clinic, National Cancer Center, Goyang, Gyeonggi-do, 4Department of Pharmacology, Chonbuk National University Medical School, Jeonju, Jeonbuk, Republic of Korea
Abstract: Previous in vitro studies have demonstrated the inhibitory effect of green tea on drug transporters. Because rosuvastatin, a lipid-lowering drug widely used for the prevention of cardiovascular events, is a substrate for many drug transporters, there is a possibility that there is interaction between green tea and rosuvastatin. The aim of this study was to investigate the effect of green tea on the pharmacokinetics of rosuvastatin in healthy volunteers. An open-label, three-treatment, fixed-sequence study was conducted. On Day 1, 20 mg of rosuvastatin was given to all subjects. After a 3-day washout period, the subjects received 20 mg of rosuvastatin plus 300 mg of epigallocatechin-3-gallate (EGCG), a major ingredient of green tea (Day 4). After a 10-day pretreatment of EGCG up to Day 14, they received rosuvastatin (20 mg) plus EGCG (300 mg) once again (Day 15). Blood samples for the pharmacokinetic assessments were collected up to 8 hours after each dose of rosuvastatin. A total of 13 healthy volunteers were enrolled. Compared with the administration of rosuvastatin alone, the concomitant use at Day 4 significantly reduced the area under the concentration–time curve from time 0 to the last measurable time (AUClast) by 19% (geometric mean ratio 0.81, 90% confidence interval [CI] 0.67–0.97) and the peak plasma concentration (Cmax) by 15% (geometric mean ratio 0.85, 90% CI 0.70–1.04). AUClast or Cmax of rosuvastatin on Day 15 was not significantly different from that on Day 1. This study demonstrated that co-administration of EGCG reduces the systemic exposure of rosuvastatin by 19%, and pretreatment of EGCG can eliminate that effect of co-administration of EGCG.
Keywords: rosuvastatin, green tea, EGCG, pharmacokinetics, drug interaction, drug transporter
Introduction
Green tea (Camellia sinensis) is one of the most popular beverages in Asia and is gaining popularity worldwide.1 Recently, green tea has attracted considerable attention because of its health benefits ranging from improvement of metabolic syndrome to cancer prevention.2–4 Of note, many studies have reported that green tea reduces the risk of cardiovascular diseases.5–7 Given that many people aware of its health benefits consume green tea on a regular basis, there is a possibility that the ingredients in green tea might interact with other drugs on many levels of pharmacokinetics. Green tea is highly rich in catechins that have been reported to inhibit drug transporters and thereby interfere with the transport of many drugs across the cell membrane. Catechins in green tea include epicatechin, epigallocatechin (EGC), epicatechin-3-gallate (ECG) and epigallocatechin-3-gallate (EGCG),8 of which EGCG is the most abundant and biologically active. ECG and EGCG can inhibit or promote drug absorption or elimination by interacting with the intestinal and hepatic organic anion transport peptides (OATPs) responsible for drug absorption from the intestinal lumen and removal from the bloodstream. An in vitro study9 has reported that ECG and EGCG decrease the uptake of the model substrate estrone-3-sulfate by OATP1B1, OATP2B1, and OATP1A2 and increase its uptake by OATP1B3. Another in vitro study10 also reported the inhibitory effects of green tea on transport by transporters such as OATP1B1, OATP1B3, organic cation transporter (OCT) 1, OCT2, multidrug and toxic compound extrusion (MATE) 1, MATE2-K, and P-glycoprotein (P-gp). These studies suggest that green tea catechins ECG and EGCG can alter the pharmacokinetic profile of a drug that is transported by one of these transporters.
Despite in vitro evidence, only a few drugs have been reported to have significant interaction with green tea in vivo. Misaka et al11 reported that green tea ingestion greatly reduces the plasma concentrations of nadolol by inhibiting OATP1A2-mediated uptake. Werba et al1 reported that the plasma concentrations of simvastatin increase with concomitant administration of green tea, mainly via inhibition of intestinal CYP3A4 and P-gp and hepatic OATP1B1.
In this study, we investigated the effect of green tea on the pharmacokinetics of rosuvastatin in healthy volunteers by concomitant administration of green tea ingredient EGCG with or without pretreatment of EGCG. Rosuvastatin is an HMG-CoA reductase inhibitor used for treating hypercholesterolemia in patients with cardiovascular disease. In vitro studies demonstrated that rosuvastatin is a substrate for many drug transporters, including OATP1B1, OATP1B3, OATP2B1, OATP1A2, Na+/taurocholate cotransporting polypeptide (NTCP), P-gp, breast cancer resistance protein (BCRP), and multidrug resistance-associated protein 2 (MRP2).12–15 In enterocytes, rosuvastatin is absorbed from the intestinal lumen via OATP2B1 and OATP1A2 and pumped out via BCRP, MRP2, and P-gp; in hepatocytes, rosuvastatin is transported into the cytoplasm via OATP1B1, OATP1B3, OATP2B1, and NTCP and excreted into bile via BCRP, P-gp, and MRP2. Because many of these transporters (ie, OATP1B1, OATP1B3, OATP2B1, OATP1A2, and P-gp) are reported to be targets of EGC and EGCG, there is a great possibility that rosuvastatin and these green tea catechins interact on many levels of the pharmacokinetics of rosuvastatin. However, detailed information on in vivo pharmacokinetics of rosuvastatin is lacking in case of concomitant administration of rosuvastatin and green tea compounds.
We conducted an open-label, three-treatment, fixed-sequence study as follows. Initially, rosuvastatin was given alone; after a 3-day washout period, rosuvastatin was concomitantly given with a single dose of EGCG; finally, EGCG was given for 10 days as pretreatment, and then rosuvastatin and EGCG were given concomitantly. In each treatment, plasma concentrations of rosuvastatin were measured to determine its systemic exposure. We also stratified our analysis per genetic polymorphisms of OATP1B1, OATP2B1, and BCRP, because single-nucleotide variants of these genes alter the pharmacodynamic or pharmacokinetic properties of rosuvastatin.16–19
Materials and methods
Study population
This study included healthy Korean male or female volunteers aged 20–55 years. Exclusion criteria were any marked medical or medication history and any abnormal findings on physical examination, 12-lead electrocardiogram, or clinical laboratory tests. Subjects were also excluded from the study if they had taken any agent or food that was reported to inhibit or induce transporter OATP or BCRP in the previous 2 weeks.
Study design
To determine the effect of green tea extract on the pharmacokinetics of rosuvastatin, we conducted an open-label, three-treatment, fixed-sequence study (Figure 1). Clinical trials were conducted at Chonbuk National University Hospital. The institutional review boards of Konkuk University Medical Center (IRB No KUH1280061) and Chonbuk National University Hospital (IRB No CUH 2015-03-014) approved the study protocol, and written informed consent was obtained from all subjects before study enrollment. All procedures were performed in accordance with the recommendations of the Declaration of Helsinki. Furthermore, the study was conducted in compliance with the current Good Clinical Practices and other applicable laws and regulatory requirements currently established in South Korea.
 | Figure 1 Study design. |
All the study subjects received a 20-mg rosuvastatin tablet with 150 mL of water by oral route on Day 1. After a 3-day washout period, they received 300 mg of EGCG followed by 20 mg of rosuvastatin 1 hour later. From Day 5 through 14, subjects only received 300 mg of EGCG. On Day 15, just like Day 4, they received 300 mg of EGCG followed by 20 mg of rosuvastatin 1 hour later. EGCG was administered as two capsules of Teavigo™, a commercially available, caffeine-free, 94% pure crystalline EGCG (Healthy Origins, Pittsburgh, PA, USA).20,21 Food or drinks containing caffeine, grapefruit, or alcohol were not permitted throughout the study. Smoking was also not allowed during the study period.
Pharmacokinetic analysis
Blood samples (8 mL) were collected predose and 0.5, 1, 2, 4, 6, and 8 hours after the administration of rosuvastatin for pharmacokinetic analysis. Plasma was obtained by centrifugation at 3,000 rpm for 10 minutes and transferred into polypropylene tubes containing 0.2 M sodium acetate (pH 4.0). The plasma samples were stored at −70°C until they were analyzed. Rooibos™,22 an automated schedule broadcast software, was used to manage the pharmacokinetic blood sampling process.
Plasma concentrations of rosuvastatin were determined by a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method using rosuvastatin-d6 as an internal standard. Briefly, plasma (90 μL, added 0.2 M sodium acetate), rosuvastatin-d6 (10 μL at 1 μg/mL in 50% methanol), and methanol (500 μL) were mixed thoroughly and centrifuged at 13,200 rpm for 5 minutes. After evaporating the supernatant, the residue was reconstituted with 100 μL of 50% methanol, centrifuged at 13,200 rpm for 5 minutes, and injected into the LC-MS/MS system. The lower limit of quantification (LLOQ) for rosuvastatin was 0.5 ng/mL with a linear calibration range of 0.5–100 ng/mL. Each analytical batch had six quality control (QC) samples of known values, ie, two QC samples each for low, intermediate, and high concentrations. The analytical results were accepted only when more than four out of six QC sample concentrations were determined within 15% of known values. Intra- and interday accuracies were 95.5%–104.2%; intra- and interday precisions varied with <6.6 CV%.
The peak plasma concentration (Cmax) and time to Cmax (ie, tmax) were directly obtained from the observed values. The area under the concentration–time curve (AUC) from time 0 to the last measurable time (AUClast) was calculated using the trapezoidal rule. The elimination rate constant (λz) was determined from the slope of the terminal log-linear portion of the plasma concentration–time curve. The area under the concentration–time curve extrapolated to infinity (AUCinf) was calculated as AUClast + last nonzero plasma concentration/λz. Clearance corrected for bioavailability (CL/F) was obtained by dose divided by AUCinf. Noncompartmental analyses were performed using Phoenix WinNonlin® version 6 (Certara, Raleigh, NC, USA).
Genotyping
Genotypes were determined by identifying single-nucleotide polymorphisms (SNPs) in SLCO2B1, SLCO1B1, and ABCG2 encoding for OATP2B1, OATP1B1, and BCRP, respectively. SNPs were determined using the Pyrosequencing Assay performed on the AB 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA), PyroMark Vacuum Prep Tool (Biotage AB, Uppsala, Sweden), and PyroMark Q96 ID (Biotage AB). Briefly, genomic DNA was extracted from 200 μL of whole blood drawn from each volunteer with an Exgene Blood SV kit (Geneall BioTechnology, Seoul, Korea). Then, a polymerase chain reaction (PCR) mixture (total volume, 20 μL) was prepared with 1 μL of 10 pmol primer (forward and reverse primer, respectively), 10 μL of premix Taq polymerase, 7.0 μL of DNase-free water, and 1 μL of genomic DNA. Genotyping assays were performed to identify the following SNPs: SLCO1B1 521T>C (rs4149056), ABCG2 421C>A (rs2231142), and SLCO2B1 935G>A (rs12422149). SNP primers were designed by PyroMark Assay Design 2.0 software (Biotage AB). The assays were conducted using the following PCR protocol: initial denaturation at 95°C for 15 minutes; 45 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 72°C for 40 seconds; and final extension at 72°C for 10 minutes. Then, 3 μL of streptavidin sepharose beads (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was added to 40 μL of binding buffer (Biotage AB) and mixed with 20 μL of PCR product and 20 μL water for 5 minutes at room temperature using a Microplate Orbital Shaker (Finepcr, Seoul, Korea). The beads containing the immobilized templates were captured on the filter probes after the vacuum was applied and then washed with 70% ethanol for 10 seconds, denaturation solution (Biotage AB) for 10 seconds, and washing buffer (Biotage AB) for 10 seconds. The vacuum was then released, and the beads were released into a PyroMark Q96 Plate Low (Biotage AB) containing 40 μL of annealing buffer (Biotage AB) and 0.5 μM of sequencing primer. Pyrosequencing reactions were performed using the PyroMark Gold Q96 Reagents (Biotage AB), which contained the enzyme, substrate, and nucleotides. The assays were performed on the PyroMark ID (Biotage AB), the sample genotype was determined using the PyroMark ID 1.0 software (Biotage AB), and data analyses were performed with Power Marker software version 3.25 (Statgen, Raleigh, NC, USA).
Statistical analysis
To compare Cmax, AUClast, and AUCinf of rosuvastatin between single administration (Day 1) and concomitant administration with EGCG (Day 4 or Day 15), we performed a mixed-effects analysis, in which the subject was a random effect and the treatment was a fixed effect. The analysis was conducted with the values log transformed, and the geometric mean ratios and 90% CI were obtained by exponentiating the mean difference of the log-transformed values between treatments. To compare Tmax and CL/F of rosuvastatin between single administration (Day 1) and concomitant administration with EGCG (Day 4 or Day 15), the Wilcoxon signed rank test was used. The Mann–Whitney U test was used to compare the differences in Cmax and AUClast between concomitant administration of rosuvastatin and EGCG (Day 4 or Day 15) and single administration of rosuvastatin (Day 1) across different genotypes (wild-type vs variant genotype). The Mann-Whitney U test was also used to compare the values of Cmax and AUClast between genotypes (wild-type vs variant genotype) in each treatment period. IBM SPSS Statistics™ 21 (version 21; Datasolution Inc., Seoul, Korea) was used for statistical analysis. Data are reported as mean ± standard deviation unless indicated otherwise. The two-sided level of statistical significance was set at 0.05.
Results
Study participants
This study enrolled two male and 11 female volunteers. All the subjects completed the study. Their ages were 26.8±4.0 years and weights were 56.2±11.7 kg. The baseline characteristics are presented in Table 1.
 | Table 1 Subject baseline characteristics on start of study |
Safety profiling
Thirteen adverse events were reported in eight out of 13 subjects. The adverse events were five cases of nausea, three cases of headache, three cases of abdominal pain, and two cases of diarrhea. All the adverse events were mild in intensity and resolved without any treatment. Because all the adverse events occurred from Day 5 through 14, the period of single administration of EGCG, they were believed to be related to the dose of EGCG used in this study.
Pharmacokinetics
Table 2 summarizes the pharmacokinetic parameters of rosuvastatin alone and concomitant rosuvastatin–EGCG administration. A concomitant administration of EGCG with rosuvastatin, without any pretreatment with EGCG, significantly reduced the systemic exposure of rosuvastatin. When rosuvastatin was given alone on Day 1, its plasma concentration peaked at 4 hours after the dose. The plasma concentrations of rosuvastatin peaked at 2 hours post dose after the concomitant administration of EGCG (Day 4). There was no significant difference in Tmax between rosuvastatin–EGCG and rosuvastatin alone on Day 4. Compared with the rosuvastatin-only regimen, the single-dose concomitant administration of rosuvastatin and EGCG on Day 4 significantly reduced AUClast by 19% (geometric mean ratio 0.81, 90% CI 0.67–0.97) and AUCinf by 20% (geometric mean ratio 0.80, 90% CI 0.66–0.96), although decreases in Cmax were not statistically significant (geometric mean ratio 0.85, 90% CI 0.70–1.04) (Figure 2, Table 2).
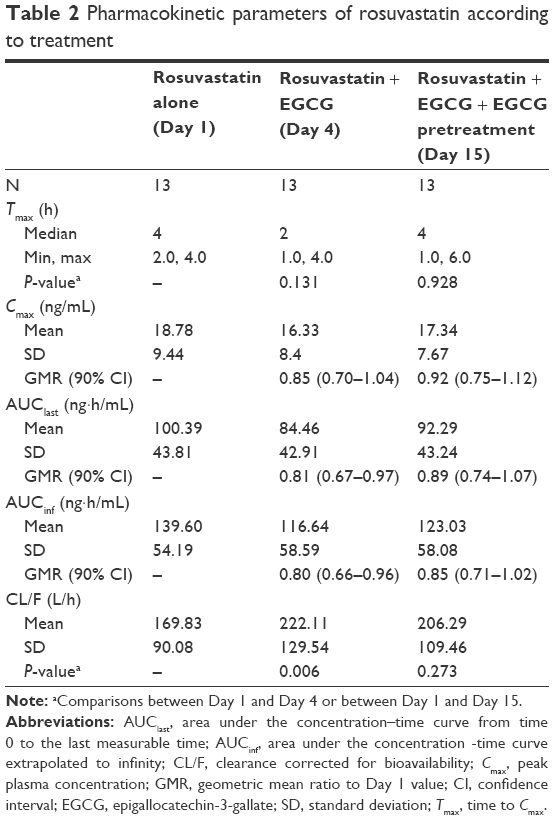 | Table 2 Pharmacokinetic parameters of rosuvastatin according to treatment |
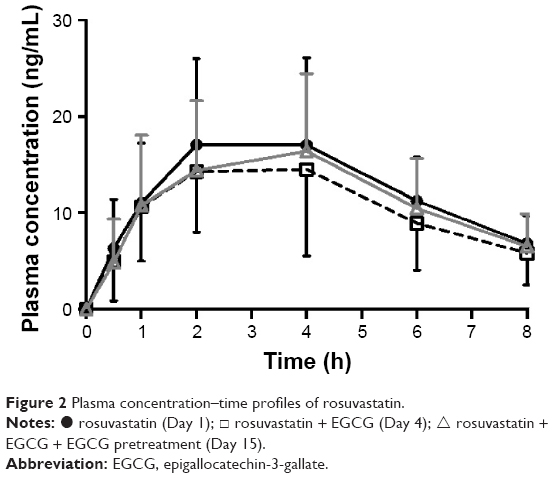 | Figure 2 Plasma concentration–time profiles of rosuvastatin. |
In contrast to the single-dose rosuvastatin–EGCG administration, a 10-day pretreatment of EGCG did not cause any significant changes in the pharmacokinetics of rosuvastatin. The plasma concentrations of rosuvastatin peaked at the median of 4 hours post dose, similar to the administration of rosuvastatin alone (Figure 2, Table 2). There was no significant difference in AUClast, AUCinf, or Cmax of rosuvastatin between rosuvastatin alone and rosuvastatin–EGCG on Day 15 (for AUClast, geometric mean ratio 0.89, 90% CI 0.74–1.07; for AUCinf, geometric mean ratio 0.85, 90% CI 0.71–1.02; and for Cmax, geometric mean ratio 0.92, 90% CI 0.75–1.12). Cmax, AUClast, and AUCinf in individual subjects are plotted in Figure 3.
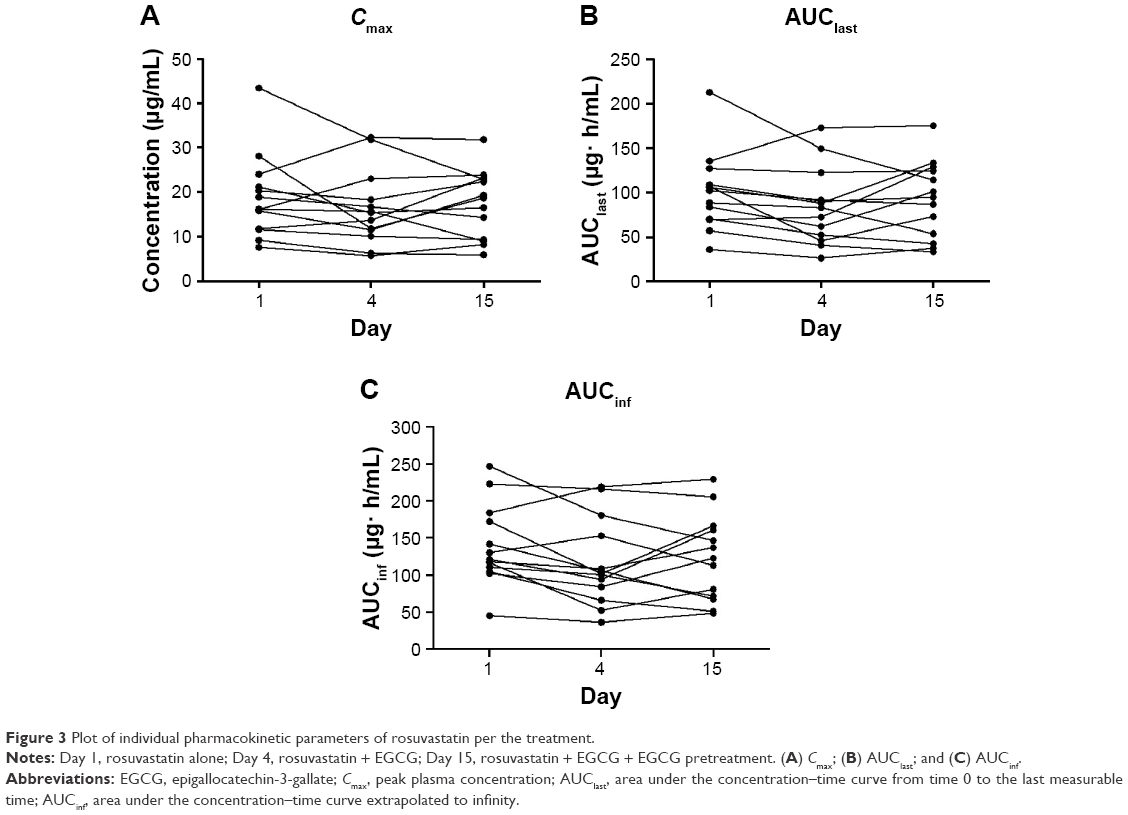 | Figure 3 Plot of individual pharmacokinetic parameters of rosuvastatin per the treatment. |
Overall, these pharmacokinetic data indicate that a single-day concomitant use of EGCG and rosuvastatin significantly reduces the systemic exposure of rosuvastatin and that a long-term pretreatment with EGCG does not significantly alter the pharmacokinetics of rosuvastatin concomitantly given with EGCG.
Effect of single-nucleotide variants of drug transporters on EGCG–rosuvastatin interaction
The pharmacokinetics of rosuvastatin is influenced by genetic polymorphisms of molecular channels responsible for its transcellular transport.16–19 To determine whether genetic variants of BCRP and OATP2B1 modify the pharmacokinetic interaction between EGCG and rosuvastatin, we compared the changes from the single administration of rosuvastatin (Day 1) in AUClast and Cmax of rosuvastatin between genotypes of the respective genes. The result is summarized in Table 3. There was no significant difference in the degree of pharmacokinetic changes between the variants of BCRP or OATP2B1. We did not test for OATP1B1 c.521T>C because this variant was found only in one subject.
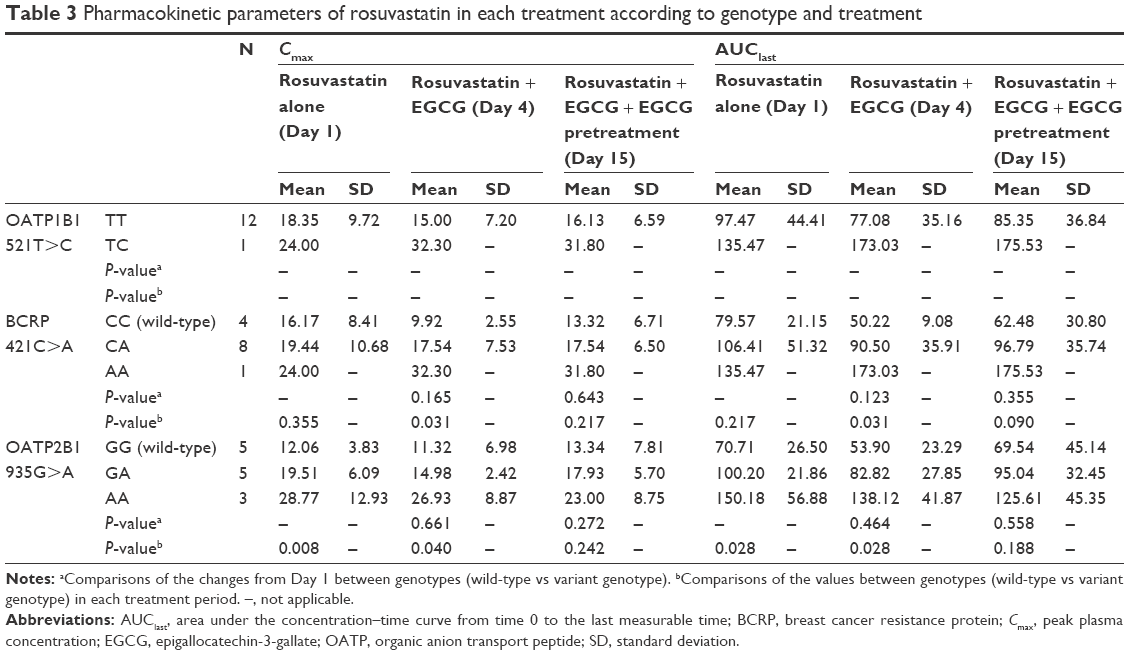 | Table 3 Pharmacokinetic parameters of rosuvastatin in each treatment according to genotype and treatment |
When we compared the AUClast and Cmax of rosuvastatin between the genotypes of BCRP within each treatment, variant genotype (CA or AA) showed significantly higher Cmax and AUClast than the wild-type (CC) on Day 4, which was consistent with previous studies.16,18 In the comparison of the AUClast and Cmax of rosuvastatin between the genotypes of OATP2B1 within each treatment period, variant genotype (GA or AA) showed significantly higher Cmax and AUClast than the wild-type (GG) on Day 1 and Day 4.
Discussion
Food or beverages have a potential to interact with a variety of medications and to alter their pharmacokinetic properties. Given their beneficial effect on cardiovascular health, green tea and its ingredients are likely to be consumed by patients with cardiovascular diseases taking multiple medications. This possibility raises concerns over the interaction between green tea and cardiovascular drugs.
In this study, we investigated the pharmacokinetics of rosuvastatin with or without concomitant administration of EGCG. A single-dose, concomitant administration of EGCG and rosuvastatin led to a significant reduction in the systemic exposure of rosuvastatin, whereas a prolonged pretreatment with EGCG did not change the pharmacokinetics of rosuvastatin. We believe that the reduction in the systemic exposure of rosuvastatin with a single dose of EGCG mainly resulted from the inhibition of intestinal uptake transporters OATP2B1 or OATP1A2, because inhibition of hepatic transporters (ie, OATP1B1 or OATP2B1) or intestinal P-gp, an efflux transporter, would have increased the plasma concentration of rosuvastatin. Regarding the pharmacokinetics of rosuvastatin after the 10-day treatment with EGCG, a plausible explanation is that EGCG inhibits both absorption and elimination of rosuvastatin, leading to no significant difference in plasma concentrations of rosuvastatin. EGCG inhibits intestinal OATP1A2 and OATP2B1, which carry out drug absorption, and hepatic OATP1B1 and OATP2B1, which move drugs from the bloodstream into the cell. Low bioavailability of EGCG23 and its accumulation with multiple-dose administration24 may be responsible for the difference in the pharmacokinetics of rosuvastatin between single-dose and pretreatment with EGCG. The plasma concentrations of EGCG after single-dose administration are not likely to be high enough to inhibit hepatic uptake; with multiple doses of pretreatment, the plasma concentrations of EGCG may have risen enough to inhibit hepatic uptake of rosuvastatin. Previous studies also support this speculation, as a prolonged use or multiple doses of green tea increased the plasma concentrations of simvastatin, probably through the inhibition of hepatic OATP1B1.1 An alternative explanation is that prolonged use of EGCG may have caused an upregulation of transporters in enterocytes, resulting in increased uptake of rosuvastatin.
Unlike previous studies on green tea–drug interaction, we used a predetermined dose of a green tea ingredient instead of green tea itself. The dose of EGCG (300 mg) used in this study amounts to at least twice as high as EGCG in a cup of green tea, given that a 3-g bag of green tea steeped in 150 mL of hot water three to eight times releases 90–150 mg of EGCG.25 Given that the IC50 value of EGCG on OATP2B1 inhibition was 101 μM (46.30 mg/L)9 and the Cmax after oral administration of EGCG 200 mg was 73.7±25.3 mg/L,26 a smaller amount of EGCG could have effectively inhibited OATP2B1. In addition, many participants in this study reported several adverse events that are believed to be caused by a high dose of EGCG. Further study is needed to determine whether a smaller amount of green tea could alter the pharmacokinetics of rosuvastatin. Meanwhile, pure EGCG may differ from green tea itself in the extent of inhibition of transporters. Roth et al9 reported that only gallated forms of catechins, namely ECG and EGCG, can inhibit OATP1A2, OATP1B1, and OATP2B1. Therefore, green tea may exert greater inhibitory effect on drug transporters than does pure EGCG because green tea contains both ECG and EGCG.
Our study is also unique in that we studied the pharmacokinetics of rosuvastatin after both single-dose and prolonged treatment of EGCG, whereas previous studies only evaluated the effect of green tea after prolonged use. Although people who are aware of its health benefit tend to drink green tea on a regular basis rather than occasionally, we also wanted to find out if occasional intake of green tea also affects the pharmacokinetics of rosuvastatin.
There are several limitations in our study. Regarding the pharmacokinetic profiling, we followed plasma concentrations of rosuvastatin up to 8 hours after administration, which may have been too short for its known half-life. However, the geometric mean ratios determined from AUCinf were comparable to those determined from AUClast, suggesting that sampling up to 8 hours after dosing was sufficient to reflect the systemic exposure of rosuvastatin, although it was not sufficient to completely define the elimination phase. In addition, our sampling method, ie, at 1, 2, and 4 hours after dosing, may not be sufficient to determine Tmax, given the fact that Tmax of rosuvastatin is typically between 2 and 4 hours after dosing. Another limitation is that we were only able to recruit 13 volunteers for this study. A future study with a larger sample size is needed to confirm our findings and better define the effect of genetic polymorphisms on green tea–rosuvastatin interaction. Because genetic polymorphisms of BCRP affect its lipid-lowering efficacy and pharmacokinetics,17,18 we tried to find out if these polymorphisms also modify the interaction between rosuvastatin and EGCG. Although the result was not statistically significant, a study involving more participants is needed to yield a definitive answer. The significant difference in AUClast and Cmax across OATP2B1 genotypes also needs to be confirmed using a large sample size.
Acknowledgments
This work was supported by Konkuk University Medical Center Research Grant 2014.
Disclosure
The authors report no conflicts of interest in this work.
References
Werba JP, Misaka S, Giroli MG, et al. Overview of green tea interaction with cardiovascular drugs. Curr Pharm Des. 2015;21(9):1213–1219. | ||
Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9(6):429–439. | ||
Amiot MJ, Riva C, Vinet A. Effects of dietary polyphenols on metabolic syndrome features in humans: a systematic review. Obes Rev. 2016;17(7):573–586. | ||
Kuriyama S, Shimazu T, Ohmori K, et al. Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: the Ohsaki study. JAMA. 2006;296(10):1255–1265. | ||
Hartley L, Flowers N, Holmes J, et al. Green and black tea for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;6:CD009934. | ||
Pang J, Zhang Z, Zheng TZ, et al. Green tea consumption and risk of cardiovascular and ischemic related diseases: a meta-analysis. Int J Cardiol. 2016;202:967–974. | ||
Wang ZM, Zhou B, Wang YS, et al. Black and green tea consumption and the risk of coronary artery disease: a meta-analysis. Am J Clin Nutr. 2011;93(3):506–515. | ||
Graham HN. Green tea composition, consumption, and polyphenol chemistry. Prev Med. 1992;21(3):334–350. | ||
Roth M, Timmermann BN, Hagenbuch B. Interactions of green tea catechins with organic anion-transporting polypeptides. Drug Metab Dispos. 2011;39(5):920–926. | ||
Knop J, Misaka S, Singer K, et al. Inhibitory effects of green tea and (−)-epigallocatechin gallate on transport by OATP1B1, OATP1B3, OCT1, OCT2, MATE1, MATE2-K and P-glycoprotein. PLoS One. 2015;10(10):e0139370. | ||
Misaka S, Yatabe J, Muller F, et al. Green tea ingestion greatly reduces plasma concentrations of nadolol in healthy subjects. Clin Pharmacol Ther. 2014;95(4):432–438. | ||
Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130(6):1793–1806. | ||
Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36(10):2014–2023. | ||
Varma MV, Rotter CJ, Chupka J, et al. pH-sensitive interaction of HMG-CoA reductase inhibitors (statins) with organic anion transporting polypeptide 2B1. Mol Pharm. 2011;8(4):1303–1313. | ||
Windass AS, Lowes S, Wang Y, Brown CD. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J Pharmacol Exp Ther. 2007;322(3):1221–1227. | ||
Lee HK, Hu M, Lui S, Ho CS, Wong CK, Tomlinson B. Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics. 2013;14(11):1283–1294. | ||
Tomlinson B, Hu M, Lee VW, et al. ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin. Clin Pharmacol Ther. 2010;87(5):558–562. | ||
Zhang W, Yu BN, He YJ, et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373(1–2):99–103. | ||
Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet. 2012;5(2):257–264. | ||
Richards JC, Lonac MC, Johnson TK, Schweder MM, Bell C. Epigallocatechin-3-gallate increases maximal oxygen uptake in adult humans. Med Sci Sports Exerc. 2010;42(4):739–744. | ||
Ullmann U, Haller J, Bakker GC, Brink EJ, Weber P. Epigallocatechin gallate (EGCG) (TEAVIGO) does not impair nonhaem-iron absorption in man. Phytomedicine. 2005;12(6–7):410–415. | ||
Lee JH, Chong WS, Park JK, Park TS, Chae SW, Kim MG. Rooibos™: automated schedule broadcast software for clinical pharmacology studies. Transl Clin Pharmacol. 2016;24:127–131. | ||
Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, elimination of tea polyphenols in rats. Drug Metab Dispos. 1997;25(9):1045–1050. | ||
Chow HH, Cai Y, Hakim IA, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res. 2003;9(9):3312–3319. | ||
Yang CS, Lambert JD, Ju J, Lu G, Sang S. Tea and cancer prevention: molecular mechanisms and human relevance. Toxicol Appl Pharmacol. 2007;224(3):265–273. | ||
Chow HH, Cai Y, Alberts DS, et al. Phase I pharmacokinetic study of tea polyphenols following single-dose administration of epigallocatechin gallate and polyphenon E. Cancer Epidemiol Biomarkers Prev. 2001;10(1):53–58. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.