")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Edaravone and Acetovanillone Upregulate Nrf2 and PI3K/Akt/mTOR Signaling and Prevent Cyclophosphamide Cardiotoxicity in Rats
Authors Hassanein EHM, Abd El-Ghafar OAM , Ahmed MA, Sayed AM, Gad-Elrab WM, Ajarem JS, Allam AA, Mahmoud AM
Received 17 September 2020
Accepted for publication 11 November 2020
Published 30 November 2020 Volume 2020:14 Pages 5275—5288
DOI https://doi.org/10.2147/DDDT.S281854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Emad HM Hassanein,1 Omnia AM Abd El-Ghafar,2 Marwa A Ahmed,3 Ahmed M Sayed,4 Wail M Gad-Elrab,5 Jamaan S Ajarem,6 Ahmed A Allam,7 Ayman M Mahmoud7,8
1Department of Pharmacology and Toxicology Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt; 2Department of Pharmacology and Toxicology, Faculty of Pharmacy, Nahda University, Beni-Suef, Egypt; 3Department of Pharmacology, Faculty of Medicine, Assiut University, Assiut, Egypt; 4Biochemistry Laboratory, Chemistry Department, Faculty of Science, Assiut University, Assiut, Egypt; 5Human Anatomy & Embryology Department Faculty of Medicine, Al-Azhar University, Assiut, Egypt; 6Zoology Department, College of Science, King Saud University, Riyadh, Saudi Arabia; 7Zoology Department Faculty of Science, Beni-Suef University, Beni-Suef, Egypt; 8Biotechnology Department, Research Institute of Medicinal and Aromatic Plants, Beni-Suef University, Beni-Suef, Egypt
Correspondence: Ayman M Mahmoud
Physiology Division, Zoology Department, Faculty of Science, Beni-Suef University, Salah Salim Street, Beni-Suef 62514, Egypt
Tel +20 1278355478
Email [email protected]
Jamaan S Ajarem
Zoology Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
Email [email protected]
Introduction: Cyclophosphamide (CP) causes redox imbalance and its use is associated with marked cardiotoxicity that limits its clinical applications. The present study investigated the protective effects of acetovanillone (AV) and edaravone (ED) against CP-induced oxidative stress and cardiac damage, emphasizing the role of PI3K/Akt/mTOR and Nrf2 signaling.
Materials and Methods: Rats received either AV (100 mg/kg) or ED (20 mg/kg) orally for 10 days and CP (200 mg/kg) on day 7. At day 11, the rats were sacrificed, and samples were collected for analysis.
Results: AV and ED ameliorated serum troponin I, CK-MB, LDH, AST and ALP, and prevented cardiac histological alterations in CP-intoxicated rats. Both treatments decreased cardiac lipid peroxidation and enhanced GSH, SOD and cytoglobin in CP-induced rats. AV and ED downregulated Keap1, whereas increased the expression of PI3K, Akt, mTOR and Nrf2 in the heart of rats received CP. Additionally, the binding modes of AV and ED to Keap1 were pinpointed in silico using molecular docking simulations.
Conclusion: AV and ED prevent CP cardiotoxicity by attenuating oxidative stress and tissue injury, and modulating cytoglobin, and PI3K/Akt/mTOR and Keap1/Nrf2 signaling. Therefore, AV and ED may represent promising agents that can prevent cardiac injury in patients receiving CP.
Keywords: chemotherapy, acetovanillone, edaravone, oxidative stress, cardiotoxicity, Nrf2, mTOR
Introduction
Cyclophosphamide (CP) is a cytotoxic alkylating phosphoramide ester extensively used as an anti-neoplastic and immunosuppressive agent in organ transplantation and systemic lupus erythematosus. CP has the ability to modulate DNA synthesis and prevent cellular proliferation.1,2 Despite the therapeutic effects of CP, pulmonary injury, hepatotoxicity, nephrotoxicity, bone marrow suppression, cardiotoxicity, genotoxicity and other adverse effects are associated with its use. The incidence of CP cardiotoxic effects remains high and was found to be dose-related.3 Exposure to high doses of CP leads to acute cardiotoxic effects, including extravasation of toxic metabolites, myocyte damage and diastolic contractile dysfunction.4 Tachyarrhythmias, myocarditis, hypotension, pericardial disease and heart failure represent the common manifestations of CP cardiotoxicity. These manifestations typically present within the first 48 h after the administration of CP.5,6 Acute heart failure occurs in 7–33% of patients receiving more than 150 mg/kg CP.7 Numerous studies have shown a surplus increase in reactive oxygen species (ROS) following CP administration.8–11 Excess ROS can damage several cellular biomolecules, including proteins, lipids and DNA, resulting in cell death. Additionally, acrolein, the toxic metabolite of CP, can induce toxicity through altering the antioxidant defense system manifested by the depletion of cellular reduced glutathione (GSH) and superoxide dismutase (SOD).12 Subsequently, a massive increase in lipid peroxidation (LPO), nitric oxide (NO) and protein carbonyls occurs as a result of CP administration, culminating in tissue injury.8–11 Such effects can interfere with the heart regulatory mechanisms and increase the risk of cardiovascular diseases. Thus, agents with radical-scavenging activity could be effective against CP-induced oxidative injury and cardiotoxicity.
The nuclear factor erythroid 2-related factor 2 (Nrf2) is an antioxidant intracellular defense system widely distributed in various tissues. Under physiological conditions, Nrf2 is sequestered in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1) which facilitates the ubiquitination and subsequent proteolysis of Nrf2. Upon exposure to ROS or electrophilic chemicals, Nrf2 is released from Keap1, translocates into the nucleus and binds to the antioxidant response element (ARE), resulting in coordinated antioxidant and anti-inflammatory response by stimulating the expression of many cytodefensive genes.13 Studies have demonstrated a decline in Nrf2 signaling in CP-intoxicated rodents.8–11 Therefore, activation of Nrf2 signaling is an effective strategy to counteract CP-induced oxidative stress and cardiotoxicity and other adverse effects. In this context, Nrf2 activation enhanced antioxidant defenses and attenuated oxidative injury, inflammation, and cell death in the liver and kidney of CP-intoxicated rats,8–11 making it a valuable therapeutic target to prevent cardiac injury associated with the use of CP. Moreover, the PI3K/Akt/mTOR is another signaling pathway that has been reported to impede cell survival process upon its downregulation.14,15 Dysregulation of this signaling has also been implicated in cardiomyopathy.16
Acetovanillone (AV) is a small molecule extracted from Picrorhiza kurroa and has been reported to exert antioxidant, anti-inflammatory and anti-apoptosis activities.17 It is a potent NADPH oxidase (NOX) inhibitor that attenuates excessive ROS generation in different disease models.18–21 The beneficial effects of AV have been attributed to its ability to suppress ROS generation via inhibiting NOX activity and enhancement of the cellular antioxidant defenses.21,22 Edaravone (ED; 3-methyl-1-phenyl-2-pyrazolin-5-one) is a low molecular weight agent with potent radical-scavenging properties that protect the cells from death.23 ED has a neuroprotective activity mediated via its ability to suppress oxidative stress,24,25 and protected against bleomycin-induced lung injury26 and amitriptyline-induced cardiotoxicity27 in murine models. Given the potent antioxidant properties of AV and ED, this study explored their protective effect against CP cardiotoxicity, pointing to the possible involvement of PI3K/Akt/mTOR and Nrf2 signaling.
Materials and Methods
Drugs and Chemicals
AV, ED and CP were obtained from Sigma-Aldrich (USA). Assay kits of cardiac troponin I (CTnI) was purchased from Lifespan BioSciences (USA). Creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) kits and TRIzol were supplied by Thermo Fisher Scientific (USA). Aspartate aminotransferase (AST) and alkaline phosphatase (ALP) kits were purchased from BioDiagnostics (Cairo, Egypt). Primary antibodies for Nrf2, cytoglobin, PI3K, Akt and mTOR were obtained from Santa Cruz (USA). Primers were purchased from Vivantis Technologies (Malaysia) and SYBR green master mix was obtained from Fermentas (USA).
Experimental Animals and Treatments
Forty-eight male Wistar rats weighing 180–210 g were purchased from the central animal house, Faculty of Medicine, Assiut University (Egypt). The Animals were housed in controlled temperature, humidity and 12 h light‐dark cycles, and supplied a standard laboratory diet of known composition and water ad libitum. All experiments were conducted in line with the guidelines of the National Institutes of Health (NIH publication No. 85–23, revised 2011) and approved by the Animal Care and Use Committee of Assiut University (IRB No. 17,300,462).
The animals were allocated into six groups (n = 8) as follows:
Group I (Control): received 0.5% carboxymethyl cellulose (CMC) via oral gavage for 10 days and a single intraperitoneal (i.p.) injection of saline at day 7.
Group II (AV): received 100 mg/kg AV19 for 10 days.
Group III (ED): Rats received 20 mg/kg ED28 for 10 days.
Group IV (CP): received 0.5% CMC for 10 days and a single dose of CP (200 mg/kg) at day 7.29
Group V (AV + CP): received 100 mg/kg AV for 10 days and a single dose of CP (200 mg/kg) at day 7.
Group VI (ED + CP): received 20 mg/kg ED for 10 days and a single dose of CP (200 mg/kg) at day 7.
AV and ED were dissolved in 0.5% CMC and administered orally, and CP was injected i.p. At day 11, all rats were sacrificed under pentobarbital anesthesia and blood was collected via cardiac puncture. Serum was prepared by centrifugation at 4000 rpm for 10 min. The animals were immediately dissected, and the heart was removed, washed in cold phosphate-buffered saline (PBS) and samples were fixed in neutral buffered formalin. Other samples were kept frozen at −80°C in RNAlater whereas others were homogenized in cold PBS (10% w/v), centrifuged at 10,000 rpm for 10 min at 4°C and the supernatant was collected and kept frozen at −80°C.
Measurement of Cardiac Function Biomarkers
Serum CTn1 was estimated by a specific ELISA kit and the activities of LDH, CK-MB, AST and ALP were determined following the manufacturers’ instructions.
Histology and Immunohistochemistry
Heart specimens, fixed in 10% neutral buffered formalin for 48 h, were dehydrated, cleared, embedded in paraffin, sectioned (4–5 µm) and stained with hematoxylin and eosin (H&E) stain according to the previously described method.30 The sections were examined blindly under light microscope (Leica Q 500 MCO, Germany).
Other sections were used for the immunohistochemical staining of PI3K, Akt, mTOR, Nrf2 and cytoglobin. Briefly, the sections were deparaffinized and treated with 3% hydrogen peroxide (H2O2) for 10 min to inactivate endogenous peroxidases, heated in 10 mM citrate buffer at 121°C for 30 min for antigen retrieval and then blocked by 5% bovine serum albumin (BSA) in Tris-buffered saline. The slides were incubated with the primary antibodies overnight at 4°C and then washed and incubated with horseradish peroxidase (HRP)‐conjugated secondary antibodies for 30 min at room temperature. The sections were subjected to diaminobenzidine staining and hematoxylin counterstaining. The percentage of area occupied by brown color was measured in randomly selected six fields in each slide using ImageJ (NIH, USA).
Determination of LPO, GSH and SOD
Malondialdehyde (MDA), a marker of LPO, was assayed in the heart homogenate as previously described.31 The antioxidants GSH and SOD were assayed according to the methods of Ellman32 and Marklund and Marklund,33 respectively.
Gene Expression Analysis
The effect of AV and ED on mRNA abundance of Keap1 and Nrf2 genes was determined by qRT-PCR using the ABI 7500 real-time PCR system (Applied Biosystems, USA). Briefly, total RNA was isolated using TRIzol reagent. The isolated RNA was quantified using a nanodrop and reverse transcribed into cDNA. The obtained cDNA was amplified using SYBR green master mix and the primers listed in Table 1 in a total reaction volume of 20 μL. The amplification data were analyzed using the 2−ΔΔCt method34 and normalized to GAPDH as a housekeeping gene.
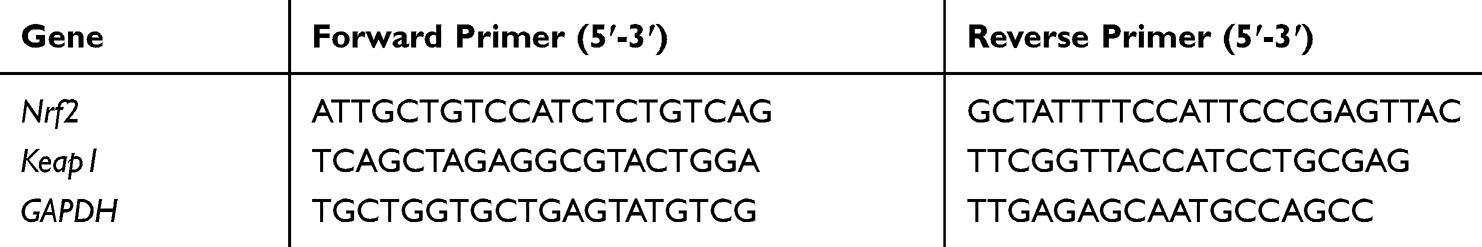 |
Table 1 Primer Used for qRT-PCR |
Molecular Docking
The binding mode of AV and ED with Keap1 was determined by molecular docking using Autodock vina 1.5.6.35 The crystal structure of Keap1 and its (1S,2R)‐2‐[(1S)‐1‐[(1,3‐dioxo‐2,3‐dihydro‐1H‐isoindol‐2‐yl)methyl]‐1,2,3,4‐tetrahydroisoquinolin‐2-carbonyl]cyclohexane‐1‐carboxylic acid (compound (S, R, S)) was obtained from protein data bank with PDB ID: 4l7b.36 The water molecules and ligand were removed and the binding pocket of Keap1 was used for docking against AV and ED. PyMoL 1.7.6 software was used to visualize the docking models of AV and ED with Keap1.
Statistical Analysis
The results were expressed as mean ± SEM and all the statistical comparisons were performed using one-way ANOVA followed by Tukey’s post-hoc on GraphPad Prism 7.0 (GraphPad software, USA). Statistical significance was considered for a P value less than 0.05.
Results
AV and ED Ameliorate Heart Injury in CP-Intoxicated Rats
The ameliorative effect of AV and ED on CP cardiotoxicity was assessed through determination of CTn1, CK-MB, LDH, AST and ALP (Figure 1). CP induced a significant elevation of serum CTn1 (Figure 1A), CK-MB (Figure 1B), LDH (Figure 1C), AST (Figure 1D) and ALP (Figure 1E) when compared with the control rats (P<0.001). Oral administration of either AV or ED decreased serum CTn1, CK-MB, LDH, AST and ALP in CP-intoxicated rats. Treatment with ED decreased serum CK-MB (Figure 1B) and LDH (Figure 1C) significantly (P<0.001) in CP-administered rats when compared with AV. Normal rats received either AV or ED exhibited non-significant changes in all assayed heart function markers (Figure 1).
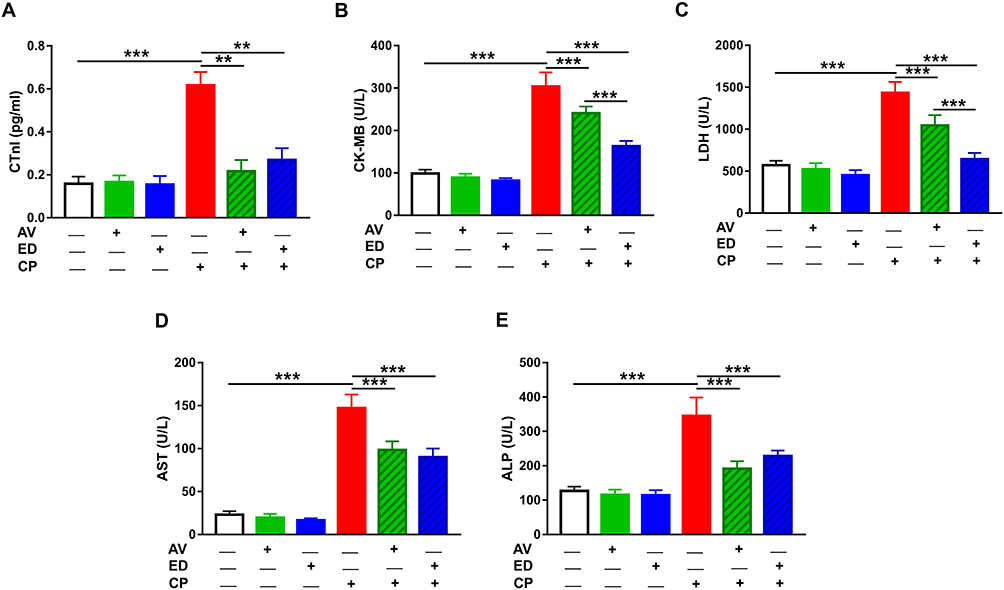 |
Figure 1 AV and ED ameliorate heart function in CP-intoxicated rats. Treatment with AV and ED decreased serum (A) CTnI, (B) CK-MB, (C) LDH, (D) AST and (E) ALP in CP-intoxicated rats. Data are mean ± SEM, (n = 8). **P<0.01 and ***P<0.001. |
AV and ED Prevent CP-Induced Heart Injury in Rats
The cardioprotective effect of AV and ED on CP-induced cardiac injury was further confirmed by the histological findings. Examination of H&E-stained sections revealed well-organized and intact cardiomyocytes with normal nuclei and cytoplasmic striation in the control (Figure 2A), and AV- (Figure 2B) and ED-supplemented rats (Figure 2C). Administration of CP resulted in histological alterations, including degenerative changes, fragmented myofibrils, nuclear pyknosis, increased intercellular spaces between adjacent cardiomyocytes and many congested blood vessels (Figure 2D–F). CP-intoxicated rats treated with AV (Figure 2G) or ED (Figure 2H) exhibited an improvement in the histological architecture of the heart with fewer degenerative changes and congestions (Table 2).
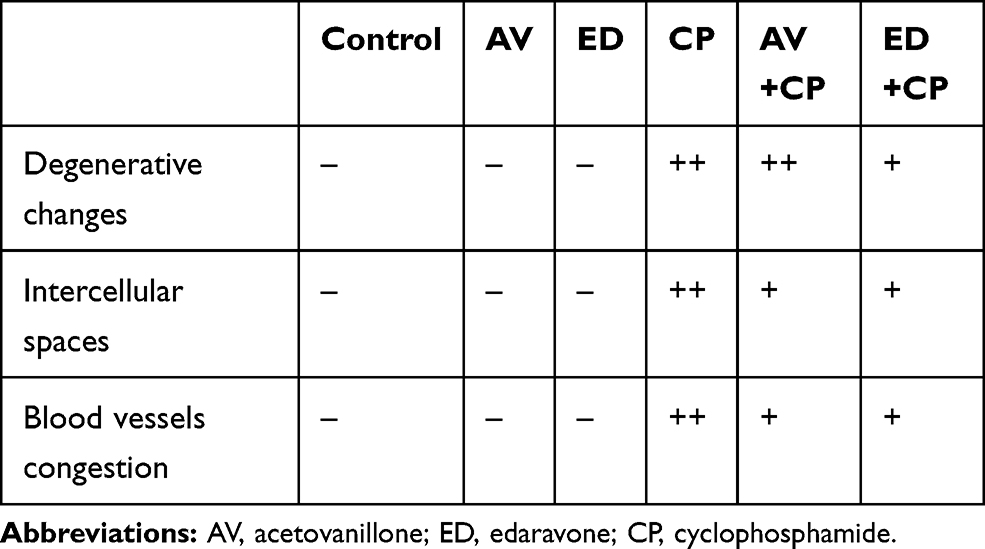 |
Table 2 Heart Histopathological Score Lesions |
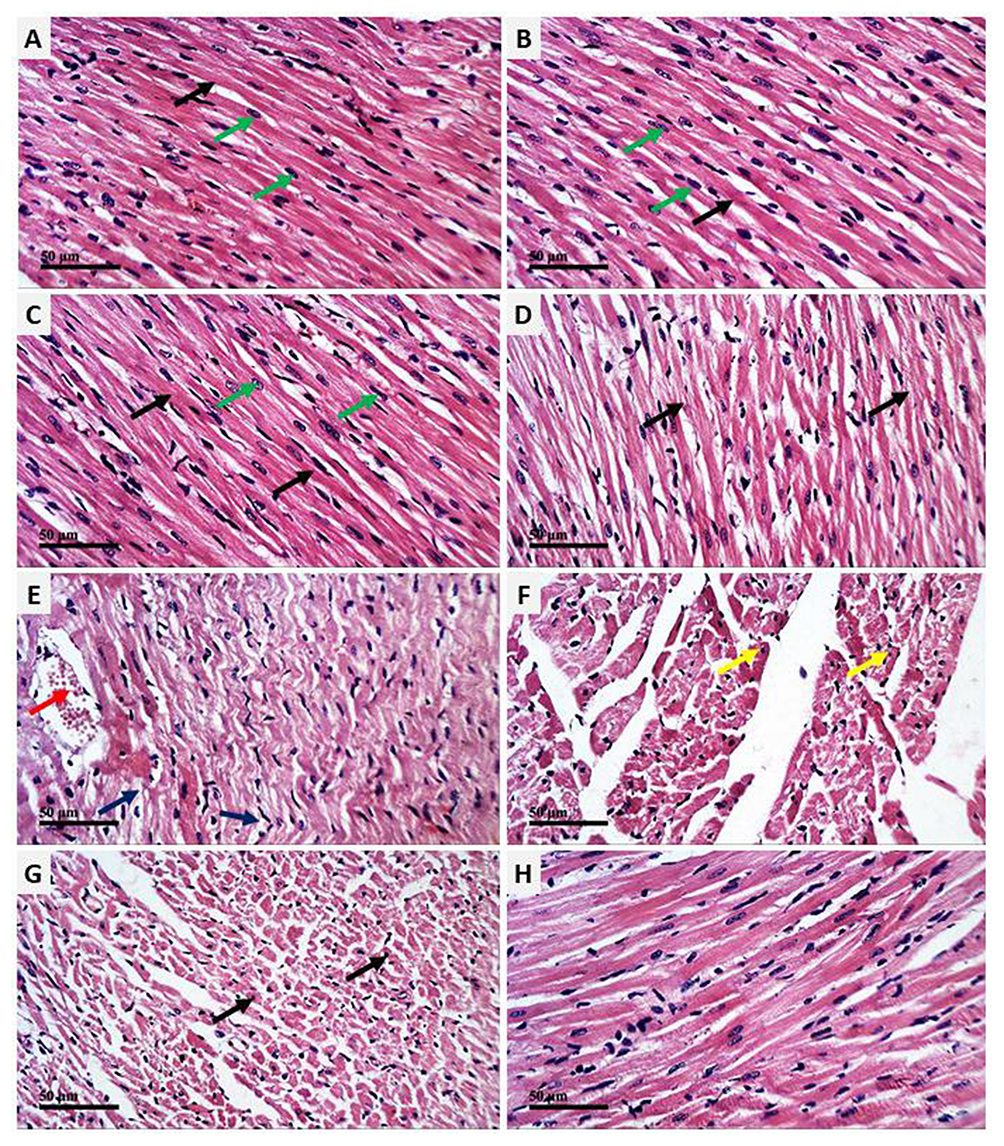 |
Figure 2 AV and ED prevent CP-induced cardiac injury in rats. Photomicrographs of the heart tissue sections of control (A) and rats treated with AV (B) and ED (C) showing normal structure of the cardiomyocytes with normal nuclei (green arrow) and cytoplasmic striation (black arrow), CP-intoxicated rats (D–F) showing degenerative changes (blue arrow), fragmented myofibrils (black arrow), nuclear pyknosis (yellow arrow), increased intercellular spaces between adjacent cardiomyocytes and many congested intermuscular blood vessels (red arrow), and CP-intoxicated rats treated with AV (G) and ED (H) showing improved histological architecture of the heart with fewer degenerative changes and congestions (arrow). (H&E, Scale bar = 50 µm). |
AV and ED Attenuate Cardiac Oxidative Stress in CP-Induced Rats
Given the role of oxidative stress in mediating the deleterious effects of CP, we evaluated the effects of AV and ED on cardiac MDA and the antioxidants GSH and SOD in the heart of rats (Figure 3). MDA showed a significant increase in heart of CP-treated rats when compared with the control group (Figure 3A; P<0.001). In contrast, GSH and SOD were significantly declined in the heart of CP-intoxicated rats as represented in Figure 3B and C, respectively. Oral supplementation of either AV or ED decreased cardiac MDA and increased GSH and SOD in CP-induced rats. Of note, both treatment agents did not alter cardiac redox balance in normal rats.
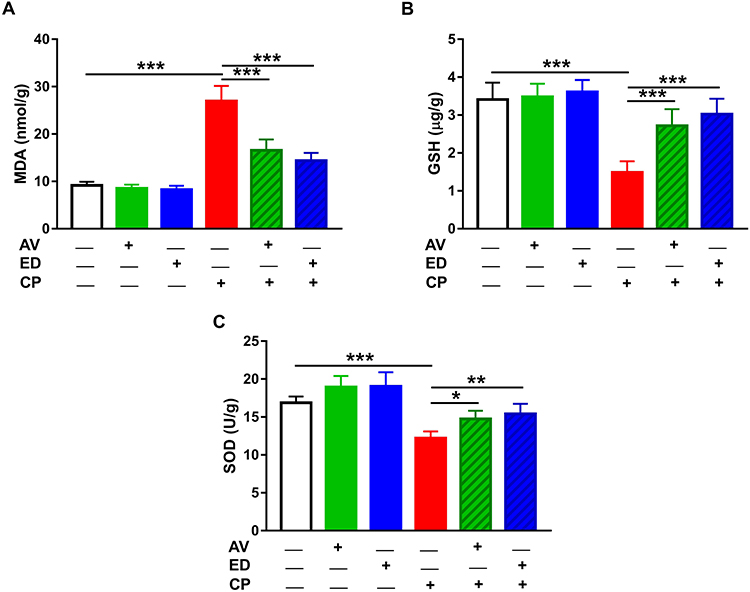 |
Figure 3 AV and ED attenuate cardiac oxidative stress in CP-induced rats. AV and ED decreased cardiac MDA (A) and increased GSH (B) and SOD (C) in CP-intoxicated rats. Data are mean ± SEM, (n = 8). *P<0.05, **P<0.01 and ***P<0.001. |
AV and ED Upregulate Keap1/Nrf2 Signaling Pathway in the Heart of CP-Induced Rats
The effect of AV and ED on the Nrf2 signaling was assessed by evaluating the expression levels of Keap1 and Nrf2 in the heart of normal and CP-intoxicated rats (Figure 4). In addition, molecular docking simulations were performed to explore the binding mode of AV and ED with Keap1 (Figure 5). The results showed a significant increase in the mRNA abundance of cardiac Keap1 (Figure 4A) in CP-intoxicated rats when compared with the control group (P<0.001). While AV and ED did not affect the expression levels of AV and ED in normal rats, both treatments downregulated Keap1 mRNA in rats that received CP. Nrf2 gene and protein expression levels exhibited a different pattern as shown in Figure 5B–D. Nrf2 mRNA and protein were decreased significantly in the heart of rats administered with CP. Treatment with AV and ED resulted in a significant increase in Nrf2, both mRNA and protein, in the heart of both normal and CP-intoxicated rats.
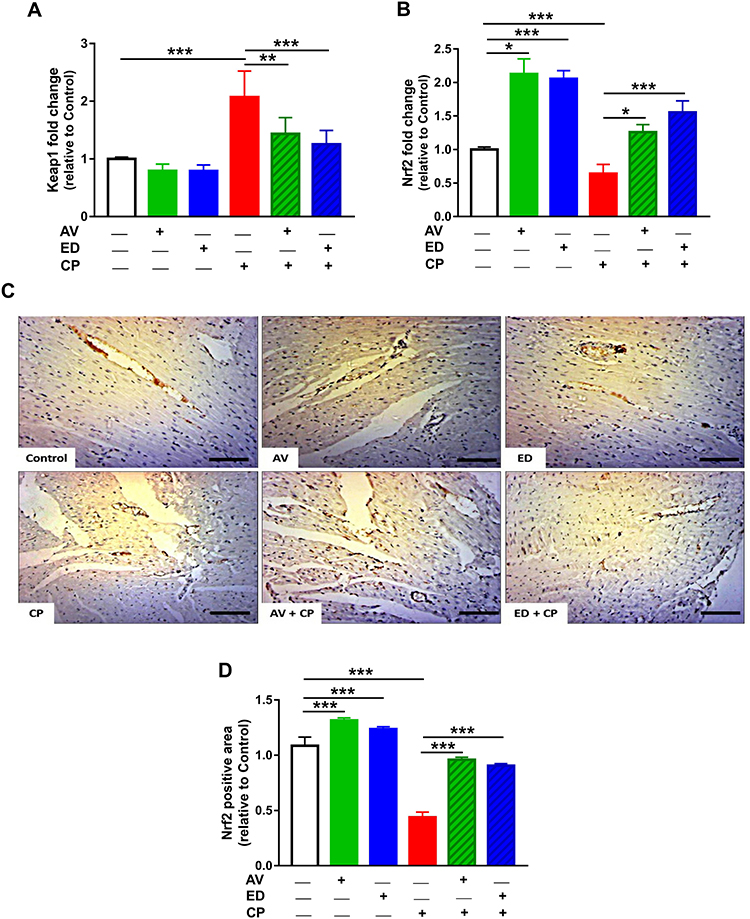 |
Figure 4 AV and ED upregulate Keap1/Nrf2 signaling pathway in the heart of CP-induced rats. AV and ED downregulated Keap1 mRNA (A) and increased Nrf2 gene (B) and protein expression (C, D) in rats [Scale bar = 100µm]. Data are mean ± SEM, (n = 8). *P<0.05, **P<0.01 and ***P<0.001. |
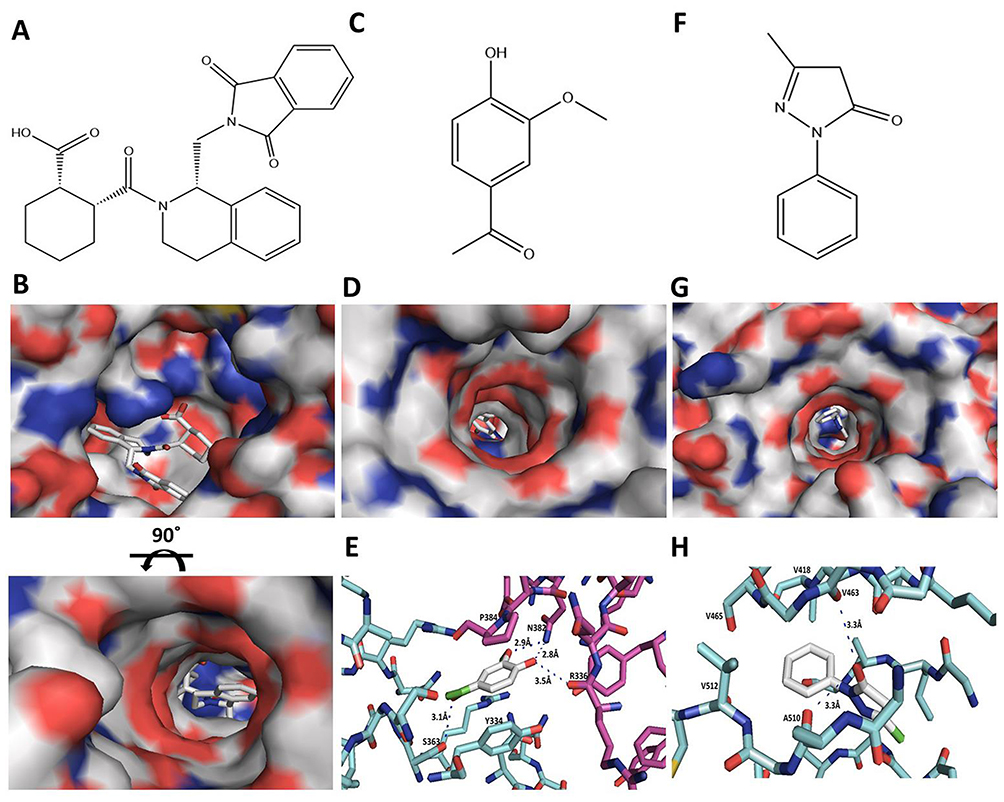 |
Figure 5 Molecular interactions of AV and ED with the Keap1. (A, B) Chemical structure of SRS (A) and surface map of the SRS complex with Keap1 (B). (C–E) Chemical structure of AV (C) surface map of the docking model of AV with the binding site of Keap1 (D) and the amino acid residues on Keap1 within a 5 A° contact radius of AV (E). (F–H) Chemical structure of ED (F) surface map of the docking model of ED with the binding site of Keap1 (G) and the amino acid residues on Keap1 within a 5 A° contact radius of ED (H). |
Molecular docking analysis showed that AV docks into the determined active site of Keap1 and forms four hydrogen bonds with the binding site (R336, S363 and N382) of Keap1 at the dimeric interface (Figure 5). The benzene ring of AV exhibits hydrophobic interaction with the side chains of P384 and Y334. The binding affinity was −5.96±0.27 kcal mol−1. ED forms two hydrogen bonds with V463 and A510 of Keap1. The benzene ring of ED showed a tight fit in the hydrophobic pocket of Keap1 (V418, V463, V465 and V512). The binding affinity was −6.23±0.10 kcal mol−1 (Figure 5).
AV and ED Upregulate PI3K/Akt/mTOR Signaling in the Heart of CP-Induced Rats
Immunohistochemical staining was used to evaluate the effect of AV and ED on the expression levels of PI3K, Akt and mTOR in the heart of both normal and CP-intoxicated rats. Oral administration of AV and ED upregulated cardiac PI3K (Figure 6A and B), Akt (Figure 6C and D) and mTOR (Figure 6E and F) in normal rats. CP administration significantly decreased the expression levels of PI3K, Akt and mTOR in the heart tissue, an effect that was significantly reversed in rats treated with either AV or ED. The effect of ED on PI3K and TOR levels in CP-intoxicated rats was significant when compared with AV (P<0.001).
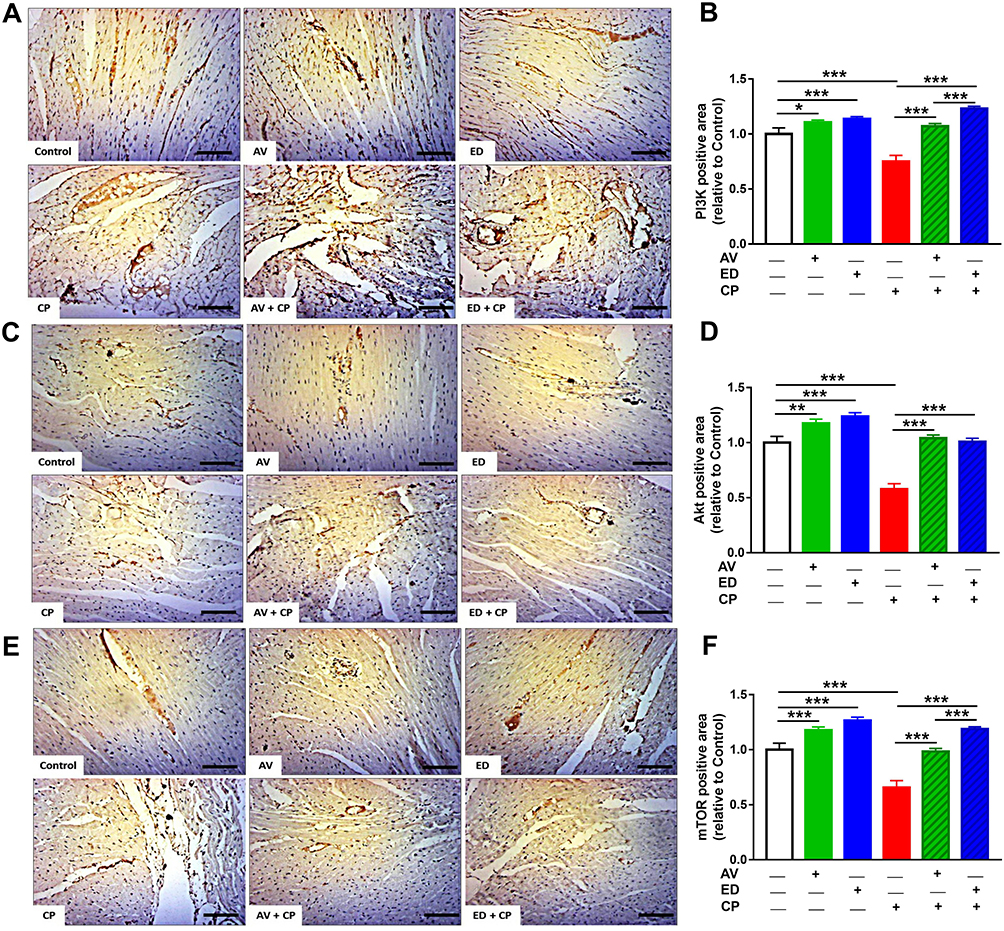 |
Figure 6 AV and ED upregulate PI3K/Akt/mTOR signaling in the heart of CP-induced rats. Treatment with AV or ED increased the expression of PI3K (A, B), Akt (C, D) and mTOR (E, F) in the heart of rats. [Scale bar = 100µm]. Data are mean ± SEM, (n = 8). *P<0.05, **P<0.01 and ***P<0.001. |
AV and ED Increase Cardiac Cytoglobin Expression in CP-Induced Rats
Treatment with AV significantly increased the expression of cardiac cytoglobin in the heart of normal rats when compared with the control (Figure 7; P<0.001). CP administration resulted in a significant downregulation of cardiac cytoglobin expression. On the other hand, CP-intoxicated rats treated with AV or ED exhibited a significant increase in cardiac cytoglobin. When compared to ED, AV increased cardiac cytoglobin significantly (P<0.01) in CP-intoxicated rats.
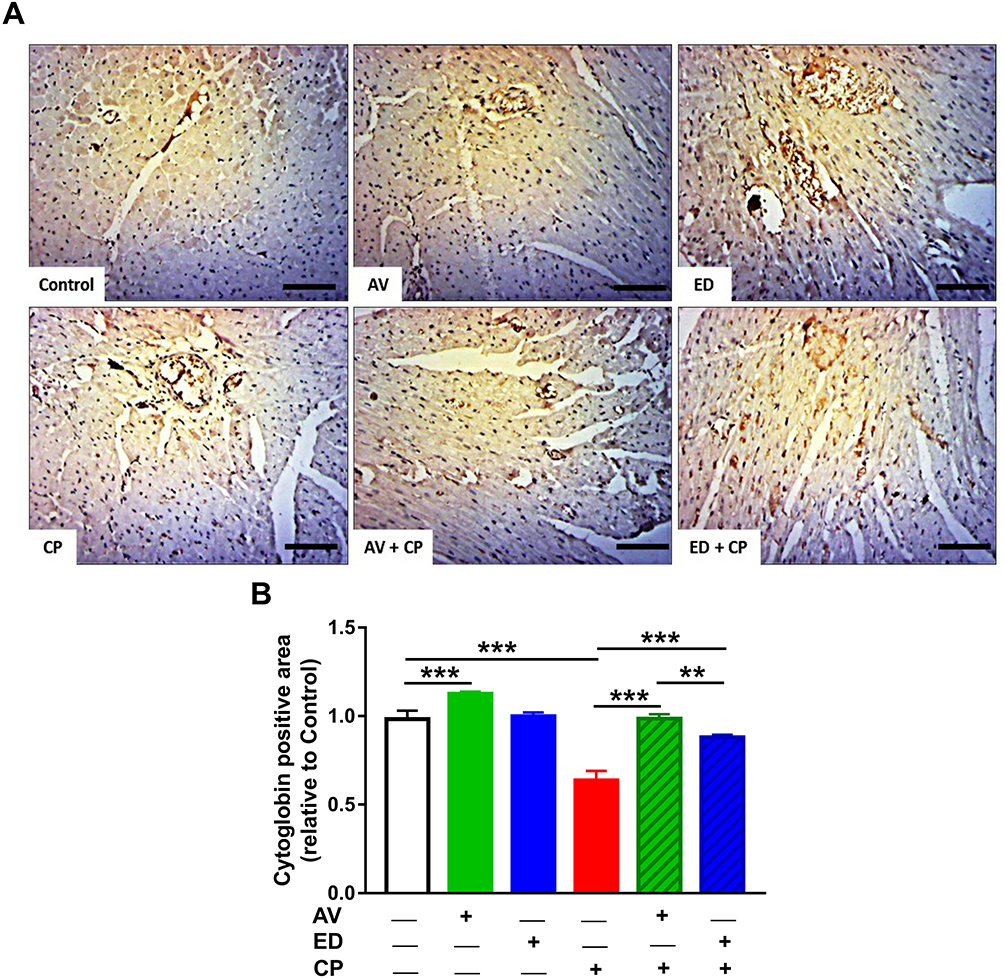 |
Figure 7 AV and ED increase cardiac cytoglobin expression in CP-induced rats. Treatment with AV or ED increased the expression of cytoglobin in the heart of rats. (A) Representative images showing immunohistochemical staining of cytoglobin in the heart of rats. [Scale bar = 100µm]. (B) image analysis of the expression of cytoglobin in the heart of rats. Data are mean ± SEM, (n = 8). **P<0.01 and ***P<0.001. |
Discussion
Cardiotoxicity is a life-threatening complication of chemotherapy and CP is well known to cause cardiac adverse effects.3,37 Given the role of oxidative stress in mediating its adverse effects, we investigated the protective effects of AV and ED on CP cardiotoxicity in rats with an emphasis on PI3K/Akt/mTOR and Nrf2 signaling.
CP administration caused cardiac injury manifested by the increased serum CTnI, CK-MB, LDH, AST and ALP along with the histological alterations, including degenerative changes, fragmented myofibrils, nuclear pyknosis and congested blood vessels. Similar findings have been reported in previous studies where CTnI, CK-MB and LDH were increased in rodents following the administration of CP.38,39 Treatment of the CP-intoxicated rats with AV or ED ameliorated the circulating levels of all assayed cardiac function markers and effectively prevented the histological alterations in the heart tissue. The cardioprotective effects of AV and ED have been demonstrated in different studies. For instance, AV showed protective effects against cisplatin cardiotoxicity,40 isoproterenol-induced cardiac hypertrophy41 and diabetic cardiomyopathy.20 ED has also shown protective effects in animal models of methamphetamine-,42 valproic acid-43 and doxorubicin-induced cardiotoxicity.44 In these studies, the cardioprotective effects of AV and ED were attributed mainly to their ability to attenuate oxidative stress. Therefore, we investigated the effect of AV and ED on LPO and the antioxidants GSH and SOD in the heart of CP-intoxicated rats. The LPO marker MDA was significantly increased following CP administration whereas GSH and SOD were declined, demonstrating oxidative stress. The cellular mechanism of CP toxicity is so far mediated by oxidative stress caused by excessive ROS generation and the principle alkylating metabolites phosphoramide mustard and acrolein.12 Acrolein binds covalently to lipids and proteins, leading to the formation of free radicals.12 In addition, elevated ROS levels induce cell injury through oxidizing the cellular macromolecules and depleting antioxidant defenses by disrupting their protein confirmation.45 Accordingly, ROS and MDA were increased and cellular antioxidants were decreased in different tissues of rats received CP,8,9,11,46 adding support to the role of oxidative stress in the adverse effects of CP. AV and ED supplementation effectively decreased LPO and boosted the antioxidant defenses in the heart of CP-intoxicated rats. The ability of AV and ED to attenuate oxidative stress in different models of cardiac injury has been previously reported.20,40–44 In cisplatin-intoxicated rats, oral supplementation of 600 mg/L AV for 5 days decreased cardiac LPO and increased GSH.40 AV increased cardiac GSH in isoproterenol-induced rats41 and serum SOD and catalase in diabetic rats.20 Similarly, ED decreased ROS and MDA and increased the activity of antioxidant enzymes in the heart of valproic acid-43 and methamphetamine-induced rats.42 These studies along with the findings of this study demonstrate the potent antioxidant activities of both AV and ED.
NOX is a major enzymatic source of ROS generation in the cardiovascular system.47 NOX-derived ROS, such as superoxide, contributes to cardiovascular pathogenesis through activating other enzymatic systems and triggering ROS production. Besides the oxidation of cellular macromolecules and depletion of antioxidants, ROS produced by NOX can cause mitochondrial DNA damage and oxidation of components of the membrane permeability transition pore and opening of the redox-sensitive mitochondrial ATP-sensitive K+ channel, resulting in mitochondrial uncoupling and more ROS production.47–49 Given the role of NOXs in cardiovascular disease, it is noteworthy assuming that inhibition of NOX-derived ROS plays a role in the cardioprotective effect of AV and ED.
Nrf2, a redox-sensitive transcription factor controlling the expression of antioxidant enzymes, has been reported to decrease in different tissues of CP-intoxicated rats.8–11 In addition, Nrf2 deficiency exaggerated cardiotoxicity induced by the anticancer drug doxorubicin in mice.50 In contrast, upregulation of Nrf2 protected against CP hepato- and nephrotoxicity,8–11 and negatively regulated oxidative stress and cardiac dysfunction induced by transverse aortic constriction.51 We assumed that Nrf2 is involved in the cardioprotective effect of AV and ED. Therefore, we evaluated the effect of AV and ED on Keap1 and Nrf2 expression in the heart of rats and conducted an in silico investigation of their binding modes with Keap1. mRNA abundance of Keap1 was increased whereas Nrf2 gene and protein expression levels were downregulated in the heart of CP-intoxicated rats. Although activated by oxidative stress, Nrf2 was declined in CP-intoxicated rats which could be attributed to sustained and prolonged ROS generation. Accordingly, previous studies have demonstrated suppressed Nrf2 signaling under conditions of excessive ROS generation and oxidative stress both in vitro and in vivo.52–54 Both AV and ED decreased Keap1 mRNA and upregulated the expression of Nrf2 in the heart of CP-induced rats, pointing to the role of Keap1/Nrf2 signaling in their cardioprotective effect. These findings added support to previous studies showing the possible role of Nrf2 upregulation in the beneficial effects of AV and ED against different disease conditions. In rat models of cisplatin cardiotoxicity40 and quinolinic acid neurotoxicity,55 AV has been reported to upregulate Nrf2 mRNA levels and suppress oxidative stress. ED has also shown an ability to upregulate Nrf2 signaling and subsequently attenuate oxidative injury in experimental models of traumatic brain injury56 and chlorpyrifos neurotoxicity.57 Our study introduces new information that Nrf2 signaling mediated, at least in part, the protective effects of AV and ED against CP cardiotoxicity. Interestingly, both agents upregulated Nrf2 expression in the heart of normal rats. Additionally, the molecular docking simulations revealed the ability of AV and ED to bind to Keap1. Both agents exhibited binding affinities and form hydrogen bonds with the side chains of the charged amino acids in vicinity of the binding pocket of Keap1. The benzene ring of AV and ED promotes the hydrophobic interaction with the hydrophobic residues of the binding sites of Keap1. These polar and hydrophobic interactions permit a tight fit of AV and ED in the binding site of Keap1. Thus, these in silico findings suggest the value of AV and ED as promising modulators of Keap1 activity.
The PI3K/Akt/mTOR signaling is another pathway whose activation was associated with attenuation of oxidative stress.14 Activation of the PI3K/Akt/mTOR pathway has been reported to activate Nrf2 signaling, resulting in upregulation of antioxidants. For instance, PI3K/Akt/mTOR-mediated Nrf2/HO-1 signaling protected against oxidative stress induced by oxidized low-density lipoprotein in endothelial cells.58 mTOR is an atypical serine/threonine kinase belongs to the family of the PI3K related kinases and exerts its cellular functions through assembly with adaptor proteins to form mTORC1 and mTORC2.59 mTOR is involved in the regulation of the embryonic development of the cardiovascular system and maintenance of cardiac structure and function. In this context, mTOR knockout in mice provoked derangements in cardiac fatty acid metabolism, cardiac dysfunction, massive apoptosis, fibrosis and mitochondrial abnormalities and dysfunction.60,61 The Akt is one of the main regulators of mTOR where its activation activates mTOR or inhibits the endogenous mTORC1 inhibitor PRAS40.62 Previous studies have demonstrated the inhibitory effect of the anticancer drugs on the PI3K/Akt/mTOR pathway.63,64 Acute cardiac dysfunction induced by doxorubicin was associated with the inhibition of mTOR signaling.63 In addition, doxorubicin-induced myocardial apoptosis was associated with the downregulation of the PI3K/Akt/mTOR pathway.64 On the other hand, activation of this signaling pathway contributed to the protection against drug-induced cardiac injury.63,64 The current study showed the downregulation of PI3K, Akt and mTOR expression in the heart of rats administered with CP. In accordance, the study of Albayrak et al65 demonstrated the downregulation of PI3K, Akt and mTOR expression in the kidney of mice received CP every 2 days for 3 consecutive weeks. Given the role of the PI3K/AKT/mTOR pathway in maintenance of the cardiac structure and function and its suppression by CP, it would be intriguing to know whether the modulation of this pathway is involved in the cardioprotective effects of AV and ED. Our results revealed the positive effect of AV and ED on the PI3K/Akt/mTOR signaling pathway where both agents increased the cardiac levels of PI3K, Akt and mTOR in normal and CP-intoxicated rats. These findings clearly support the notion that upregulation of the PI3K/Akt/mTOR signaling plays a role in the protective effect of AV and ED against CP cardiotoxicity.
The upregulation of cytoglobin expression is another effect we thought to participate in the cardioprotective activity of AV and ED. Cytoglobin is an intracellular respiratory globin ubiquitously expressed almost in all tissues.66 It has been shown to prevent oxidative stress through scavenging excess ROS,67 thereby maintaining physiological ROS levels.68 Cytoglobin protected the cardiac progenitor cells against oxidative stress and promoted their survival.69 The direct interaction between cytoglobin and PI3K/Akt/mTOR signaling in the heart has not been presented before. Given the role of PI3K/Akt/mTOR signaling in tumorigenesis and the progression of cancer,22 numerous studies have demonstrated its interaction with cytoglobin.70,71 In L929 fibroblast cell line, overexpression of cytoglobin downregulated cell proliferation, migration and tumor growth through repressing the PI3K/Akt/mTOR pathway and inducing apoptosis.71 In addition, positive correlation between PI3K/Akt activation and low expression of cytoglobin has been associated with tumor progression in glioma.70 Here, CP administration resulted in downregulation of cytoglobin expression in the heart of rats which might be attributed to surplus and sustained ROS generation. Treatment with AV and ED resulted in a pronounced upregulation of cytoglobin in the cardiac tissue. Therefore, the positive regulation of cytoglobin is involved in the cardioprotective effect of AV and ED against CP toxicity. It is worth mentioning that numerous studies have discussed the antioxidant activity of AV and ED, but our study introduced a new information on their effect on cardiac cytoglobin expression, and shed light on the possible role of cytoglobin in the pathogenesis of CP cardiotoxicity.
Conclusion
The present findings provide a compelling evidence for the protective effect of AV and ED against CP cardiotoxicity. The cardioprotective efficacy of AV and ED was associated with their ability to upregulate Nrf2 and PI3K/Akt/mTOR signaling, and cytoglobin, resulting in attenuation of oxidative stress and tissue injury. Therefore, AV and ED might represent promising cardioprotective agents in patients on chemotherapy, pending further investigations and clinical studies to explore the exact mechanism(s) underlying their beneficial effects.
Acknowledgment
The authors acknowledge King Saud University, Riyadh, Saudi Arabia, for funding this work through Researchers Supporting Project (RSP-2019/149).
Disclosure
The authors declare that there are no potential conflicts of interest.
References
1. Moignet A, Hasanali Z, Zambello R, et al. Cyclophosphamide as a first-line therapy in LGL leukemia. Leukemia. 2014;28(5):1134–1136. doi:10.1038/leu.2013.359
2. Stork CM, Schreffler SM. Cyclophosphamide. In: Wexler P, editor. Encyclopedia of Toxicology (Third Edition). Oxford: Academic Press; 2014:1111–1113.
3. Iqubal A, Iqubal MK, Sharma S, et al. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: old drug with a new vision. Life Sci. 2019;218:112–131. doi:10.1016/j.lfs.2018.12.018
4. Kurauchi K, Nishikawa T, Miyahara E, Okamoto Y, Kawano Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res Notes. 2017;10(1):406. doi:10.1186/s13104-017-2726-2
5. Shanholtz C. Acute life-threatening toxicity of cancer treatment. Crit Care Clin. 2001;17(3):483–502. doi:10.1016/S0749-0704(05)70196-2
6. Morandi P, Ruffini PA, Benvenuto GM, La Vecchia L, Mezzena G, Raimondi R. Serum cardiac troponin I levels and ECG/echo monitoring in breast cancer patients undergoing high-dose (7 g/m(2)) cyclophosphamide. Bone Marrow Transplant. 2001;28(3):277–282. doi:10.1038/sj.bmt.1703132
7. Braverman AC, Antin JH, Plappert MT, Cook EF, Lee RT. Cyclophosphamide cardiotoxicity in bone marrow transplantation: a prospective evaluation of new dosing regimens. J Clin Oncol. 1991;9(7):1215–1223. doi:10.1200/JCO.1991.9.7.1215
8. Aladaileh SH, Abukhalil MH, Saghir SAM, et al. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules. 2019;9(8):346. doi:10.3390/biom9080346
9. ALHaithloul HAS, Alotaibi MF, Bin-Jumah M, Elgebaly H, Mahmoud AM. Olea europaea leaf extract up-regulates Nrf2/ARE/HO-1 signaling and attenuates cyclophosphamide-induced oxidative stress, inflammation and apoptosis in rat kidney. Biomed Pharmacother. 2019;111:676–685. doi:10.1016/j.biopha.2018.12.112
10. Kamel EM, Mahmoud AM, Ahmed SA, Lamsabhi AM. A phytochemical and computational study on flavonoids isolated from Trifolium resupinatum L. and their novel hepatoprotective activity. Food Funct. 2016;7(4):2094–2106. doi:10.1039/C6FO00194G
11. Mahmoud AM, Germoush MO, Alotaibi MF, Hussein OE. Possible involvement of Nrf2 and PPARγ up-regulation in the protective effect of umbelliferone against cyclophosphamide-induced hepatotoxicity. Biomed Pharmacother. 2017;86:297–306. doi:10.1016/j.biopha.2016.12.047
12. Moghe A, Ghare S, Lamoreau B, et al. Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci. 2015;143(2):242–255. doi:10.1093/toxsci/kfu233
13. Satta S, Mahmoud AM, Wilkinson FL, Yvonne Alexander M, White SJ. The role of Nrf2 in cardiovascular function and disease. Oxid Med Cell Longev. 2017;2017:9237263. doi:10.1155/2017/9237263
14. Zeng J, Zhao H, Chen B. DJ-1/PARK7 inhibits high glucose-induced oxidative stress to prevent retinal pericyte apoptosis via the PI3K/AKT/mTOR signaling pathway. Exp Eye Res. 2019;189:107830. doi:10.1016/j.exer.2019.107830
15. Wang X, Pan J, Liu D, et al. Nicorandil alleviates apoptosis in diabetic cardiomyopathy through PI3K/Akt pathway. J Cell Mol Med. 2019;23(8):5349–5359. doi:10.1111/jcmm.14413
16. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114(3):549–564. doi:10.1161/CIRCRESAHA.114.302022
17. Sun Y, Gong F, Yin J, et al. Therapeutic effect of apocynin through antioxidant activity and suppression of apoptosis and inflammation after spinal cord injury. Exp Ther Med. 2017;13(3):952–960. doi:10.3892/etm.2017.4090
18. Kapoor M, Sharma N, Sandhir R, Nehru B. Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion hippocampus injury in rat brain. Biomed Pharmacother. 2018;97:458–472. doi:10.1016/j.biopha.2017.10.123
19. Francis S, Laurieri N, Nwokocha C, Delgoda R. Treatment of rats with apocynin has considerable inhibitory effects on arylamine N-acetyltransferase activity in the liver. Sci Rep. 2016;6:26906. doi:10.1038/srep26906
20. Gimenes R, Gimenes C, Rosa CM, et al. Influence of apocynin on cardiac remodeling in rats with streptozotocin-induced diabetes mellitus. Cardiovasc Diabetol. 2018;17(1):15.
21. Hou L, Sun F, Huang R, Sun W, Zhang D, Wang Q. Inhibition of NADPH oxidase by apocynin prevents learning and memory deficits in a mouse Parkinson’s disease model. Redox Biol. 2019;22:101134. doi:10.1016/j.redox.2019.101134
22. Tan YC, Abdul Sattar M, Ahmeda AF, et al. Apocynin and catalase prevent hypertension and kidney injury in Cyclosporine A-induced nephrotoxicity in rats. PLoS One. 2020;15(4):e0231472. doi:10.1371/journal.pone.0231472
23. Homma T, Kobayashi S, Sato H, Fujii J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp Cell Res. 2019;384(1):111592. doi:10.1016/j.yexcr.2019.111592
24. Lin H, Ma X, Wang BC, et al. Edaravone ameliorates compression-induced damage in rat nucleus pulposus cells. Life Sci. 2017;189:76–83. doi:10.1016/j.lfs.2017.09.024
25. He F, Cao Y-P, Che F-Y, Yang L-H, Xiao S-H, Liu J. Inhibitory effects of edaravone in β-amyloid-induced neurotoxicity in rats. Biomed Res Int. 2014;2014:370368. doi:10.1155/2014/370368
26. Tajima S, Bando M, Ishii Y, et al. Effects of edaravone, a free-radical scavenger, on bleomycin-induced lung injury in mice. Eur Respir J. 2008;32(5):1337–1343. doi:10.1183/09031936.00164407
27. Basol N, Aygun H, Gul SS. Beneficial effects of edaravone in experimental model of amitriptyline-induced cardiotoxicity in rats. Naunyn Schmiedebergs Arch Pharmacol. 2019;392(11):1447–1453. doi:10.1007/s00210-019-01683-6
28. Liu J, Jiang Y, Zhang G, Lin Z, Du S. Protective effect of edaravone on blood-brain barrier by affecting NRF-2/HO-1 signaling pathway. Exp Ther Med. 2019;18(4):2437–2442.
29. Shokrzadeh M, Chabra A, Naghshvar F, Ahmadi A, Jafarinejhad M, Hasani-Nourian Y. Protective effects of melatonin against cyclophosphamide-induced oxidative lung toxicity in mice. Drug Res. 2015;65(6):281–286.
30. Bancroft JD, Gamble M. Theory and Practice of Histological Techniques. Elsevier health sciences; 2008.
31. Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86(1):271–278. doi:10.1016/0003-2697(78)90342-1
32. Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. doi:10.1016/0003-9861(59)90090-6
33. Marklund SL. Superoxide dismutase isoenzymes in tissues and plasma from New Zealand black mice, nude mice and normal BALB/c mice. Mutat Res. 1985;148(1–2):129–134. doi:10.1016/0027-5107(85)90216-7
34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
35. Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
36. Jnoff E, Albrecht C, Barker JJ, et al. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem. 2014;9(4):699–705. doi:10.1002/cmdc.201300525
37. Goldberg MA, Antin JH, Guinan EC, Rappeport JM. Cyclophosphamide cardiotoxicity: an analysis of dosing as a risk factor. Blood. 1986;68(5):1114–1118. doi:10.1182/blood.V68.5.1114.1114
38. Omole JG, Ayoka OA, Alabi QK, et al. Protective effect of kolaviron on cyclophosphamide-induced cardiac toxicity in rats. J Evid Based Integr Med. 2018;23:2156587218757649. doi:10.1177/2156587218757649
39. El-Sheikh AA, Abdelzaher WY, Gad AA, Abdel-Gaber SA. Purine versus non-purine xanthine oxidase inhibitors against cyclophosphamide-induced cardiac and bone marrow toxicity in rats. Hum Exp Toxicol. 2020;39(3):249–261. doi:10.1177/0960327119883412
40. El-Sawalhi MM, Ahmed LA. Exploring the protective role of apocynin, a specific NADPH oxidase inhibitor, in cisplatin-induced cardiotoxicity in rats. Chem Biol Interact. 2014;207:58–66. doi:10.1016/j.cbi.2013.11.008
41. Saleem N, Prasad A, Goswami SK. Apocynin prevents isoproterenol-induced cardiac hypertrophy in rat. Mol Cell Biochem. 2018;445(1):79–88. doi:10.1007/s11010-017-3253-0
42. Koohsari M, Shaki F, Jahani D. Protective effects of edaravone against methamphetamine-induced cardiotoxicity. Braz Arch Biol Technol. 2016;59. doi:10.1590/1678-4324-2016160093
43. Emekli-Alturfan E, Alev B, Tunali S, et al. Effects of edaravone on cardiac damage in valproic acid induced toxicity. Ann Clin Lab Sci. 2015;45(2):166–172.
44. Hassan MQ, Akhtar MS, Afzal O, et al. Edaravone and benidipine protect myocardial damage by regulating mitochondrial stress, apoptosis signalling and cardiac biomarkers against doxorubicin-induced cardiotoxicity. Clin Exp Hypertens. 2020;42(5):381–392. doi:10.1080/10641963.2019.1676770
45. Smathers RL, Galligan JJ, Stewart BJ, Petersen DR. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem Biol Interact. 2011;192(1–2):107–112. doi:10.1016/j.cbi.2011.02.021
46. Mahmoud AM, Al Dera HS. 18β-glycyrrhetinic acid exerts protective effects against cyclophosphamide-induced hepatotoxicity: potential role of PPARγ and Nrf2 upregulation. Genes Nutr. 2015;10(6):41. doi:10.1007/s12263-015-0491-1
47. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. doi:10.1161/01.RES.87.10.840
48. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. doi:10.1084/jem.192.7.1001
49. Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116(3):531–549. doi:10.1161/CIRCRESAHA.116.303584
50. Li S, Wang W, Niu T, et al. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev. 2014;2014:748524. doi:10.1155/2014/748524
51. Xing Y, Niu T, Wang W, et al. Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS One. 2012;7(9):e44899. doi:10.1371/journal.pone.0044899
52. Abd El-Twab SM, Hussein OE, Hozayen WG, Bin-Jumah M, Mahmoud AM. Chicoric acid prevents methotrexate-induced kidney injury by suppressing NF-kappaB/NLRP3 inflammasome activation and up-regulating Nrf2/ARE/HO-1 signaling. Inflamm Res. 2019;68(6):511–523. doi:10.1007/s00011-019-01241-z
53. Mahmoud AM, Germoush MO, Al-Anazi KM, Mahmoud AH, Farah MA, Allam AA. Commiphora molmol protects against methotrexate-induced nephrotoxicity by up-regulating Nrf2/ARE/HO-1 signaling. Biomed Pharmacother. 2018;106:499–509. doi:10.1016/j.biopha.2018.06.171
54. Mahmoud AM, Wilkinson FL, McCarthy EM, et al. Endothelial microparticles prevent lipid-induced endothelial damage via Akt/eNOS signaling and reduced oxidative stress. FASEB J. 2017;31(10):4636–4648. doi:10.1096/fj.201601244RR
55. Cruz-Álvarez S, Santana-Martínez R, Avila-Chávez E, et al. Apocynin protects against neurological damage induced by quinolinic acid by an increase in glutathione synthesis and Nrf2 levels. Neuroscience. 2017;350:65–74. doi:10.1016/j.neuroscience.2017.03.011
56. Zhang M, Teng CH, Wu FF, et al. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp Ther Med. 2019;18(1):467–474.
57. Shou L, Bei Y, Song Y, et al. Nrf2 mediates the protective effect of edaravone after chlorpyrifos-induced nervous system toxicity. Environ Toxicol. 2019;34(5):626–633. doi:10.1002/tox.22728
58. Yuan W, Chang H, Liu X, Wang S, Liu H, Xuan H. Brazilian green propolis inhibits Ox-LDL-stimulated oxidative stress in human umbilical vein endothelial cells partly through PI3K/Akt/mTOR-mediated Nrf2/HO-1 pathway. Evid Based Complement Alternat Med. 2019;2019:5789574. doi:10.1155/2019/5789574
59. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.017
60. Zhang D, Contu R, Latronico MV, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120(8):2805–2816. doi:10.1172/JCI43008
61. Tamai T, Yamaguchi O, Hikoso S, et al. Rheb (Ras homologue enriched in brain)-dependent mammalian target of rapamycin complex 1 (mTORC1) activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J Biol Chem. 2013;288(14):10176–10187. doi:10.1074/jbc.M112.423640
62. Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903–915. doi:10.1016/j.molcel.2007.03.003
63. Zhu W, Soonpaa MH, Chen H, et al. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009;119(1):99–106. doi:10.1161/CIRCULATIONAHA.108.799700
64. Lee BS, Oh J, Kang SK, et al. Insulin protects cardiac myocytes from doxorubicin toxicity by Sp1-mediated transactivation of survivin. PLoS One. 2015;10(8):e0135438. doi:10.1371/journal.pone.0135438
65. Albayrak G, Sonmez PK, Akogullari D, Uluer ET. Cyclophosphamide inhibits PI3K/AKT/mTOR signaling pathway in mice kidneys. Proceedings. 2018;2(25):1587. doi:10.3390/proceedings2251587
66. Ludemann J, Verissimo KM, Dreger K, Fago A, Schneider I, Burmester T. Globin E is a myoglobin-related, respiratory protein highly expressed in lungfish oocytes. Sci Rep. 2019;9(1):280. doi:10.1038/s41598-018-36592-w
67. Ou L, Li X, Chen B, et al. Recombinant human cytoglobin prevents atherosclerosis by regulating lipid metabolism and oxidative stress. J Cardiovasc Pharmacol Ther. 2018;23(2):162–173. doi:10.1177/1074248417724870
68. Latina A, Viticchie G, Lena AM, et al. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene. 2016;35(12):1493–1503. doi:10.1038/onc.2015.222
69. Zhang S, Li X, Jourd’heuil FL, et al. Cytoglobin promotes cardiac progenitor cell survival against oxidative stress via the upregulation of the NFkappaB/iNOS signal pathway and nitric oxide production. Sci Rep. 2017;7(1):10754. doi:10.1038/s41598-017-11342-6
70. Xu HW, Huang YJ, Xie ZY, et al. The expression of cytoglobin as a prognostic factor in gliomas: a retrospective analysis of 88 patients. BMC Cancer. 2013;13:247. doi:10.1186/1471-2407-13-247
71. Demirci S, Doğan A, Apdik H, et al. Cytoglobin inhibits migration through PI3K/AKT/mTOR pathway in fibroblast cells. Mol Cell Biochem. 2018;437(1):133–142. doi:10.1007/s11010-017-3101-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.