")
Back to Journals » Journal of Inflammation Research » Volume 14
Dysregulation of DPP4-CXCL12 Balance by TGF-β1/SMAD Pathway Promotes CXCR4+ Inflammatory Cell Infiltration in Keloid Scars
Authors Chen Z , Gao Z, Xia L, Wang X, Lu L, Wu X
Received 24 June 2021
Accepted for publication 18 August 2021
Published 26 August 2021 Volume 2021:14 Pages 4169—4180
DOI https://doi.org/10.2147/JIR.S326385
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
ZongAn Chen,1,* Zhen Gao,1,* LingLing Xia,1 XiaoQing Wang,2,* LiMing Lu,3,* XiaoLi Wu1,*
1Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Orthopedics, Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, People’s Republic of China; 3Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: XiaoLi Wu
Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, People’s Republic of China
Tel +86-2163089567
Email [email protected]
Purpose: Recent studies have confirmed the important role of chronic inflammation in keloid; however, mechanism of chronic inflammation in keloid tissue remains largely unclear, especially the dynamic of infiltrated inflammatory cells.
Patients and Methods: Tissue and blood samples collected from keloid patients and healthy subjects were studied by immunohistochemistry and flow cytometry. Fibroblasts from keloid scars and normal skin were isolated by enzymic digestion.
Results: We found that CXCL12 expression was elevated which was correlated with decreased dipeptidyl peptidase-4 (DPP4) expression in keloid scars relative to mature scars. In vitro studies suggested that autocrine transforming growth factor β 1 (TGF-β 1) in keloid-derived fibroblasts negatively regulated DPP4 expression which inhibited the reduction of extracellular CXCL12 levels by DPP4. Furthermore, immunofluorescence showed that most fibroblasts in keloid scars were DPP4lowTGFβ 1high compared with DPP4highTGFβ 1low fibroblasts in normal skin tissue, which facilitated extracellular CXCL12 accumulation in fibroblasts in keloid scars. Furthermore, we found that most circulating leukocytes in peripheral blood and tissue-infiltrated inflammatory cells in keloid scars expressed the C-X-C motif chemokine receptor 4 (CXCR4) instead of CXCR7, indicating that the chemotaxis driven by CXCL12 is likely to be mediated mainly by CXCR4.
Conclusion: Our study indicated that the TGF-β/DPP4/CXCL12 axis may contribute to chronic inflammation in keloid scars by recruiting inflammatory cells through the CXCR4 receptor.
Keywords: keloid, chemokines, inflammation, DPP4, CXCL12, TGF-β 1
Graphical Abstract:
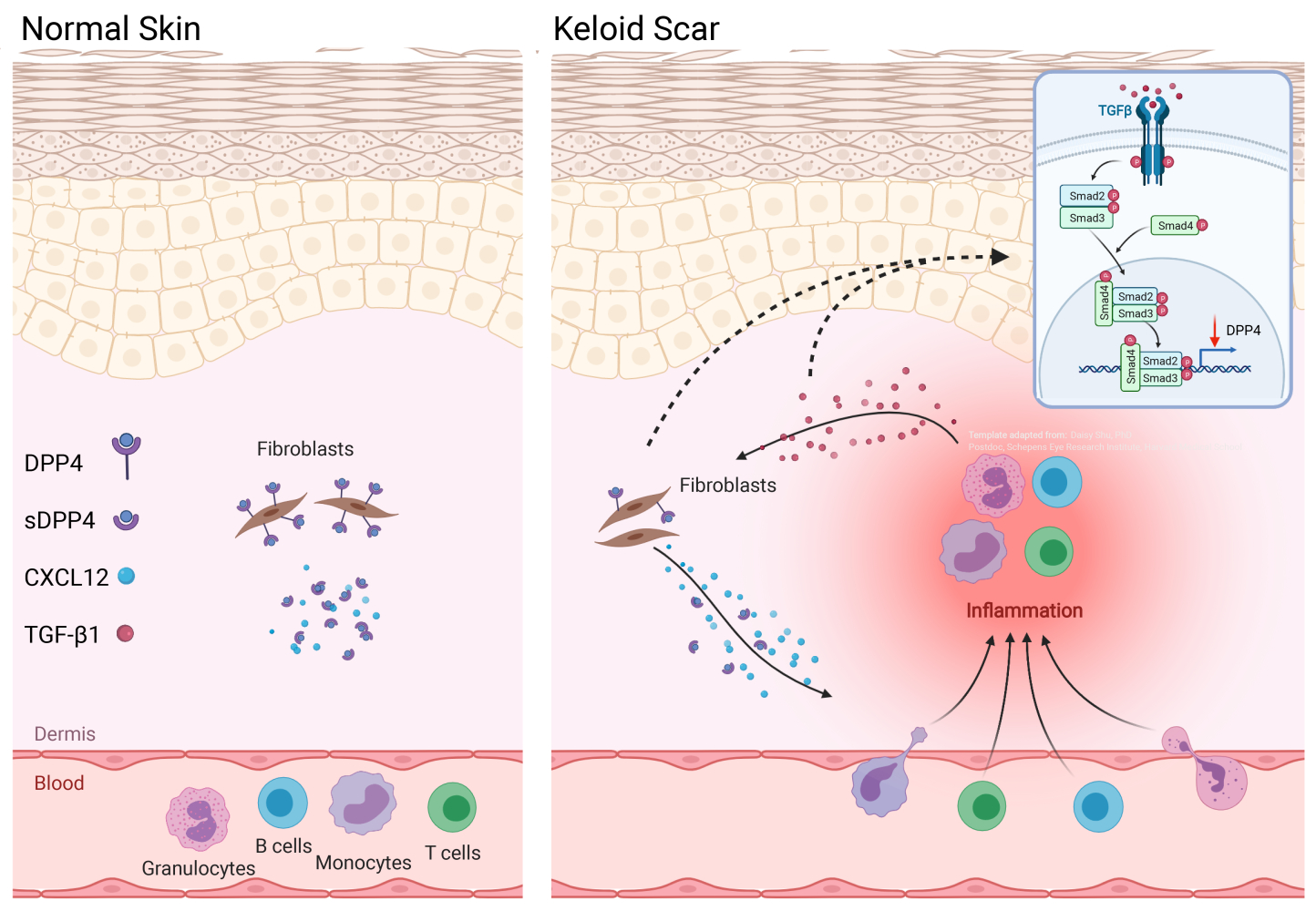
Introduction
Keloid disease is a fibro-proliferative disease characterized by over-deposition of extracellular matrix (ECM) in scar tissue with aggressive margins beyond the original wound boundary.1 This disease is characterized by chronic inflammatory infiltration, especially in lesions in which multiple immunocytes aggregate.2 The inflammatory microenvironment has attracted much research attention, and various targeted strategies have been developed to address this issue, and the resulting findings may provide some insights into the research and treatment of keloid disease.
Chronic inflammation is believed to play a critical role in the pathogenesis of keloid disease.3–5 Chemokines and their receptors are major mediators of peripheral blood cell recruitment to modulate immune surveillance and the inflammatory response by coordinating leukocyte and progenitor cell homeostasis.6,7 Recent studies have revealed that interleukin 17 (IL17) or thymic stromal lymphopoietin (TSLP) enhances the expression of the C-X-C motif chemokine ligand 12 (CXCL12) in keloid scars.8,9 CXCL12 and its receptors, C-X-C motif chemokine receptor 4 (CXCR4) and 7 (CXCR7), have been reported to be broadly involved in wound healing and skin disorders, where inflammatory and immune responses are supported by the homeostasis of monocytes, granulocytes and lymphocytes.10–12 Although a pattern of CXCL12 overexpression has been identified in keloid scars, its potential role in local tissue inflammation, especially the expression pattern of its receptors, remains unclear.
Post-translational modification of chemokines is an essential regulatory mechanism for enhancing or dampening the inflammatory response.6,13 Dipeptidyl peptidase-4 (DPP4), also known as CD26, is a type II integral transmembrane glycoprotein that post-translationally regulates several chemokines by removing dipeptides from the N-termini of peptide chains.13 N-terminal truncation of CXCL12 by DPP4 decreases its activity and attenuates the interaction with its receptor CXCR4.6,14 Recent studies have reported that DPP4 deficiency increases the accumulation of CXCL12 and promotes Th1 inflammation.7,15,16 Hence, the question of whether DPP4 plays a role in the regulation of CXCL12-mediated cell tracking in keloids has arises.
In this study, we aimed to explore the role and regulation of inflammation in keloid pathogenesis, especially the factors that induce inflammatory cell infiltration, and we found that transforming growth factor β1 (TGF-β1) induces CXCL12 accumulation through inhibition of DPP4 expression in fibroblasts. We further explored the expression pattern of CXCR4 and CXCR7 on both circulating and tissue-infiltrated immune cell subsets.
Materials and Methods
Subjects and Sample Collection
Samples were collected from patients receiving surgery at the Department of Plastic and Reconstruction Surgery, Shanghai Ninth People’s Hospital. Written informed consent was obtained according to protocols approved by the Institutional Review Board of Shanghai Ninth People’s Hospital. In this study, none of the patients had received any treatment for at least one year. The diagnosis of keloids was performed by three clinicians and confirmed by postsurgical pathological diagnosis. Skin samples from more than 1 cm outside the keloid scar were used as a normal skin control (nonlesion). Mature scars collected from age-matched healthy subjects without history of keloids or hypertrophic scars undergoing plastic surgery during the same period were used as a negative control (control). Atrophic or flat scars with duration of one year and without hyperplasia or pruritus were considered mature scars. And this study was conducted in accordance with the Declaration of Helsinki. Detailed demographic characteristics of the patients and healthy subjects are listed in Supplemental Table S1.
Cell Culture
Fibroblasts derived from keloid scars (KF) and normal human skin (NHDF) were routinely cultured in DMEM supplemented with 2 μM glutamine, 4.5 g/L D-glucose, 1% penicillin/streptomycin (Lonza, Walkersville, MD), and 10% fetal bovine serum (Life Technologies, Carlsbad, USA) in a humidified incubator with 5% CO2 at 37°C.
RNA Extraction and Real-Time Quantitative PCR
Total RNA was extracted with TRIzol Reagent (Takara, Beijing, China) according to the manufacturer’s protocol. Messenger RNA (mRNA) was reverse-transcribed to cDNA using RT Master Mix (Takara, Beijing, China). Target mRNA levels were quantified by real-time PCR using SYBR Green PCR Master Mix (Takara) and a ViiA™ 6 System (Life Technologies). The primers used in this study are listed in Supplemental Table S2.
Immunohistochemistry and Immunofluorescence
Sections (4 μm) of formalin-fixed, paraffin-embedded samples were prepared and stained by applying a conventional avidin-biotin complex technique.17 The primary antibodies used are listed in Supplemental Table S3. Sections were incubated with primary antibodies and appropriate conjugated secondary antibodies. Immunohistochemistry images were acquired with an inverted microscope. Immunofluorescence images were acquired with a confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Flow Cytometry Analysis
Flow cytometry analysis was conducted as previously described.18 Briefly, cells were first stained with viability stain (BD Biosciences, San Jose, USA) in DPBS. After three washes with stain buffer (BioLegend, San Diego, USA.), cells were stained with conjugated primary antibodies. Data were acquired with an LSRFortessa flow cytometer (BD Biosciences) and were analyzed with FlowJo (Tree Star Inc., Ashland, USA.). The antibodies used in this study are listed in Supplemental Table S3. In this study, peripheral blood monocytes were collected from healthy subjects of the same age and sex as the patients.
Western Blot Analysis
Protein was extracted with 1% SDS lysis buffer at 100°C for 15 minutes. The protein concentration was measured using the bicinchoninic acid assay (Life Technologies) according to the manufacturer’s instructions. Protein samples were adjusted to the same concentration, separated by SDS–PAGE, and electrotransferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with primary antibodies at 4°C overnight (Supplemental Table S3) and secondary antibodies at room temperature for 1.5 hours. Immunoreactive bands were detected using chemiluminescence reagents.
DPP4 Activity Assay
DPP4 activity was assayed using a DPP4 activity assay kit (Sigma-Aldrich, St. Louis, USA) according to the manufacturer’s instructions. Briefly, DPP4 cleaves a nonfluorescent substrate, H-Gly-Pro-AMC, to release a fluorescent product, 7-amino-4-methyl coumarin (AMC) (ex = 360/em = 460 nm). One unit of DPP4 is the amount of enzyme that will hydrolyze the DPP4 substrate to yield 1.0 mmol of AMC per minute at 37°C. The enzyme activity of 100 mg of tissue sample homogenized in 1 mL of DPP4 Assay Buffer was measured in duplicate.
Enzyme-Linked Immunosorbent Assay
The concentration of CXCL12 in lysates, plasma and cell culture supernatant was determined using a specific ELISA kit (R&D Systems) according to the manufacturer’s instructions. Briefly, samples were diluted adequately and then incubated with antibodies in a precoated 96-well plate. After a two-hour incubation, the plate was washed and incubated with streptavidin-HRP for 45 minutes at room temperature. After the washing step, substrate solution was added to each well, and the plate was incubated for 20 minutes at room temperature in the dark. Stop solution was added, and the optical density of each well was measured at 450 nm and then corrected by subtracting the absorbance at 630 nm. Duplicate readings for each standard and sample were averaged, and the resulting differential absorbance values were used to calculate the concentration in each sample.
Statistical Analysis
Statistical significance was determined using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA). Student’s t-test was used for paired data, and differences were deemed significant at four levels: ns= P>0.05, *P < 0.05, **P < 0.01, ***P < 0.001. Pearson correlation analysis was performed, and a P value < 0.05 was considered statistically significant.
Results
Enhanced CXCL12 Expression and Decreased DPP4 Expression in Keloid Scars
Immunohistochemistry was performed to compare the expression profiles of CXCL12 and DPP4 at different sites of keloid scarring, including the central keloid scar (midlesion), the periphery of the keloid scar (perilesion), healthy skin tissue from keloid patients (nonlesion), and normal scar tissue from healthy subjects (control) (Figure 1A). Our results showed a high level of CXCL12 accumulation and a decreased level of DPP4 in the dermis of keloid scars (midlesion and perilesion) compared with that in healthy skin tissue from keloid patients and normal scars (Supplemental Figure S1a). Quantification analysis of tissue homogenate revealed that lower DPP4 activity was correlated with a higher level of CXCL12 in keloids (Figure 1B). Moreover, no difference was found in the plasma between keloid patients and healthy subjects (Supplemental Figure S1b).
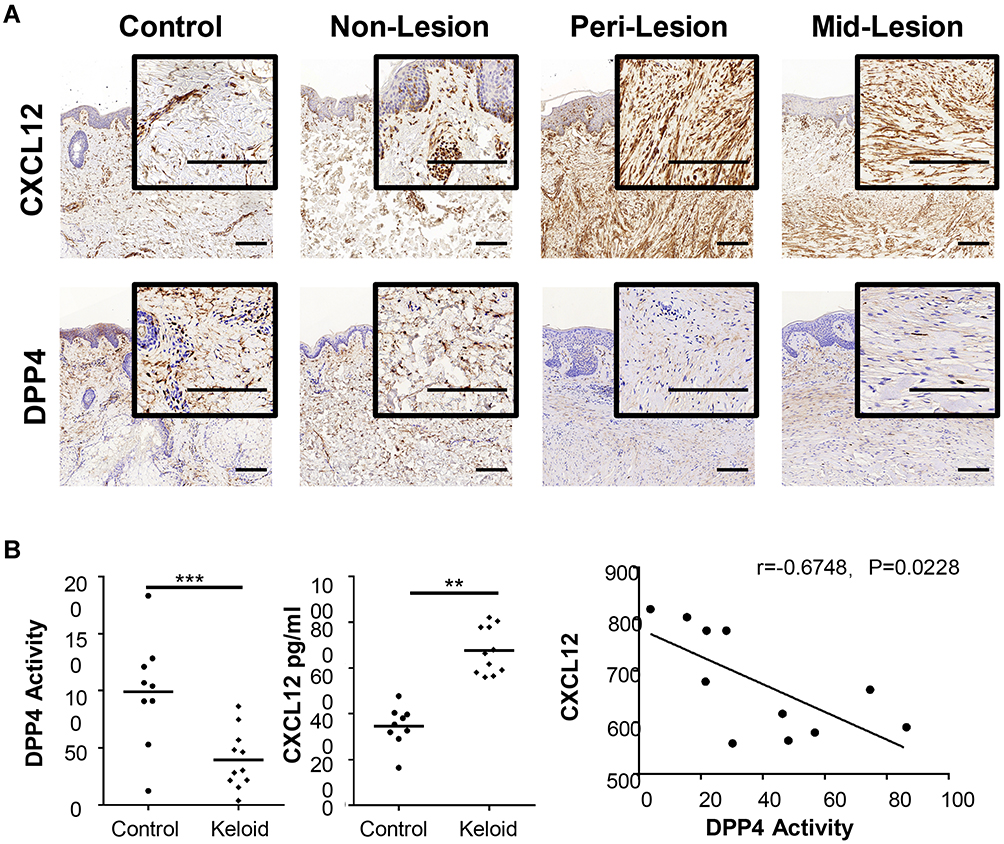 |
Figure 1 Expression of CXCL12 and DPP4 in keloids and mature scars. (A) Immunohistochemical staining of CXCL12 and DPP4 in mature scar tissue (n=11) and various sites of keloid scarring (n=12). Bars=200μm. (B) Quantification of DPP4 activity (microunit/mL) and CXCL12 (pg/mL) in tissue homogenates from healthy subjects (n=9) and keloid patients (n=11). Correlation analysis of DPP4 activity and CXCL12 was performed for each group. **P < 0.01, ***P < 0.001. Abbreviations: Mid-lesion, the central site of the keloid scar; Peri-lesion, the periphery of the keloid scar; Non-lesion, healthy skin tissue from keloid patients; Control, mature scar tissue from healthy subjects. |
Inhibition of DPP4 Increased Extracellular CXCL12 without Inducing CXCL12 Expression
We found downregulated expression of DPP4, a critical extracellular enzyme that degrades CXCL12, in keloid-derived fibroblasts (KFs) compared with that in normal human dermal fibroblasts (NHDFs) (Figure 2A and Supplemental Figure S2a). Treatment of NHDFs with linagliptin, a selective DPP4 activity inhibitor, elevated extracellular CXCL12 levels without increasing intracellular CXCL12 expression (Figure 2B, C and Supplemental Figure S2b). Consistent with previous reports, these results indicate that DPP4 exerts potent degrading activity against CXCL12 and that inhibition of DPP4 resulted in the accumulation of CXCL12.6,14
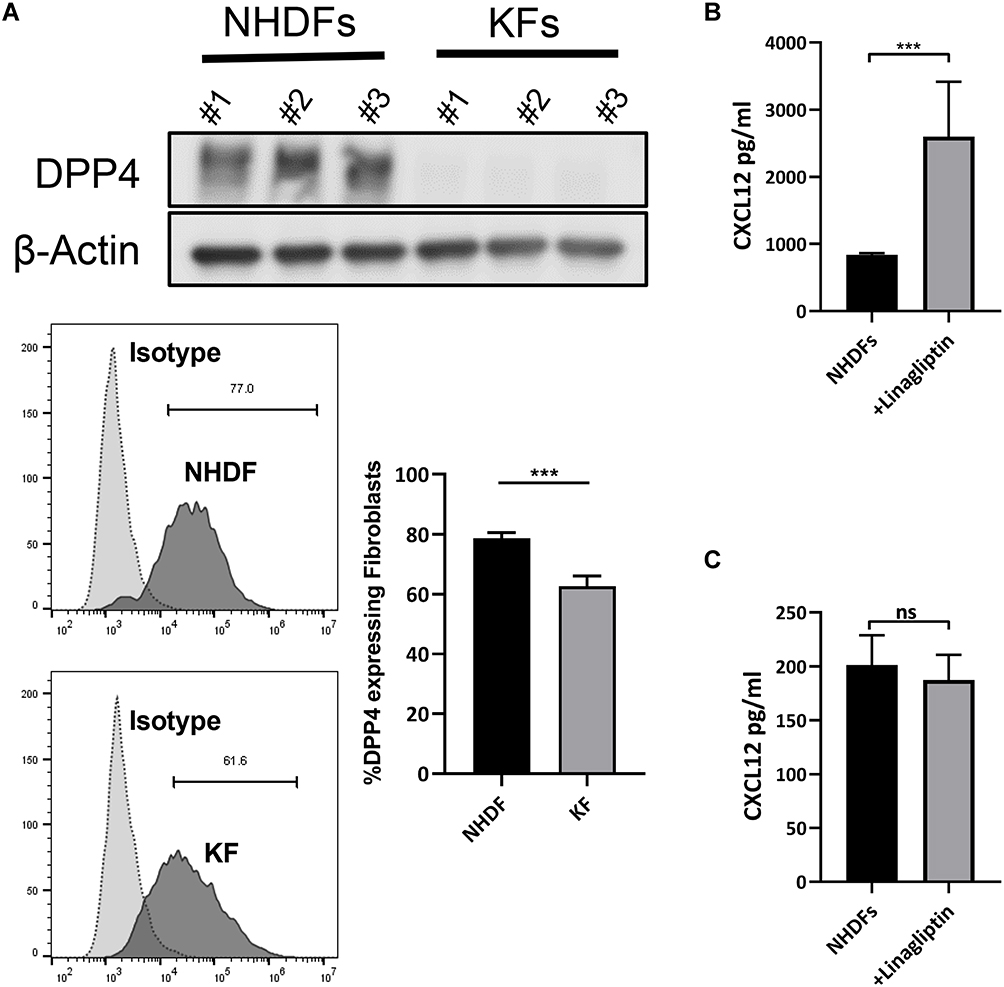 |
Figure 2 Inhibition of DPP4 increased extracellular CXCL12 without inducing CXCL12 expression. (A) Western blot and flow cytometry analyses of DPP4 in normal human dermal fibroblasts (NHDF) (n=3) and keloid-derived fibroblasts (KFs) (n=3). (B) ELISA analyses of CXCL12 in the culture supernatant of NHDFs (n=3) after inhibition of DPP4 activity. (C) ELISA analyses of CXCL12 in cell lysate of NHDFs (n=3) after linagliptin treatment (10nM). ns= P>0.05, ***P < 0.001. |
TGF-β1 Increased Extracellular CXCL12 Levels without Inducing CXCL12 Expression
We next investigated the potential mechanism by which CXCL12 accumulation and DPP4 deficiency occur in keloid tissue. Given the knowledge of TGF-β signaling in fibrosis and a previous report of TGF-β signaling in tumor fibroblast activation, we speculated that TGF-β signaling contributes to CXCL12 accumulation in keloids.19,20 First, we confirmed increased TGF-β1 expression in KFs compared with that in NHDFs (Figure 3A, B, and Supplemental Figure S2c). Furthermore, there were no significant differences in CXCL12 expression (Figure 3A). In addition, in vitro TGF-β1 treatment increased CXCL12 levels in the cell culture supernatant of NHDFs (Figure 3C). However, real-time PCR and ELISA analysis indicated that TGF-β1 treatment did not influence the expression of CXCL12 in NHDFs (Figure 3D). These results suggest that TGF-β1 upregulates extracellular CXCL12 indirectly rather than by directly inducing its expression.
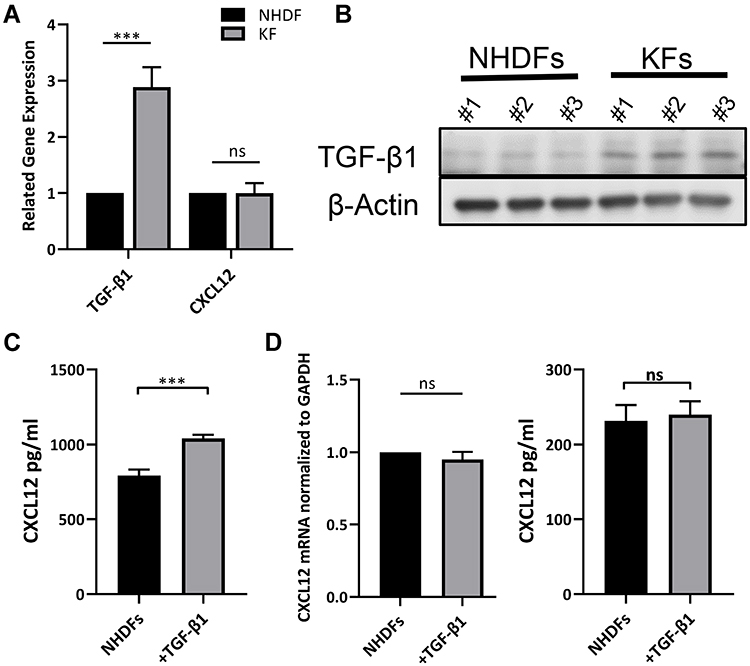 |
Figure 3 TGF-β1 increased extracellular CXCL12 without inducing CXCL12 expression. (A) Detection of TGF-β1 and CXCL12 expression in NHDFs (n=3) and KFs (n=3) by real-time PCR analyses. (B) Western blot analyses of TGF-β1 in NHDFs (n=3) and KFs (n=3). (C) Expression of CXCL12 in the cell supernatant of NHDFs (n=3) after TGF-β1 treatment (10ng/mL) was measured by ELISA. (D) Expression of CXCL12 in the cell lysate of NHDFs (n=3) after TGF-β1 treatment (10ng/mL) was measured by real-time PCR and ELISA; ns= P>0.05, ***P < 0.001. ns= P>0.05, ***P < 0.001. |
TGFβ1 Induced CXCL12 Accumulation Through Inhibition of DPP4 Expression
TGF-β1 treatment in NHDFs reduced DPP4 expression (Figure 4A and Supplemental Figure S2d). Vactosertib, a highly selective TGF-β (ALK4/ALK5) inhibitor, reversed TGF-β-induced inhibition of DPP4 through suppression of smad2 phosphorylation (Figure 4B). KFs treated with an siRNA against TGF-β1 or vactosertib showed increased DPP4 expression and reduced extracellular CXCL12 levels (Figure 4C and D). Immunofluorescence staining revealed a significantly higher number of TGF-β1+DPP4low fibroblasts in keloid lesions than in mature scars and nonlesions (Figure 4E).
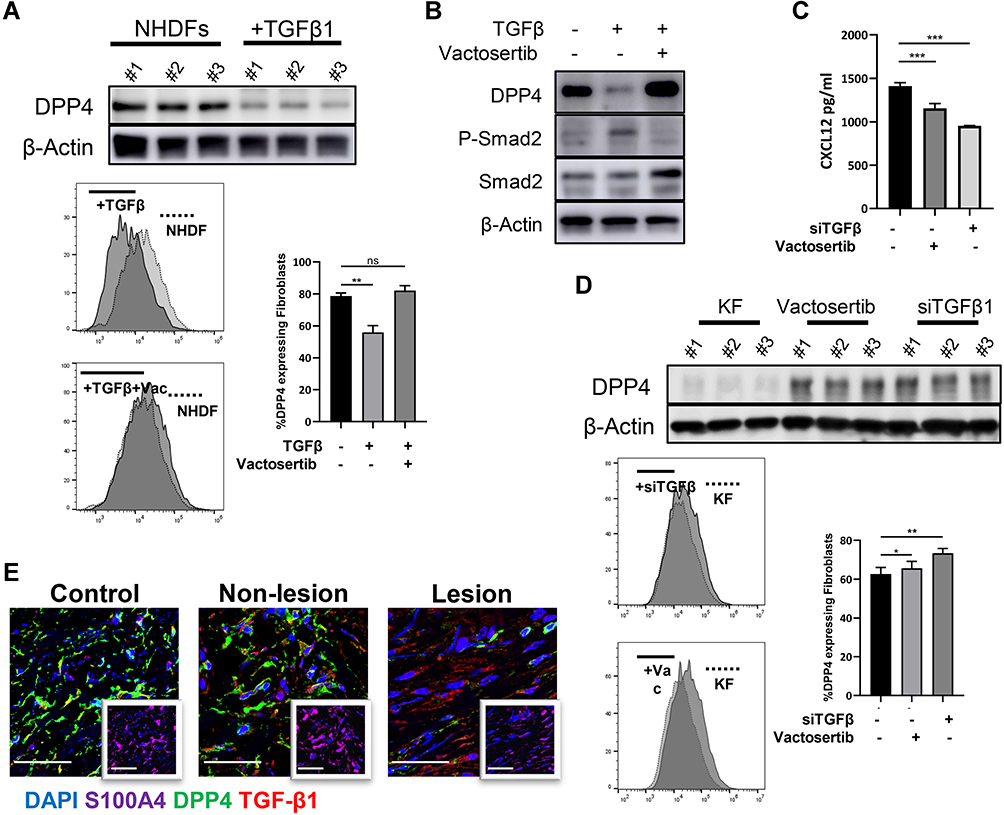 |
Figure 4 TGF-β induced CXCL12 accumulation through inhibition of DPP4 expression. (A) Western blot and flow cytometry analyses of DPP4 in NHDFs (n=3) after TGF-β1 treatment (10ng/mL). (B) The phosphorylation of smad2 in NHDFs after TGF-β treatment (10ng/mL) with or without vactosertib (500nM) was detected by Western blot. (C) ELISA analyses of DPP4 in KFs (n=3) after TGF-β1 treatment with or without vactosertib (500nM). (D) Western blot and flow cytometry analyses of DPP4 in KFs (n=3) after vactosertib (500nM) or siTGF-β1 treatment. (E) Immunofluorescence analysis of DPP4 and TGF-β1 expression in keloid tissue (n=12). Immunofluorescence sections prepared from the keloid tissue specimens. Fibroblasts were stained with anti-S100A4 antibody and are shown in the small window. Bars=50μm. ns= P>0.05, *P < 0.05, **P < 0.01, ***P < 0.001. |
Infiltrated CXCR4+ Inflammatory Cells in Keloid Scars
To examine the potential role of the chemokine-receptor axis in tissue inflammation, immunohistochemical analysis of CXCR4 and CXCR7 was performed on keloid lesions (Figure 5A). Semiquantitative analysis of CXCR4 revealed that the proportion of CXCR4+ cells was significantly higher in keloid lesions than in adjacent normal skin (nonlesion) and mature scars (control) (Figure 5B). CXCR7 staining was mostly located in the vascular endothelium in control samples, nonlesion samples and keloid scars. There was no apparent expression of CXCR7 in keloid tissue.
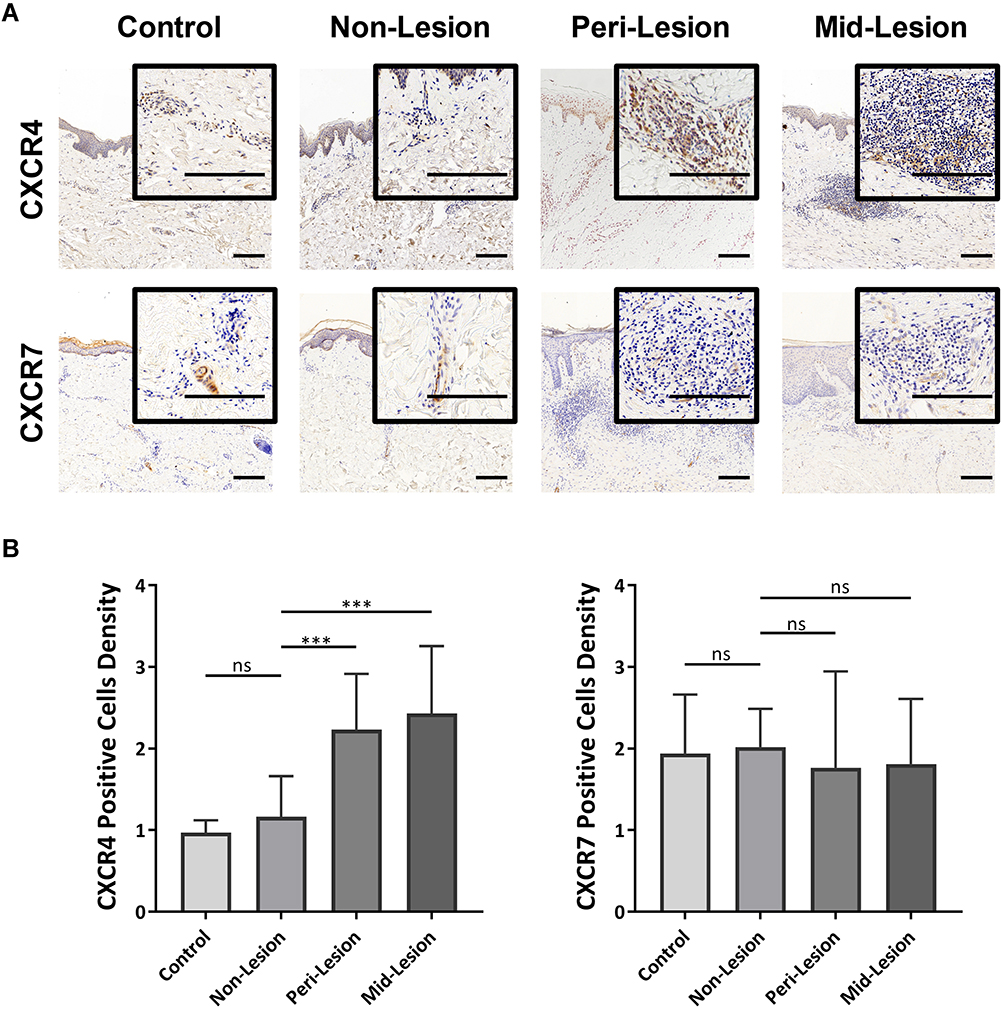 |
Figure 5 Infiltrated CXCR4+ inflammatory cells in keloid scars. (A) Immunohistochemical staining of CXCR4 and CXCR7 in mature scar tissue (n=11) and various sites of keloid scarring (n=12). The areas where inflammatory cells had infiltrated are amplified and shown in the small window. Bars=200μm. (B) Statistical analysis of CXCR4 and CXCR7 expression. ns= P>0.05, ***P < 0.001. |
Most Circulating Immune Cells and Infiltrated Inflammatory Cells in Keloid Scars Express CXCR4
CXCR4 was expressed in several inflammatory cell types in the keloid lesions, including CD3+ T cells, CD20+ B cells, CD68+ macrophages, and CD117+ mast cells (Figure 6A). Analysis of circulating cells revealed that nearly half of the lymphocytes and 40% of the granulocytes expressed CXCR4 in the healthy subjects and keloid patients, including CD4+ (52.57 ± 5.258 and 53.44 ± 6.658) and CD8+ T cells (34.45 ± 9.006 and 43.14 ± 9.105), CD20+ B cells (58.11 ± 13.224 and 72.93 ± 11.726), CD14+ monocytes (77.60 ± 8.517 and 83.37 ± 4.305), neutrophils (37.47 ± 8.405 and 35.19 ± 9.868), basophils (29.58 ± 11.130 and 29.90 ± 9.536) and eosinophils (25.10 ± 6.794 and 27.13 ± 5.958) (Figure 6B–E). There was a significantly higher number of CXCR4+CD8+ T cells and a slightly increased number of CXCR4+CD20+ B cells in the peripheral blood of keloid patients compared with that in healthy subjects (Figure 6B and C). No differences in the expression of CXCR4 on monocytes and granulocytes were found between healthy subjects and keloid patients (Figure 6D and E).
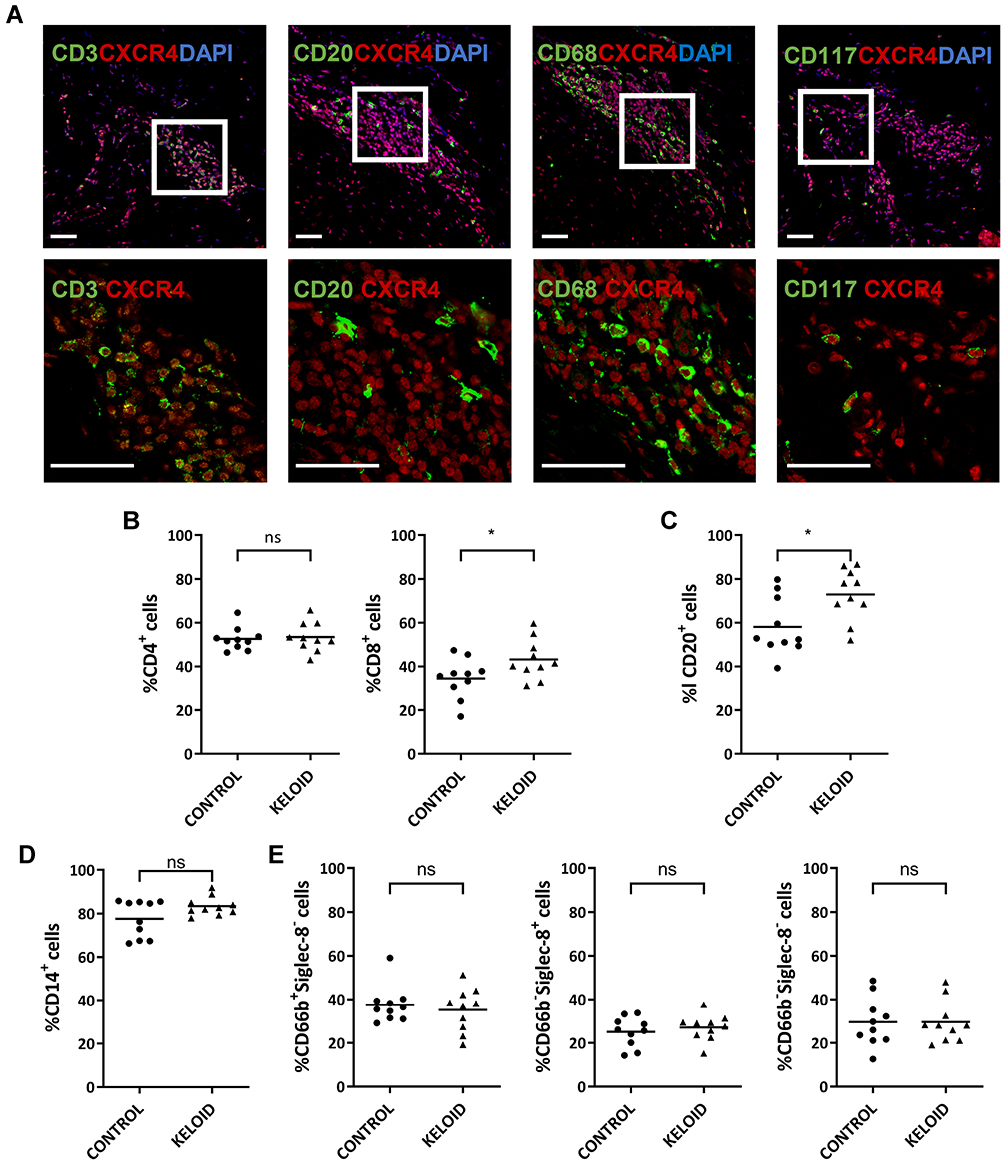 |
Figure 6 Expression of CXCR4 on circulating immune cells and keloid-resident inflammatory cells. (A) Immunofluorescence analysis of CXCR4 expression in different keloid-resident inflammatory cells (CD3+ T cells, CD20+ B cells, CD68+ macrophages and CD117+ mast cells) (n=12). Bars=50μm. (B–E) Flow cytometry analyses of CXCR4 expression in cycling CD4+ and CD8+ T lymphocytes (B), CD20+ B lymphocytes (C), CD14+ monocytes (D), and granulocytes, including CD66b+Siglec-8- neutrophils, CD66b−Siglec-8+ basophils, and CD66b−Siglect-8− eosinophils (E). Peripheral blood samples were collected from healthy subjects (Control) (n=10) and keloid patients (Keloid) (n=10); ns= P>0.05, *P < 0.05. |
However, no more than 10% of peripheral blood cells expressed CXCR7 in both healthy subjects and keloid patients, including CD4+ T cells (1.360±0.1809 and 1.640±0.3968), CD8+ T cells (4.174 ± 0.8113 and 2.639 ± 0.4926), B cells (3.575 ± 0.6961 and 2.323 ± 0.4038), neutrophils (2.049 ± 0.7106 and 0.5340 ± 0.08919), basophils (2.910 ± 0.8173 and 0.8242 ± 0.1342), and eosinophils (2.658 ± 0.6897 and 1.937 ± 0.3294) (Supplemental Figure S3). No significant differences in the expression of CXCR7 among these subsets were found between healthy subjects and keloid patients (Supplemental Figure S3).
Discussion
In this study, to explore potential mechanism of chronic inflammation in keloid tissue, we identified the role of fibroblasts in inflammatory cell chemotaxis. Our results indicated that TGF-β1 inhibited DPP4 expression in keloid fibroblasts, which increased extracellular CXCL12 levels. We also found that most circulating and tissue-infiltrated immunocytes in keloid scars expressed CXCR4 instead of CXCR7, which indicated the potential involvement of CXCL12/CXCR4 in mediating chemotaxis in chronic inflammation in keloid scars.
The important role of the CXCL12/CXCR4 axis has been well documented in keloids and many other fibrotic diseases.9,21–23 CXCL12, together with its corresponding receptors, plays a noteworthy role in inflammatory cell chemotaxis.12 Although it has been reported that CXCL12 expression is increased in the dermis of keloid scars.9 The mechanism by which CXCL12 is regulated remains unclear. The role of TGF-β1 in keloid formation has been well documented. Here, we found that exposure to TGF-β1 increased extracellular CXCL12 levels in normal skin fibroblasts. However, the extracellular CXCL12 accumulation induced by TGF-β1 was achieved by inhibiting DPP4 expression, which resulted in degradation of extracellular CXCL12 rather than the direct induction of CXCL12 expression. Inhibition of TGF-β1 in keloid fibroblasts increased DPP4 expression, leading to a decreased extracellular level of CXCL12. Although this regulation of the TGF-β/DPP4/CXCL12 axis has been reported in other studies, this is the first study to report it in keloids. Examination of clinical samples showed that most fibroblasts in keloid scars were DPP4lowTGF-β1high; in contrast, most fibroblasts in mature scars and normal skin were DPP4highTGF-β1low. It has been reported that DPP4+ fibroblasts are a major source of collagen in normal skin and wound healing.24,25 However, the role of DPP4 in abnormal microenvironments may be controversial. In human breast carcinoma-associated fibroblasts, cross-talk between autocrine TGF-β1 and CXCL12 signaling mediates fibroblast activation.19 A further study has found that TGF-β and SDF-1 autocrine signaling attenuated DPP4 expression.20 Hence, in keloid scars, the decreased DPP4 expression may be the result of tissue specific microenvironments, especially in regard to the role of TGF-β1. Altogether, we found that TGF-β1 increased the extracellular level of CXCL12 through inhibition of DPP4 expression, which may be responsible for the overexpression of CXCL12 in keloid scars.
Consequently, the question of how this CXCL12 accumulation contributes to keloid pathology arose. A previous study reported that in keloid disease, TSLP in keloid scars induces CXCR4+ fibrocyte infiltration through upregulation of CXCL12.9 However, in addition to fibrocytes, CXCR4 has been identified in many other cells, mostly immunocytes.12 Furthermore, CXCL12 has two major receptors, CXCR4 and CXCR7. Chemotaxis of inflammatory cells is dependent on both imbalanced chemokine expression and related receptor expression in target cells. In our study, early half of lymphocytes or granulocytes were CXCR4+ and less than 10% of cells expressed CXCR7, indicating that CXCR4 was more common in leukocytes. Based on our results, we inferred that circulating CD8+ T cells and CD20+ B cells in keloids are more susceptible to CXCL12 chemotaxis, given that a larger proportion of these two cells was CXCR4+ in keloid patients than in healthy subjects. In line with previous studies, distinct inflammatory cell aggregates were identified in keloid tissue.2 Consistently, we found that most of these infiltrated inflammatory cells expressed CXCR4 instead of CXCR7. Altogether, this study provides evidence that CXCL12-CXCR4-mediated cell chemotaxis plays an important role in chronic inflammation in keloids. However, because of the lack of in vivo experiments, it is difficult to conclude that CXCR4 play a more important than CXCR7 in CXCL12 chemotaxis. The establishment of an effective keloid animal model may be the key to solving this outstanding problem.
Conclusion
The present study showed that TGF-β1 increases extracellular CXCL12 levels by inhibiting DPP4 expression in keloid tissue, and that this mechanism mediates the chemotaxis of inflammatory cells via CXCR4. Nonetheless, this study is more about presenting a possibility than a conclusion, and the involvement of the CXCL12/CXCR4 axis in keloids is not yet conclusive. Furthermore, because of the lack of suitable in vivo models, we only conducted studies on cells and clinical samples. Further in vivo studies based on reliable animal models are required to elucidate the mechanism underlying the effects of this axis on keloid formation.
Abbreviations
CXCL12, C-X-C motif chemokine ligand 12; DPP4, Dipeptidyl peptidase-4; TGF-β1, transforming growth factor β1; CXCR4, C-X-C motif chemokine receptor 4; CXCR7, C-X-C motif chemokine receptor 7; ECM, extracellular matrix; IL17, interleukin 17; TSLP, thymic stromal lymphopoietin; KF, Keloid derived fibroblasts; NHDF, normal skin derived fibroblasts; mRNA, messenger RNA; PVDF, polyvinylidene difluoride.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
Written informed consent was obtained according to protocols approved by the Ethics Department of Shanghai Ninth People’s Hospital.
Acknowledgments
The graphic abstract was created with BioRender.com.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval for the version to be published, agreed to the journal the manuscript has been submitted to, and agree to be accountable for all aspects of the work.
Funding
This project was supported by the National Natural Science Foundation of China (grant numbers: 82071856, 81671579, 31370904, 81871791); Shanghai Pujiang Program (15PJD021); Program for scientific and technological innovation from the Science and Technology Commission of Shanghai Municipality (18140903800, 15401900500); Shuguang Planning of Shanghai Municipal Education Commission (16SG14); Program of Shanghai translational medicine collaborative innovation center cooperation (TM201522, TM201721).
Disclosure
The authors declare that they have no competing interests.
References
1. Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. 2017;18(3):606. doi:10.3390/ijms18030606
2. Bagabir R, Byers RJ, Chaudhry IH, Muller W, Paus R, Bayat A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol. 2012;167(5):1053–1066. doi:10.1111/j.1365-2133.2012.11190.x
3. Dohi T, Padmanabhan J, Akaishi S, et al. The interplay of mechanical stress, strain, and stiffness at the keloid periphery correlates with increased caveolin-1/ROCK signaling and scar progression. Plast Reconstr Surg. 2019;144(1):58e–67e. doi:10.1097/PRS.0000000000005717
4. Jfri A, Alajmi A. Spontaneous keloids: a literature review. Dermatology. 2018;234(3–4):127–130. doi:10.1159/000491924
5. Fujita M, Yamamoto Y, Jiang JJ, et al. NEDD4 is involved in inflammation development during keloid formation. J Invest Dermatol. 2019;139(2):333–341. doi:10.1016/j.jid.2018.07.044
6. Mortier A, Gouwy M, Van Damme J, Proost P, Struyf S. CD26/dipeptidylpeptidase IV-chemokine interactions: double-edged regulation of inflammation and tumor biology. J Leukoc Biol. 2016;99(6):955–969. doi:10.1189/jlb.3MR0915-401R
7. Barreira da Silva R, Laird ME, Yatim N, Fiette L, Ingersoll MA, Albert ML. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol. 2015;16(8):850–858. doi:10.1038/ni.3201
8. Lee SY, Kim EK, Seo HB, et al. IL-17 induced stromal cell-derived factor-1 and profibrotic factor in keloid-derived skin fibroblasts via the STAT3 pathway. Inflammation. 2020;43(2):664–672. doi:10.1007/s10753-019-01148-1
9. Shin JU, Kim SH, Kim H, et al. TSLP is a potential initiator of collagen synthesis and an activator of CXCR4/SDF-1 axis in keloid pathogenesis. J Invest Dermatol. 2016;136(2):507–515. doi:10.1016/j.jid.2015.11.008
10. Avniel S, Arik Z, Maly A, et al. Involvement of the CXCL12/CXCR4 pathway in the recovery of skin following burns. J Invest Dermatol. 2006;126(2):468–476. doi:10.1038/sj.jid.5700069
11. Rezk AF, Kemp DM, El-Domyati M, et al. Misbalanced CXCL12 and CCL5 chemotactic signals in vitiligo onset and progression. J Invest Dermatol. 2017;137(5):1126–1134. doi:10.1016/j.jid.2016.12.028
12. Strazza M, Azoulay-Alfaguter I, Peled M, et al. PLCepsilon1 regulates SDF-1alpha-induced lymphocyte adhesion and migration to sites of inflammation. Proc Natl Acad Sci U S A. 2017;114(10):2693–2698. doi:10.1073/pnas.1612900114
13. Broxmeyer HE, Capitano M, Campbell TB, Hangoc G, Cooper S. Modulation of hematopoietic chemokine effects in vitro and in vivo by dipeptidyl peptidase 4/CD26. Stem Cells Dev. 2016;25(8):575–585. doi:10.1089/scd.2016.0026
14. Wang W, Choi BK, Li W, et al. Quantification of intact and truncated stromal cell-derived factor-1alpha in circulation by immunoaffinity enrichment and tandem mass spectrometry. J Am Soc Mass Spectrom. 2014;25(4):614–625. doi:10.1007/s13361-013-0822-7
15. Tasic T, Baumer W, Schmiedl A, et al. Dipeptidyl peptidase IV (DPP4) deficiency increases Th1-driven allergic contact dermatitis. Clin Exp Allergy. 2011;41(8):1098–1107. doi:10.1111/j.1365-2222.2011.03778.x
16. Jungraithmayr W, De Meester I, Matheeussen V, Baerts L, Arni S, Weder W. CD26/DPP-4 inhibition recruits regenerative stem cells via stromal cell-derived factor-1 and beneficially influences ischaemia-reperfusion injury in mouse lung transplantation. Eur J Cardiothorac Surg. 2012;41(5):1166–1173. doi:10.1093/ejcts/ezr180
17. Narducci MG, Scala E, Bresin A, et al. Skin homing of Sezary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood. 2006;107(3):1108–1115. doi:10.1182/blood-2005-04-1492
18. Chen Z, Zhou L, Won T, Gao Z, Wu X, Lu L. Characterization of CD45RO(+) memory T lymphocytes in keloid disease. Br J Dermatol. 2018;178(4):940–950. doi:10.1111/bjd.16173
19. Kojima Y, Acar A, Eaton EN, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–20014. doi:10.1073/pnas.1013805107
20. Mezawa Y, Daigo Y, Takano A, et al. CD26 expression is attenuated by TGF-beta and SDF-1 autocrine signaling on stromal myofibroblasts in human breast cancers. Cancer Med. 2019;8(8):3936–3948. doi:10.1002/cam4.2249
21. McConnell AT, Ellis R, Pathy B, Plummer R, Lovat PE, O’Boyle G. The prognostic significance and impact of the CXCR4-CXCR7-CXCL12 axis in primary cutaneous melanoma. Br J Dermatol. 2016;175(6):1210–1220. doi:10.1111/bjd.14720
22. Speeckaert R, Ongenae K, van Geel N. Alterations of CXCL12 in serum of vitiligo patients. J Invest Dermatol. 2017;137(7):1586–1588. doi:10.1016/j.jid.2017.02.012
23. Shipe R, Burdick MD, Strieter BA, et al. Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J Allergy Clin Immunol. 2016;137(3):750–757 e3. doi:10.1016/j.jaci.2015.07.037
24. Worthen CA, Cui Y, Orringer JS, Johnson TM, Voorhees JJ, Fisher GJ. CD26 identifies a subpopulation of fibroblasts that produce the majority of collagen during wound healing in human skin. J Invest Dermatol. 2020;140(12):2515–2524.e3. doi:10.1016/j.jid.2020.04.010
25. Rinkevich Y, Walmsley GG, Hu MS, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348(6232):aaa2151. doi:10.1126/science.aaa2151
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.