")
Back to Journals » International Medical Case Reports Journal » Volume 10
Dyschromatosis symmetrica hereditaria with cutaneous lupus erythematosus and hyperthyroidism
Authors Al-Saif F, Alhumidi A, Alhallaf RA
Received 16 January 2017
Accepted for publication 24 February 2017
Published 2 May 2017 Volume 2017:10 Pages 149—152
DOI https://doi.org/10.2147/IMCRJ.S132489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Fahad Al-Saif,1 Ahmed Alhumidi,2 Rama Ayed Alhallaf1
1Dermatology Department, 2Pathology Department, College of Medicine, King Saud University, Riyadh, Saudi Arabia
Abstract: Dyschromatosis symmetrica hereditaria (DSH) is a rare genodermatosis characterized by various sizes of both hyper- and hypopigmented macules arranged in reticulated patterns on the face and the dorsal aspects of the extremities. There are also cutaneous and extracutaneous abnormalities, but they are rare. As far as we know, DSH associated with immune-mediated conditions has not been reported. We report the first case of DSH, which is associated with cutaneous lupus erythematosus and hyperthyroidism.
Keywords: dyschromatosis, genodermatosis, lupus, reticulated, autoimmune
Introduction
Dyschromatosis symmetrica hereditaria (DSH), initially known as reticulate acropigmentation of Dohi, is a rare pigmentary genodermatosis that was initially reported in the Japanese population. It is characterized by the presence of both hyper- and hypopigmented macules that are arranged in a reticular pattern and distributed symmetrically over the face, trunk, and distal limbs, especially the dorsa of the hands and feet.1,2 DSH usually starts in infancy or early childhood and persists for life.3 DSH is caused by heterozygous mutations of adenosine deaminase acting on RNA1 (ADAR1) gene.4 In general, systemic involvement is uncommon, but patients with neurological abnormalities, psoriasis, intracranial hemangiomas, Parry–Romberg syndrome, and thalassemia have been reported3,5–7 However, to our knowledge, no cases have reported the coexistence of multiple autoimmune abnormalities and DSH in a patient. We, therefore, present the case of a 21-year-old female with the coexistence of DSH and multiple autoimmune abnormalities. The patient has provided written informed consent to publish this case report with images.
Case report
A 21-year-old female with a history of progressive asymptomatic hypo- and hyperpigmented skin lesions distributed over her face, neck, upper, and lower extremities for 12 years presented to our clinic with a 1-year history of photosensitivity and red scaly lesions over the face. She reported that her symptoms were associated with mild migratory joint pain, but with no associated redness, swelling, or muscle pain. There were no other remarkable systemic symptoms. She had a history of nodular goiter and hyperthyroidism (previously treated with total thyroidectomy). She is currently on l-thyroxine. Her family history was positive for both consanguinity and similar pigmentary lesions in a distant cousin.
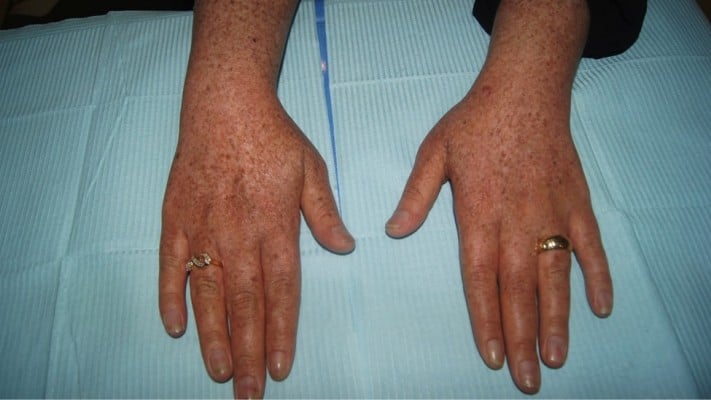
On examination, she had multiple erythematous, indurated plaques with adherent scales on some lesions over the cheeks and lips (Figure 1). There were extensive freckle-like brown macules on her face, neck, and upper chest (Figure 2). Moreover, she had both reticulated hyper- and hypopigmented macules over upper and lower limbs, especially over the dorsa of her hands and feet (Figure 3). Two scars were noted consisting of a surgical scar over the base of the neck and a trauma scar over her left cheek. Scalp examination revealed a 2×1 cm, hairless plaque and decreased hair density all over her scalp (Figure 4). No mucosal lesions, facial telangiectasia, or palmer pits were noted. Joint swelling and tenderness were not appreciated.
 | Figure 1 Freckle-like brown macules and erythematous, indurated plaques with adherent scales. |
 | Figure 2 Reticulate hyperpigmented and hypopigmented macules on neck and upper chest. |
 | Figure 3 Reticulate hyperpigmented and hypopigmented macules on dorsa of hands. |
 | Figure 4 Discoid-like patch over the scalp. |
For the histopathological examination, biopsies were taken from both hypo- and hyperpigmented lesions (Figures 5 and 6). Hyperpigmented lesions showed an abundance of melanin pigment in keratinocytes and melanocytes with the presence of a few scattered melanophages in hyperpigmented macules. In contrast, hypopigmented lesions showed a reduction in melanin and number of melanocytes as determined by Melan A immunostaining. Moreover, face biopsy revealed focal vascular interface reaction, which is a finding that can be seen in many conditions such as cutaneous lupus erythematosus (CLE).
 | Figure 5 H&E stain, original magnification ×400. Note: Biopsy specimen taken from a hyperpigmented macule shows abundance of melanin pigment in keratinocytes and melanocytes and a few scattered melanophages. Abbreviation: H&E, hematoxylin and eosin. |
 | Figure 6 H&E stain, original magnification ×200. Note: Biopsy specimen taken from a hypopigmented macule shows reduced melanin and number of melanocytes. Abbreviation: H&E, hematoxylin and eosin. |
Investigations revealed normal laboratory findings, including complete blood count, basic metabolic panel, hepatic function profile, prothrombin time, and partial thromboplastin time. Urinalysis showed leukocytes and red blood cells. C-reactive protein was 18 mg/dL (reference range 0–10). Antinuclear antibody titer was 1:1280 with coarse speckled pattern. Anti-double-stranded DNA was 17.79 IU/ mL (reference indicates <200 IU/mL is negative). Anti-Sjögren’s syndrome-related antigens A and B (SS-A and -B), Smith, and ribonucleoprotein antibodies were not detected. Complement and aldolase levels were normal. Thyroid panels showed both high thyroid-stimulating hormone (5.3 mIU/L, reference range 0.250–5) and free thyroxine (22.090 pmol/L, reference range 12–22) levels.
CLE was established through clinical features, and we started total sunblock, topical steroids, and 200 mg daily hydroxychloroquine. We referred the patient to the Rheumatology and Endocrinology Department for further investigation and treatment.
Discussion
DSH is a genodermatosis characterized by autosomal dominant inheritance disorder; however, autosomal recessive inheritance and sporadic cases have also been reported.4,8 Many novel ADAR1 mutations have been described in DSH patients. Although these cases predominantly occurred in East Asian individuals, few DSH cases have been described originating elsewhere.2,4,8,9
DSH usually starts in early childhood and is characterized by the presence of multiple hyper- and hypopigmented macules in a reticular configuration and a symmetrical distribution over the extremities, especially the dorsa of the hands and feet.2,5,9 Cases with more widespread lesions involving the face, neck, and chest have been described.6 Our patient started to have skin lesions at a later age (10 years) when compared with most of the reported cases; late onset of DSH, however, has been reported in a few cases.10
Histologically, DSH is characterized by an abnormal melanosome synthesis rate and melanocyte activity with significantly reduced melanin content and melanocytes in the hypopigmented areas. In contrast, the hyperpigmented areas show melanosomes scattered sparsely in the melanocytes and many small melanosomes dispersed or aggregated in the adjacent keratinocytes and occasional pigment incontinence.11,12 Histological examination has limited capabilities to differentiate between DSH and other reticulated pigmentary disorders such as reticulate acropigmentation of Kitamura and dyschromatosis universalis hereditaria. We, however, diagnosed DSH as the most likely cause due to the overall clinical features, particularly the type and distribution of pigmentation and absence of palmar pits.
In general, DSH affects skin pigmentation; there are, however, reported sporadic cases with cutaneous and extracutaneous abnormalities. Most reported cases of extracutaneous abnormalities are neurological abnormalities such as mental deterioration, dystonia, developmental regression, autistic disorder, depression, seizure disorder, intracranial hemangiomas, and Parry–Romberg syndrome.5,6,13 On the other hand, few cutaneous dermatoses have been reported with DSH-like psoriasis and neurofibromatosis type 1.7,14
Our patient was diagnosed with DSH, CLE, and previously treated hyperthyroidism. Clustering of multiple autoimmune disorders has been described as a spectrum ranging from involving one organ (such as autoimmune thyroiditis and CLE) to involving multiple organs.15 However, an extensive PubMed search revealed no association between DSH and autoimmune diseases. Although two cases of DSH have been reported from Saudi Arabia, to the best of our knowledge, our patient is the first case reported with associated CLE and hyperthyroidism.2,16
Disclosure
The authors report no conflicts of interest in this work.
References
Toyama I. Dyschromatosis symmetrica hereditaria. Jpn J Dermatol. 1929;29:95–96. | ||
Al Hawsawi K, Al Aboud K, Ramesh V, Al Aboud D. Dyschromatosis universalis hereditaria: report of a case and review of the literature. Pediatr Dermatol. 2002;19(6):523–526. | ||
Hayashi M, Suzuki T. Dyschromatosis symmetrica hereditaria. J Dermatol. 2013;40(5):336–343. | ||
Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73(3):693–699. | ||
Oyama M, Shimizu H, Ohata Y, Tajima S, Nishikawa, T. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140(3):491–496. | ||
He PP, He CD, Cui Y, et al. Refined localization of dyschromatosis symmetrica hereditaria gene to a 9.4-cM region at 1q21–22 and a literature review of 136 cases reported in China. Br J Dermatol. 2004;150(4):633–639. | ||
Shi BJ, Xue M, Liu Y, Jiang Y, Diao QC. First report of the coexistence of dyschromatosis symmetrica hereditaria and psoriasis: one novel TCT to A mutation in the double-RNA-specific adenosinedeaminase gene. J Eur Acad Dermatol Venereol. 2012;26(5):657–658. | ||
Zhang G, Shao M, Li Z, et al. Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Med Genet. 2016;17:14. | ||
Namitha P, Sacchidanand S. Dyschromias: a series of five interesting cases from India. Indian J Dermatol. 2015;60(6):636. | ||
Gaiewski CB, Zuneda Serafini S, Werner B, Deonizio JM. Dyschromatosis symmetrica hereditaria of late onset? Case Rep Dermatol Med. 2014;2014:639537. | ||
Nuber UA, Tinschert S, Mundlos S, Hauber I. Dyschromatosis universalis hereditaria: Familial case and ultrastructural skin investigation. Am J Med Gen A. 2004;125A(3):261–266. | ||
Kondo T, Suzuki T, Mitsuhashi Y, et al. Six novel mutations of the ADAR1 gene in patients with dyschromatosis symmetrica hereditaria: histological observation and comparison of genotypes and clinical phenotypes. J Dermatol. 2008;35(7):395–406. | ||
Dutta A, Ghosh SK, Mandal RK. Dyschromatosis symmetrica hereditaria with neurological abnormalities. Indian J Dermatol Venereol Leprol. 2014;80(6):549–551. | ||
Tan HH, Tay YK. Neurofibromatosis and reticulate acropigmentation of Dohi: a case report. Pediatr Dermatol. 1997;14(4):296–298. | ||
Lin WY, Chang CL, Fu LS, Lin CH, Lin HK. Systemic lupus erythematosus and thyroid disease: A 10-year study. J Microbiol Immunol Infect. 2015;48(6):676–683. | ||
Alfadley A, Al Ajlan A, Hainau B, Pedersen KT, Al Hoqail I. Reticulate acropigmentation of Dohi: a case report of autosomal recessive inheritance. J Am Acad Dermatol. 2000;43(1 Pt 1):113–117. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.