")
Back to Journals » OncoTargets and Therapy » Volume 12
Dual targeting of estrogen receptor α and estrogen-related receptor α: a novel endocrine therapy for endometrial cancer
Authors Mao X , Dong B, Gao M, Ruan G , Huang M, Braicu EI, Sehouli J, Sun P
Received 17 May 2019
Accepted for publication 16 July 2019
Published 20 August 2019 Volume 2019:12 Pages 6757—6767
DOI https://doi.org/10.2147/OTT.S216146
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
XiaoDan Mao,1,* Binhua Dong,1,* Min Gao,2 GuanYu Ruan,1 MeiMei Huang,1 Elena Ioana Braicu,3 Jalid Sehouli,3 PengMing Sun1
1Laboratory of Gynecologic Oncology, Fujian Provincial Maternity and Children’s Hospital, Affiliated Hospital of Fujian Medical University, Fuzhou 350001, People’s Republic of China; 2Department of Gynecology Oncology, Peking University Cancer Hospital, Beijing 100142, People’s Republic of China; 3Department of Gynecology, Campus Virchow-Klinikum, Charité Universitätmedizin Berlin, Berlin D-13353, Germany
Correspondence: PengMing Sun
Laboratory of Gynecologic Oncology, Fujian Provincial Maternity and Children’s Hospital, Affiliated Hospital of Fujian Medical University, No. 18 Daoshan Road, Fuzhou 350001, Fujian, People’s Republic of China
Tel +86 1 378 887 3900
Fax +86 5 918 755 1247
Email [email protected]
*These authors contributed equally to this work
Background: Endometrial cancer (EC) is a hormone dependent carcinoma that may involve complex molecular mechanisms. Endocrine therapy by blocking the estrogen and estrogen receptor α (ERα) has been effective in breast cancer, while it is still controversial in EC. Recently, estrogen-related receptor α (ERRα) was proven to be another endocrine therapy target.
Methods: The anti-tumor effect of selective estrogen receptor modulators (SERMs) and XCT790 (XCT) used alone or in combination were evaluated in both of ERα-positive (ERα+) and ERα-negative (ERα-) EC cells. ERα and ERRα mRNA were tested by qPCR, while the protein was detected by Western blot. The proliferation was tested by MTS and cell cycle, apoptosis rate were analyzed by flow cytometry.
Results: A relatively high dose (10 μM) of tamoxifen (TAM) suppressed the expression of ERα and ERRα in two types of EC cells. However, 10 μM raloxifene (RAL) exhibited no effect on ERα and ERRα, while 10 μM XCT down regulated ERRα specifically, but not ERα in all EC cells. When dual targeting on ERα and ERRα by combining TAM with XCT, the proliferation inhibitory effect and apoptosis reached the strongest in all EC cells (P<0.05). Moreover, the inhibitory effect of proliferation was attributed significantly to the G0/G1 arrest (P<0.05). Interestingly, the apoptosis induced by combining TAM with XCT were obviously higher in ERα+ EC cells than ERα- EC cells (P<0.05).
Conclusion: Taken together, the results indicate that dual targeting on ERα and ERRα represents a better anti-tumor effect, which provides a novel endocrine based therapy strategy for EC.
Keywords: ERα, ERRα, SERM, XCT790, anti-tumor effect
Introduction
There are an estimated 878,980 women in the United States with a previous diagnosis of uterine corpus cancer in 2018.1 The incidence of endomerial cancer (EC) is also slowly but stably increasing over the last two decades in China, with an estimated 63,400 new cases of EC and 21,800 estimated deaths in 2015.2 Breast cancer, EC, and ovarian cancer are known to be hormone-dependent cancers,3,4 with estrogen playing a predominant role in the proliferation and exacerbation.5 At present, hormonal therapies available usually aim to block estrogen and estrogen receptor (ER) binding and obtain better therapeutic outcomes in breast cancer.6 However, the management of advanced stage and recurrent EC remains controversial, as current treatments, such as endocrine therapy, yield few improvements in long-term survival rates.7
In 1983, Bokhman8 proposed that there were two different pathogenetic types of endometrial carcinoma: type Ⅰ, which accounts for 70–80% of cases and are generally ER-positive (ER+), and type Ⅱ, accounting for 20% cases and are ER-negative (ER-). Well-differentiated tumors generally express ERs and PRs and respond to hormonal therapy.9,10 Endocrine therapy targeting estrogen and ER showed a certain anti-tumor effect on ER+ ECs. However, the loss of steroid hormone receptor expression is common in patients with recurrent estrogen-related cancers, ultimately hampering the clinical utility of hormonal therapy.11 Estrogen-related receptor (ERR) α, an orphan member of the nuclear receptor superfamily, is a constitutively active receptor that shares considerable structural homology with the classical ERα and ERβ. Matsushima et al12 suggested that ERRα may serve as a novel molecular target for the EC treatment. On this basis, our previous research showed that exogenous XCT790 down regulating ERRα had a higher anti-tumor effect in ERα+ EC cells, while endogenous siRNA targeting ERRα displayed a better endocrine therapy in ERα- EC cells.13 Thus, the different anti-tumor effect exerted by ERRα down regulation depended on whether or not ERα was expressed in EC cells.
Based on the findings, we hypothesize that dual targeting of ERα and ERRα was the best treatment strategy for EC. Selective estrogen receptor modulators (SERMs) are characterized by their diverse range of agonist/antagonist effects on ER-mediated processes. Two of the most common clinically available SERMs are TAM and RAL, which are considered to act predominantly as estrogen antagonists in breast cancer cells.14,15 However, the effect of SERMs on EC remains unclear.16,17 In this study, we tested the anti-tumor effect of SERMs and/or XCT790 (specific antagonist of ERRα) on two types of EC cell lines in order to evaluate the best strategy for EC endocrine treatment.
Materials and methods
Cell culture and drug treatment
Human RL952, HEC-1A, and HEC-1B endometrial adenocarcinoma cells were obtained from the Shanghai Cell Biological Research Institute (Shanghai, China), and ECC-1 cells were acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA). RL952 and ECC-1 cells are ERα+, while HEC-1A and HEC-1B cells are ERα-. RL952 and ECC-1 cells were thawed and cultured in DMEM/F12 medium with 0.005 mg/ml insulin, 1% antibiotic–antimycotic solution, and 10% fetal bovine serum (FBS) or in RPMI-1640 medium supplemented with 10% FBS. HEC-1A and HEC-1B cells were cultured in high-glucose DMEM supplemented with 10% FBS at 37°C in 5% CO2. XCT790, TAM, and RAL were purchased from Sigma (St. Louis, MO, USA) and were dissolved in dimethyl sulfoxide (DMSO) at 25°C. Aliquots of stock solutions at 1 mM were stored at −20°C. Cells were transferred to phenol red-free medium (Invitrogen, Carlsbad, CA, USA) containing 1% serum-replacement-2 ( Sigma-Aldrich, St. Louis, MO, USA) for 24 h. Then, cells were treated for various lengths of time with TAM (10 μM), RAL (10 μM), XCT790 (XCT; 10 μM), TAM + XCT790 (T+X; 10 μM), RAL + XCT790 (R+X; 10 μM), or no drugs as a blank control.
Relative real-time quantitative PCR analysis
Total RNA was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Next, 1 µg of DNase I-treated RNA was reverse-transcribed into cDNA using a reverse transcription kit (Promega, Madison, WI, USA). The following PCR primers were used: ERα: sense, 5′-TGG GCT TAC TGA CCA ACC TG-3′; anti-sense, 5′- CCT GAT CAT GGA GGG TCA AA-3′ (99 bp); ERRα: sense, 5′-ACC GAG AGA TTG TGG TCA CCA-3′; anti-sense, 5′-CAT CCA CAC GCT CTG CAG TACT-3′ (101 bp); GADPH (control): sense, 5′-GCA CCG TCA AGG CTG AGA AC-3′; anti-sense, 5′-TGG TGA AGA CGC CAG TGGA-3′ (138 bp). Relative levels of ERα and ERRα mRNA were quantified by real-time quantitative PCR (qPCR) and calculated by the 2−ΔΔCT method.
Western blotting analysis
Western blotting was performed using standard procedures. The cell culture dish was transferred to ice, and cells were washed with ice-cold phosphate-buffered saline (PBS). Cell lysates were prepared with lysis/extraction reagent (Thermo Fisher Scientific, Waltham, MA, USA). Cells were homogenized in lysis buffer, and the supernatant was removed and conserved after centrifugation at 12,000 rpm for 15 min at 4°C. Proteins were quantified with a BCA protein assay reagent kit (Pierce, Rockford, IL, USA). Then, 30 µg of protein derived from the whole-cell lysates of cells treated with one of the five drug treatments was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Blotted membranes were incubated with anti-human ERα rabbit polyclonal antibody (Proteintech, Rosemont, IL, USA) at a dilution of 1:1000 or anti-human ERRα rabbit monoclonal antibody (Abcam, Cambridge, UK) at a dilution of 1:300 overnight at 4°C. Then, an enhanced chemiluminescence (ECL) detection system (Beyotime, Shanghai, China) was used to visualize the bands. The results were calculated based on the ratio of the densities of specific bands to that of the β-actin control.
3- (4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) analysis
All cells were plated and grown in 96-well plates at a concentration of 10,000 cells/well for 24 h. Cells were then treated with various drugs for a period of 0, 24, 48, 72 or 96 h. After the addition of MTS dye (20 µl), the 96-well plates were incubated for 1–2 h at 37°C. Then, 100 µl DMSO was added to the plates in order to terminate the MTS reaction, and the plates were subsequently analyzed by measuring the absorption at 490 nm with a microplate reader (Bio-Rad, Hercules, CA, USA). Each experiment was repeated three times to assess the consistency of the results.
Cell cycle analysis
Cells were seeded at a density of 1×105 cells/well into 6-well plates and cultured until 80% confluence. Then, cells were transferred to phenol red-free medium (Invitrogen, Carlsbad, CA, USA) containing 1% serum-replacement-2 ( Sigma-Aldrich, St. Louis, MO, USA) for 24 h and were treated with TAM, RAL, XCT, T+X, R+X, or no drugs as a control for 24 h. Cells were fixed and stained with propidium iodide (PI; 100 μg/ml) (BD Biosciences, Franklin Lakes, NJ, USA) and then analyzed by BD FACSCantoⅡ™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) for cell cycle analysis. All experiments were performed in triplicate.
Apoptosis analysis
For flow cytometric analysis of apoptosis, cells were seeded into 6-well plates and cultured until 80% confluence. Cells were released by digestion with 0.25% trypsin and harvested. After centrifugation, the cell pellets were washed twice with pre-cooled PBS. Then, cells were resuspended in buffer to 105/ml. Apoptosis was detected using the Annexin-V-FLUOS staining kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Annexin-V and PI fluorescence was measured using a FACS Canto II™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). All experiments were performed in triplicate.
Statistical analysis
All experiments were repeated a minimum of three times. Data are presented as mean ± S.E.M. Statistical analysis of differences between groups was performed with two-sided unpaired Student’s t-tests and ANOVA using SPSS statistical software (version 19.0, SPSS, Inc., Chicago, IL, USA). A value of P<0.05 was considered significant.
Results
Changes in ERα and ERRα expression following treatment of EC cells
After treatment of the EC cell lines with TAM, RAL, XCT, T+X, or R+X for 24 h, we performed qPCR analysis to determine the relative expression levels of ERα and ERRα mRNA. In RL952 cells, ERα expression was significantly decreased in the TAM and T+X groups but significantly increased in the RAL group (P<0.05), while XCT790 had no effect on ERα mRNA levels (P>0.05). In contrast, ERRα expression was significantly downregulated in all RL952 treatment groups (P<0.05). Furthermore, the combination of SERMs with XCT790 treatment provided a clear advantage in inhibiting the expression of ERRα mRNA compared with treatment with SERMs alone (P<0.05, Figure 1A). In ECC-1 cells, only TAM and T+X treatment resulted in downregulation of ERα (P<0.05), while RAL and/or XCT790 had no effect on the expression of ERα mRNA (P>0.05). ERRα mRNA levels were significantly downregulated by all treatment strategies except RAL (P<0.05). Similarly, the combination of SERM treatment plus XCT790 increased the inhibitory effect on the expression of ERRα mRNA when compared with that of SERM treatment alone (P<0.05, Figure 1B). Similar results were obtained in HEC-1A cells (Figure 1C). There was no expression of ERα mRNA in HEC-1B cells. However, the expression of ERRα mRNA was significantly suppressed by all drug treatments (P<0.05) except for RAL treatment alone. The combination of SERMs with XCT790 treatment increased the inhibition of ERRα mRNA expression when compared with that of SERM treatment alone in HEC-1B cells (P<0.05, Figure 1D). Similar changes in the expression of ERα and ERRα were also observed at the protein level following treatment of the EC cell lines (Figure 2A–D). These results suggest that TAM treatment downregulates both ERα and ERRα, while the inhibitory effect of RAL on ERα and ERRα is neutral. In contrast, XCT790 treatment results in the specific inhibition of ERRα. Compared to treatment with a single SERM, the expression of ERRα is significantly reduced by treatment with SERM+XCT. More specifically, T+X resulted in the greatest inhibitory effect on ERRα expression in all EC cell lines (P<0.05).
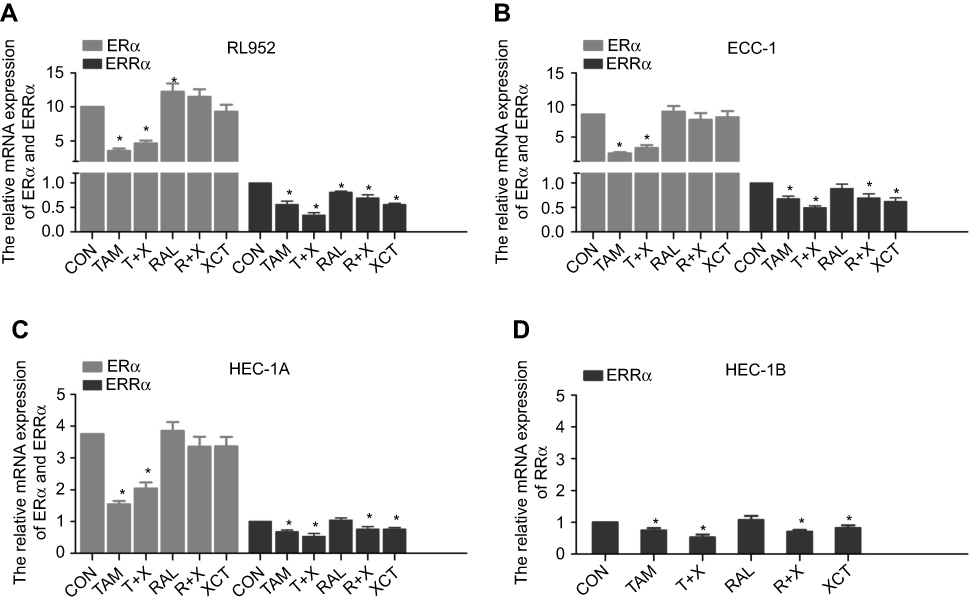 |
Figure 1 Expression of ERα and ERRα following treatment of EC cells with SERMs and/or XCT790. The mRNA levels of ERα and ERRα after treatment of RL952 (A), ECC-1 (B), HEC-1A (C), and HEC-1B (D) cells with 10 μM tamoxifen (TAM), tamoxifen combined with XCT790 (T+X), raloxifene (RAL), raloxifene combined with XCT790 (R+X), or XCT790 (XCT) for 24 h, as determined by real-time PCR. Levels in all five treatment groups were compared with those in cells treated with 0.1% DMSO as a control (CON). Data represent means ± SEM. All experiments were repeated in triplicate. *P<0.05. |
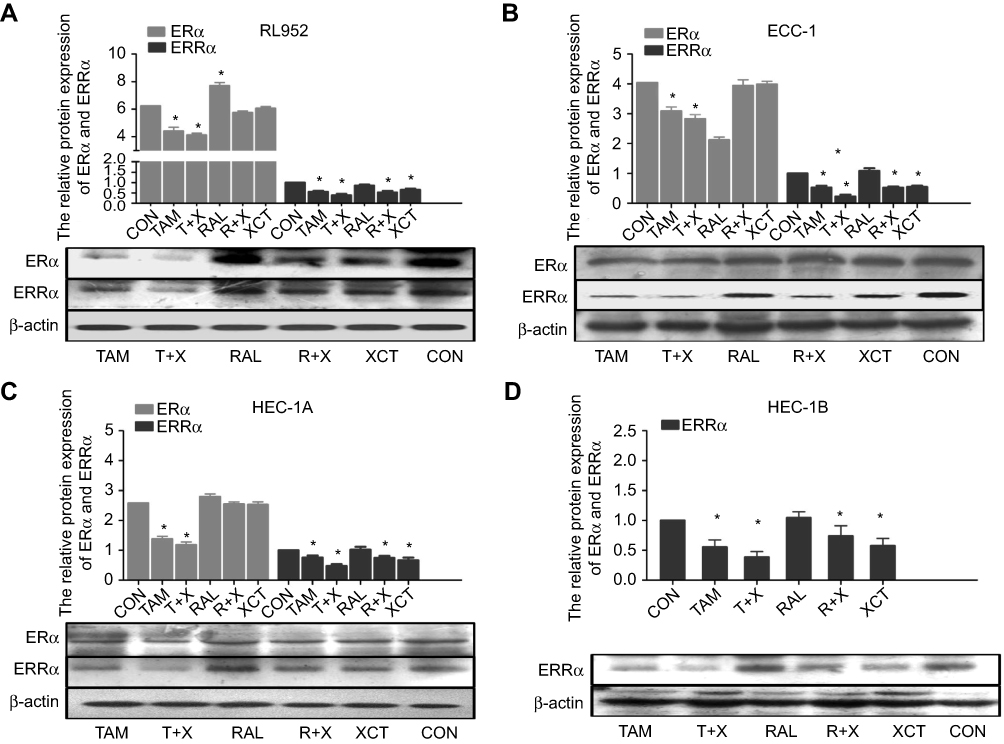 |
Figure 2 Protein expression of ERα and ERRα following treatment of EC cells with SERMs and/or XCT790. The protein levels of ERα and ERRα after treatment of RL952 (A), ECC-1 (B), HEC-1A (C), and HEC-1B (D) cells with 10 μM TAM, T+X, RAL, R+X, or XCT for 24 h as determined by Western blotting. Data represent means ± SEM. All experiments were repeated in triplicate. *P<0.05. |
Effects of treatment on EC cell proliferation
The results of the cell proliferation experiments showed that all five drug treatment strategies were effective at inhibiting the proliferation of EC cells at a concentration of 10 μM in a time-dependent manner. In RL952 cells, the extent of inhibition was greatest in the T+X group. This was followed by the TAM, XCT, and R+X groups, which all had similar rates of inhibition, with the lowest rate of inhibition in the RAL treatment group (P<0.05, Figure 3A). Similar trends were observed in ECC-1 (Figure 3B) and HEC-1B (Figure 3D) cells. In HEC-1A cells, the greatest rate of inhibition was also observed in the T+X groups; however, this was followed by the TAM and R+X groups, which exhibited similar rates of inhibition, and then the XCT group, with the RAL group again exhibiting the lowest inhibitory rate (P<0.05, Figure 3C). Thus, according to our results, all EC cells were most sensitive to T+X treatment and least sensitive to RAL treatment. To evaluate the cytotoxic effect of 10 μM T+X in cells, half maximal inhibitory concentration (IC50) was calculated. Fortunately, the IC50 were 9.30 μM, 10.2 μM, 8.23 μM and 8.06 μM in RL952, ECC-1, HEC-1A and HEC-1B for 24 hrs, respectively. By CalcuSyn software, the combination index (CI) of 10 μM T+X was also calculated, which were 2.251, 2.301,1.872 and 1.935 in RL952, ECC-1, HEC-1A and HEC-1B cells, respectively. From the CI values, TAM combined with XCT790 displayed the role of antagonism in EC cells.
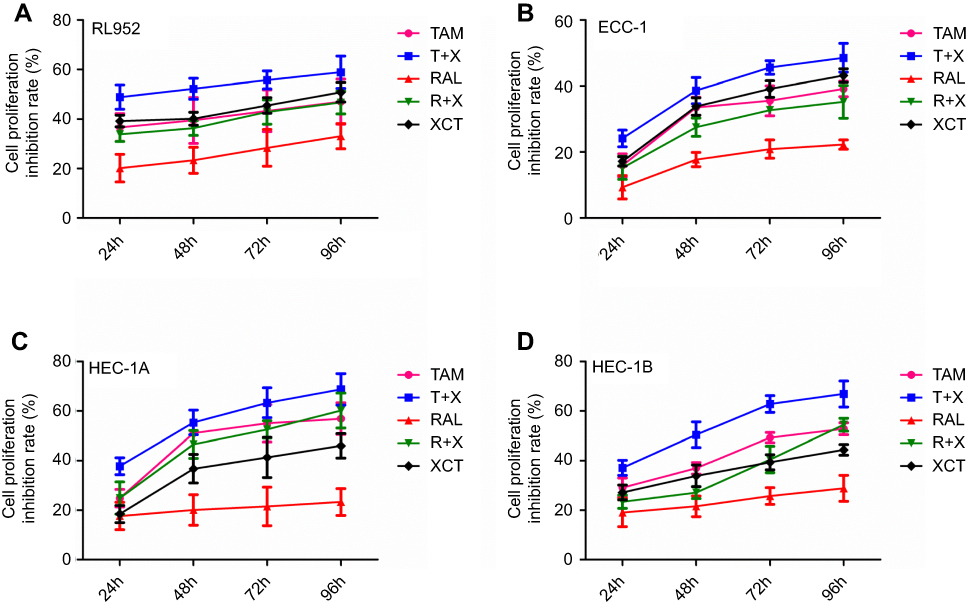 |
Figure 3 Effect of SERMS and/or XCT790 treatment on EC cell proliferation. The proliferative capacities of RL952 (A), ECC-1 (B), HEC-1A (C), and HEC-1B (D) cells were evaluated by MTS assay following TAM, T+X, RAL, R+X, or XCT treatment for 0, 24, 48, 72, or 96 h. Data represent means ± SEM. All experiments were repeated in triplicate. |
Cell cycle arrest following treatment of EC cells
The observed effects of SERMs and XCT790 on EC cell proliferation led us to evaluate the effect of these agents on the cell cycle progression of RL952 (Figure 4A), ECC-1 (Figure 4B), HEC-1A (Figure 4C), and HEC-1B (Figure 4D) cells. First, by analyzing PI staining with FCM, we investigated whether SERMs and/or XCT790 treatment would affect the distribution of cells within the three major phases of the cycle. Compared with the control group, the percentage of RL952 cells in the G0/G1 phase was significantly increased and that in the S phase was decreased after treatment with TAM, T+X, R+X, and XCT (P<0.05, Figure 4E). A similar result was obtained for ECC-1 cells, except that only the percentage of cells in the G0/G1 phase was significantly increased following TAM only treatment (P<0.05, Figure 4F). In contrast, in HEC-1A cells, the percentage of G0/G1-phase cells was significantly increased and those of S and G2/M phase cells were decreased following all treatment strategies except RAL (P<0.05, Figure 4G). Moreover, the percentage of cells in G0/G1 phase was significantly increased but that in G2/M phase was decreased in HEC-1B cells treated with all drugs except RAL (P<0.05, Figure 4H). These results suggest that the downregulation of ERRα by XCT790 and TAM mainly blocks the G1/S transition of the cell cycle in all type of EC cells. In addition, downregulation of ERα by TAM results in cell cycle arrest in the G0/G1 phase, while RAL has no effect on the cell cycle in EC cells.
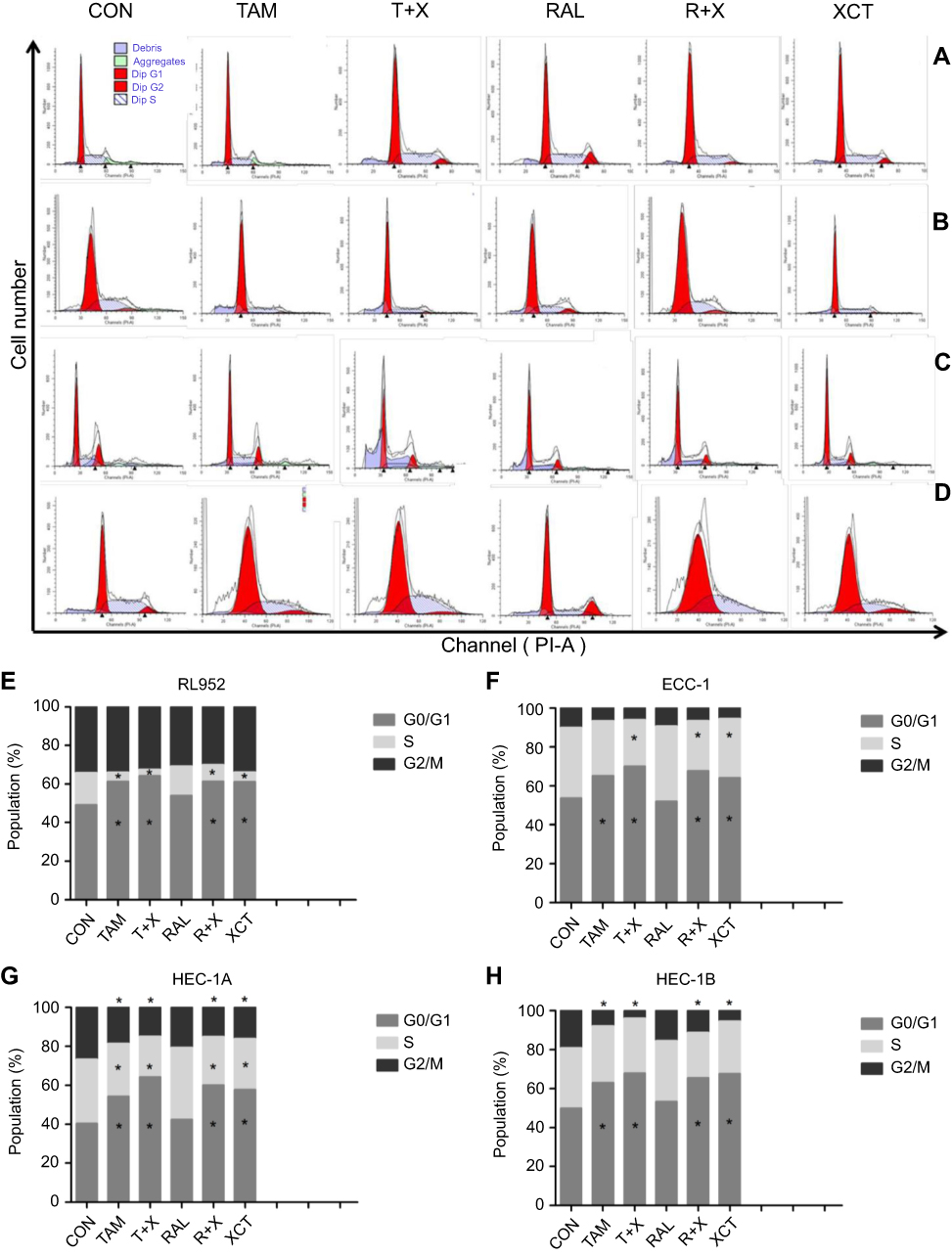 |
Figure 4 Effect of SERMS and/or XCT790 treatment on cell cycle progression of EC cells. Cell cycle changes were assessed by flow cytometry (FCM) in RL952 (A), ECC-1 (B), HEC-1A (C), and HEC-1B (D) cells treated with TAM, T+X, RAL, R+X, or XCT for 24 h. Cell distributions in each phase of the cell cycle were determined in RL952 (E), ECC-1 (F), HEC-1A (G), and HEC-1B (H) cells. All experiments were repeated in triplicate. *P<0.05. |
Effects of treatment on EC cell apoptosis
Next, rates of apoptosis were examined in RL952, ECC-1, HEC-1A, and HEC-1B cells treated with the five drug treatment strategies for 24 h (Figure 5A–D). The T+X treatment strategy led to the highest rates of apoptosis in all four EC cell lines and represented a significant increase in apoptosis over that resulting from treatment with TAM alone (P<0.05). In contrast, there was no significant impact of RAL treatment on EC cell apoptosis (Figure 5E–H). Moreover, our results suggested that ER+ EC cells are more sensitive to drug treatment, especially to treatment with XCT790 and TAM, than ER- EC cells (P<0.05).
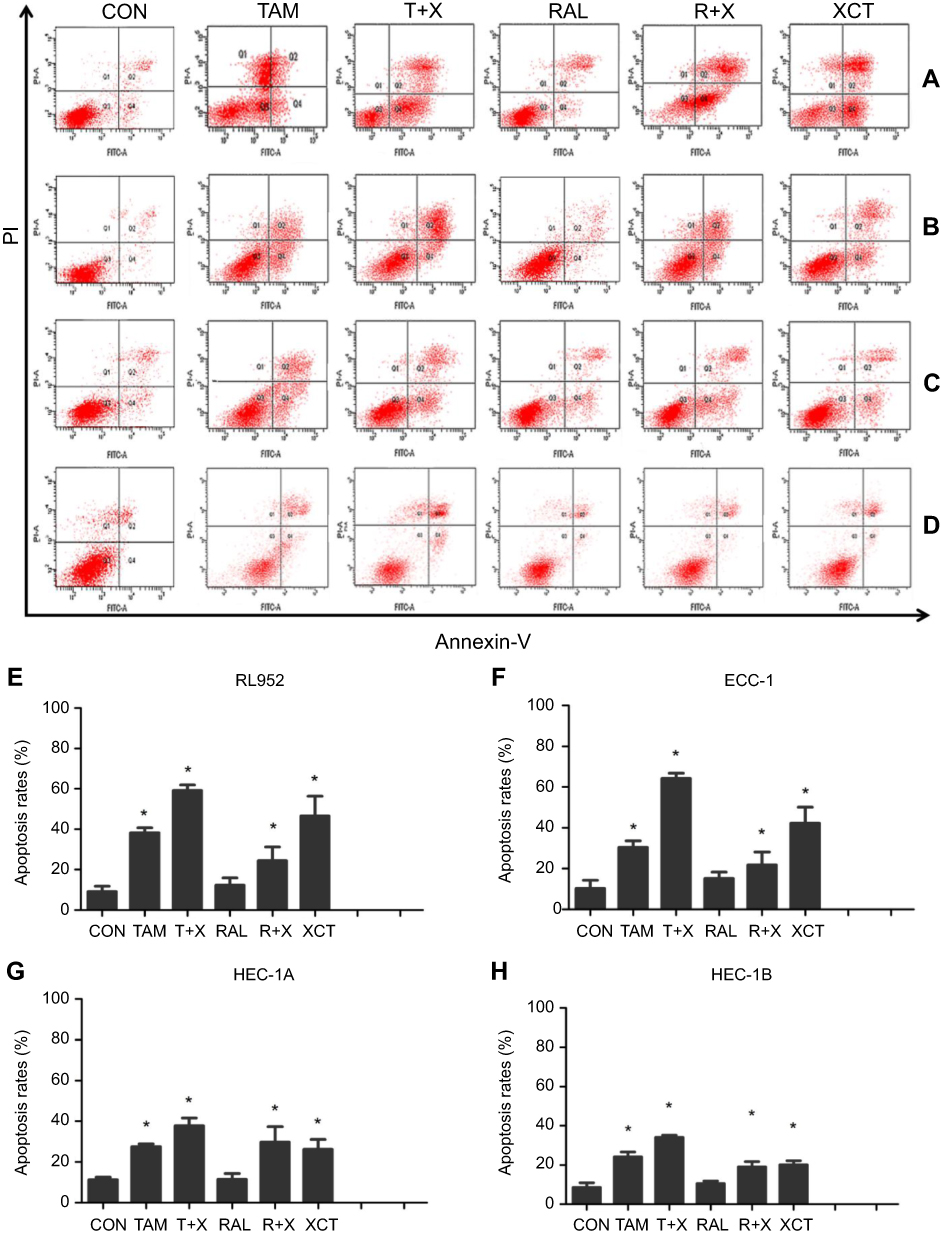 |
Figure 5 Effect of SERMS and/or XCT790 treatment on EC cell apoptosis. Cell apoptosis was assessed by flow cytometry (FCM) in RL952 (A), ECC-1 (B), HEC-1A (C), and HEC-1B (D) cells treated with TAM, T+X, RAL, R+X, or XCT for 24 h. Rates of cell apoptosis were determined in RL952 (E), ECC-1 (F), HEC-1A (G), and HEC-1B (H) cells. Data represent means ± SEM. All experiments were repeated in triplicate. *P<0.05. |
Discussion
It has been confirmed that blocking estrogen and ER exhibited anti-tumor effects in EC. In 1965, Kelley & Baker18 were the first to use progesterone to antagonize estrogen in the treatment of patients with advanced EC. Guo et al19 suggested that the specific ER antagonist ICI 182780 may be a valid approach for treating ER+ EC. With the clinical application of SERMs in endocrine therapy, some studies have considered that TAM (standard maintenance dose/0.5 µM) may stimulate endometrial hyperplasia and invasion.20,21 However, Zhou et al22 showed that a relatively high dose of TAM (50 µM) repressed proliferation and promoted apoptosis in EC cells. Thus, the effect of TAM on tumors appears to be dose-dependent. In this study, we found that treatment with 10 µM TAM attenuated the expression of ERα in all EC cells; thus, it was clear that TAM had anti-estrogen effects at high doses. Carlson et al23 showed that estrogenic compounds such as TAM increased the expression of PRs in EC, which should theoretically increase the effectiveness of EC treatment. Additionally, Whitney et al24 found that a combination of daily TAM and intermittent weekly medroxyprogesterone acetate was an active treatment for advanced or recurrent EC.
Furthermore, we also found that TAM downregulated the expression of ERRα in all EC cells. In 2001, Coward et al25 confirmed that ERRα was not directly affected by ERα antagonists such as tamoxifen. Our previous research26 showed that 17β-estradiol (17β-E2) down regulated ERRα expression in ER+ EC cells and that the down regulation of 17β-E2 in ERRα-expressing cells could be blocked by ICI 182780. Therefore, we speculate that TAM regulates ERRα in an indirect manner mediated by ERα. However, we found ERRα was also down regulated by TAM in ERα- EC cells. Suga et al27 found that the blockade of both the ERK1/2 and ER signaling pathways had a greater inhibitory effect on gynecologic tumor cell growth. Zhou et al found that the anti-tumor effect of TAM on ER+ and ER- EC cells was mediated through distinct MAPK pathways, which cross-talked with estrogen-ER signaling. Deblois et al28 suggested ERRα and PGC-1β also participated in the derepression of ERBB2 expression through competitive genomic cross-talk with ERα and, as a consequence, influenced TAM sensitivity in breast cancer cells. Hence, TAM regulating ERRα was involved in multiple signal pathways. Moreover, Thewes et al29 showed that low-dose TAM (0.1 μM/1 μM)upregulated ERRα in ER+ TAM resistant breast cancer cells, while Manna et al30 suggested that treatment with TAM caused reduction in expression of survivin, an anti-apoptotic protein, indicating the cell death-inducing potential of TAM in vitro. A high expression of nuclear ERRα was associated with a significant benefit from TAM treatment, of which the mechanism was unclear (Figure 6). However, it was obvious that there was some relationship between TAM and ERRα. In our study, a relatively high concentration of TAM (10 μM) downregulated ERRα, inhibited EC cell proliferation by blocking cell cycle transition at the G0/G1 phase and promoted EC cell apoptosis. These results again suggest that the anti-tumor effects of TAM on EC are dose-dependent.
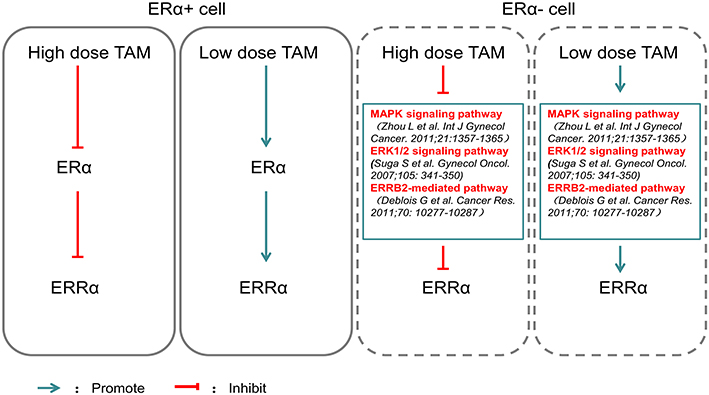 |
Figure 6 Effects of different doses of TAM on ERα and ERRα. High dose TAM inhibited ERα expression, which mediated the down regulation of ERRα, while low dose TAM exerted the opposite effect on ERα and ERRα in ERα+ EC cells. High dose TAM inhibited ERRα though these signaling pathways crosstalked with estrogen-ER signaling, while low dose TAM exerted the opposite effect on ERRα in ER- EC cell. |
RAL, a second-generation SERM, exhibited no effect on the expression of ERα or ERRα in three EC cell lines. Thus, although both TAM and RAL are SERMs, they have distinctive effects on the endometrium; however, the underlying mechanisms of their disparate effects are not yet fully understood. Shang et al31 suggested that estrogen and SERMs affect the transduction of cellular signaling pathways that govern cell growth and proliferation via downstream effectors such as PAX2. Although the application of RAL remains controversial in EC, DeMichele et al16 found that RAL users had significantly lower probability of developing EC compared with both TAM users and SERM non-users, suggesting a role for RAL in EC prevention and the individualization of SERM therapy. However, Hibner et al32 found that RAL did not inhibit the growth of EC cells in vitro and its high concentrations promoted cell growth. In our study, in comparison with the control, although RAL significantly influenced cell proliferation, there were no obvious changes in the cell proliferation inhibition rate over time. This indicates that the observed effect was related to the general toxicity of the drug.
In this study, we confirmed that the ERRα-specific antagonist XCT790 showed no effect on ERα but exhibited an anti-tumor effect on EC cell lines. XCT790 treatment exerted time-dependent inhibitory effects on the proliferation of EC cells. Bianco et al33 showed that XCT790 modulates the activity of ERRα and reduces the proliferation of various cell lines by blocking the G1/S transition of the cell cycle in an ERRα-dependent manner. This is consistent with our data showing that XCT790 blocked the G1/S transition of the cell cycle following ERRα downregulation in all EC cell lines. Rates of XCT790-induced apoptosis in ER+ EC cells were significantly higher than those in ER- EC cells, indicating that the increase in the apoptotic rate attributed to ERRα downregulation was likely mediated by ERα activity.
When combining TAM and XCT790, we achieved better anti-tumor effects than were observed with either TAM or XCT790 alone in all cells, confirming our above hypothesis. This is also in agreement with the results of Thewes et al29 who reported that combining XCT790 with TAM or fulvestrant suppressed cell viability more effectively than treatment with either TAM or fulvestrant alone in TAM-resistant breast cancer cells. However, combining RAL with XCT790, the anti-tumor effect was similar to that observed following treatment with XCT790 only, indicating that the anti-tumor effect of the R+X combination was mainly attributable to XCT790.
In general, ER+ EC cells were more sensitive to drug treatment than ER- EC cells, reflecting their associations with ERα activity. TAM exhibited a high potential for use in endocrine therapy, as it regulated ERα when used at a relatively high dose. XCT790 specifically targets ERRα, resulting in cell cycle arrest, inhibition of cell proliferation, and increased apoptosis in EC cells. Dual targeting of ERα and ERRα results in better anti-tumor effects in EC than the individual blockade of either ERα or ERRα. At present, there are few studies of the combination of XCT790 and SERMs in treating EC. Thus, in the future, the evaluation of the potential effects of TAM combined with XCT790 in vivo are necessary. Meanwhile, the new drug targeting on ERα and ERRα will be explored to apply in EC endocrine therapy.
Acknowledgments
This work was partly sponsored by the Youth Foundation of Fujian Provincial Health and Family Planning Commission (2015-1-16), the Natural Science Foundation of Fujian Province (grant nos. 2016J01491, 2017J01233) and the Innovation Fund of Fujian Provincial Maternity and Children’s Hospital (grant no. YCXQ 18-19).
Disclosure
Prof. Dr. Elena Ioana Braicu reports personal fees from Roche Pharma, Clovis, Tesaro, AstraZeneca, Immunogen, MSD, Eisai, Carrick Therapeutics and Millenium Takeda, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–296. doi:10.3322/caac.21442
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi:10.3322/caac.21338
3. Subramani R, Nandy SB, Pedroza DA, Lakshmanaswamy R. Role of growth hormone in breast cancer. Endocrinology. 2017;158:1543–1555.
4. Skrzypczak M, Merx I, Schuler-Toprak S, et al. Molecular profiling of estrogen receptor alpha and progesterone receptor transcript variants in endometrial cancer. Steroids. 2015;104:122–128.
5. Pasqualini JR. Estrogen sulfotransferases in breast and endometrial cancers. Ann N Y Acad Sci. 2009;1155:88–98.
6. Gala K, Li Q. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function. Oncogene. 2018;37(34):4692–4710. doi:10.1038/s41388-018-0273-5.
7. Ito K, Utsunomiya H, Niikura H, Yaegashi N, Sasano H. Inhibition of estrogen actions in human gynecological malignancies: new aspects of endocrine therapy for endometrial cancer and ovarian cancer. Mol Cell Endocrinol. 2011;340:161–167.
8. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15:10–17.
9. Dackus G, Jozwiak K, Sonke GS, et al. Optimal adjuvant endocrine treatment of ER+/HER2+ breast cancer patients by age at diagnosis: a population-based cohort study. Eur J Cancer. 2018;90:92–101.
10. Ruiz MP, Huang Y, Hou JY, et al. All-cause mortality in young women with endometrial cancer receiving progesterone therapy. Am J Obstet Gynecol. 2017;217:669e661–669e613. doi:10.1016/j.ajog.2017.08.007
11. Kuukasjarvi T, Kononen J, Helin H, Holli K, Isola J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J Clin Oncol. 1996;14:2584–2589. doi:10.1200/JCO.1996.14.9.2584
12. Matsushima H, Mori T, Kokabu T, et al. Anti-tumor effect of estrogen-related receptor alpha knockdown on uterine endometrial cancer. Oncotarget. 2016;7:34131–34148. doi:10.18632/oncotarget.9151
13. Sun P, Mao X, Gao M, et al. Novel endocrine therapeutic strategy in endometrial carcinoma targeting estrogen-related receptor α by XCT790 and siRNA. Cancer Manag Res. 2018;10:2521–2535. doi:10.2147/CMAR.S168043
14. Osborne CK. Tamoxifen in the treatment of breast cancer. Drug Ther (NY). 1998;339:1609–1618.
15. Fan P, Craig Jordan V. Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations. Steroids. 2014;90:44–52. doi:10.1016/j.steroids.2014.06.002
16. DeMichele A, Troxel AB, Berlin JA, et al. Impact of raloxifene or tamoxifen use on endometrial cancer risk: a population-based case-control study. J Clin Oncol. 2008;26:4151–4159. doi:10.1200/JCO.2007.14.0921
17. Nakamura K, Sawada K, Sugiyama M, et al. Efficacy of raloxifene hydrochloride for the prevention of health care problems in patients who undergo surgery for endometrial cancer: a multicenter randomized clinical trial. Int J Gynecol Cancer. 2015;25:288–295. doi:10.1097/IGC.0000000000000333
18. Kelley RM, Baker WH. The role of progesterone in human endometrial cancer. Cancer Res. 1965;25:1190–1192.
19. Guo RX, Wei LH, Zhao D, Wang JL, Li XP. [Effects of ICI182780 (Faslodex) on proliferation and apoptosis induced by 17beta-estradiol in endometrial carcinoma cells]. Beijing Da Xue Xue Bao Yi Xue Ban. 2006;38:470–474.
20. Bai JX, Yan B, Zhao ZN, et al. Tamoxifen represses miR-200 microRNAs and promotes epithelial-to-mesenchymal transition by up-regulating c-Myc in endometrial carcinoma cell lines. Endocrinology. 2013;154:635–645. doi:10.1210/en.2012-1607
21. Wood CE, Kaplan JR, Fontenot MB, Williams JK, Cline JM. Endometrial profile of tamoxifen and low-dose estradiol combination therapy. Clin Cancer Res. 2010;16:946–956. doi:10.1158/1078-0432.CCR-09-1541
22. Zhou L, Cai B, Bao W, et al. Crosstalk between estrogen receptor and mitogen-activated protein kinase signaling in the development and progression of endometrial cancer. Int J Gynecol Cancer. 2011;21:1357–1365. doi:10.1097/IGC.0b013e3182216ac9
23. Carlson JA
24. Whitney C. Phase II study of medroxyprogesterone acetate plus tamoxifen in advanced endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2004;92:4–9.
25. Coward P, Lee D, Hull MV, Lehmann JM. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci U S A. 2001;98:8880–8884.
26. Sun PM, Gao M, Wei LH, et al. An estrogen receptor alpha-dependent regulation of estrogen receptor-related receptor alpha in the proliferation of endometrial carcinoma cells. Int J Gynecol Cancer. 2006;16(Suppl 2):564–568.
27. Suga S, Kato K, Ohgami T, et al. An inhibitory effect on cell proliferation by blockage of the MAPK/estrogen receptor/MDM2 signal pathway in gynecologic cancer. Gynecol Oncol. 2007;105:341–350.
28. Deblois G, Chahrour G, Perry MC, et al. Transcriptional control of the ERBB2 AMPLICON by ERRa and PGC-1b promotes mammary gland tumorigenesis. Cancer Res. 2011;70:10277–10287.
29. Thewes V, Simon R, Schroeter P, et al. Reprogramming of the ERRalpha and ERalpha target gene landscape triggers tamoxifen resistance in breast cancer. Cancer Res. 2015;75:720–731.
30. Manna S, Bostner J, Sun Y, et al. ERRalpha is a marker of tamoxifen response and survival in triple-negative breast cancer. Clin Cancer Res. 2016;22:1421–1431.
31. Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat Rev Cancer. 2006;6:360–368.
32. Hibner M, Magrina JF, Lefler SR, et al. Effects of raloxifene hydrochloride on endometrial cancer cells in vitro. Gynecol Oncol. 2004;93:642–646.
33. Bianco S, Lanvin O, Tribollet V, et al. Modulating estrogen receptor-related receptor-alpha activity inhibits cell proliferation. J Biol Chem. 2009;284:23286–23292.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.