")
Back to Journals » Journal of Inflammation Research » Volume 14
Dual Activity of Ginsenoside Rb1 in Hypertrophic Cardiomyocytes and Activated Macrophages: Implications for the Therapeutic Intervention of Cardiac Hypertrophy
Authors Wang S, Cui Y, Xiong M, Li M, Wang P, Cui J, Du X, Chen Y , Zhang T
Received 12 March 2021
Accepted for publication 15 April 2021
Published 5 May 2021 Volume 2021:14 Pages 1789—1806
DOI https://doi.org/10.2147/JIR.S310633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Shihua Wang,1 Yimeng Cui,1 Minqi Xiong,1,2 Mei Li,1 Peiwei Wang,1,2 Jingang Cui,1,2 Xiaoye Du,1,2 Yu Chen,1– 3 Teng Zhang1,2
1Yueyang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 2Clinical Research Institute of Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 3Laboratory of Clinical and Molecular Pharmacology, Yueyang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
Correspondence: Teng Zhang; Yu Chen
Shanghai University of Traditional Chinese Medicine, 110 Ganhe Rd, Shanghai, 200437, China
Tel +86-21-55982301
; +86-21-55982302
Email [email protected]; [email protected]
Purpose: Owing to the important mechanistic implications in the pathogenesis of cardiac hypertrophy and heart failure, inflammation has been proposed as a druggable target for the treatment of cardiac hypertrophy and heart failure. Ginseng is a widely used medicinal herb for the treatment of cardiovascular disorders. As one of the major chemical components of ginseng, ginsenoside Rb1 (Rb1) contributes to the cardiovascular effects of ginseng. Meanwhile, anti-inflammatory activity of Rb1 has also been documented. The current work aims to further delineate the pharmacological implications of Rb1 in the treatment of cardiac hypertrophy.
Methods: Angiotensin II (Ang II) infusion mouse model was adopted to investigate the effects of Rb1 on cardiac hypertrophic remodeling and associated inflammation in vivo. Furthermore, the mechanisms of actions of Rb1 in modulating the hypertrophic and inflammatory responses were investigated in cardiomyocytes and macrophages, respectively.
Results: Rb1 mitigates Ang II–induced cardiac hypertrophy, cardiac inflammation and systemic inflammation in vivo. In cardiomyocytes, Rb1 directly counteracts the pro-hypertrophic effects of Ang II and phenylephrine and maintains the mitochondrial function. In lipopolysaccharide (LPS)-stimulated macrophages, Rb1 decreases the phosphorylation of mitogen-activated protein kinases (MAPKs) and mitogen-activated protein kinase kinase 1/2 (MEK1/2) and reduces the production of inflammation mediators such as interleukin (IL)-1 beta, IL-6 and tumor necrosis factor (TNF). Rb1 also suppresses the expression of pro-hypertrophic microRNA-155 (miR-155) in LPS- or Ang II-stimulated macrophages. Furthermore, in activated macrophages, miR-155 is in part accountable for the suppressive effect of Rb1 on the production of IL-6, an inflammation mediator with pro-hypertrophic functions in the heart.
Conclusion: The work here provides novel experimental evidence supporting the notion that Rb1 protects against cardiac hypertrophy in part through suppressing the inflammatory mechanisms that promotes the pathological remodeling of the heart.
Keywords: angiotensin II, inflammation, interleukin 6
Introduction
Cardiac hypertrophy is an independent risk factor for adverse clinical outcomes in patients with cardiovascular disorders.1 Sustained cardiac hypertrophy can progress to heart failure, a multifactorial clinical syndrome that constitutes a major cause of mortality and disability. In particular, cardiac hypertrophy is the most common morphological alterations associated with heart failure with preserved ejection fraction.2 Few effective therapies are currently available for the treatment of heart failure with preserved ejection fraction, which affects approximately half of heart failure patients worldwide and is plagued with poor prognosis similar to the patients with reduced ejection fraction or borderline ejection fraction.3 Therefore, it is of important clinical significance to control the progression of heart failure at the stage of cardiac hypertrophy.3,4
The clinical management is challenged by complex mechanisms underlying the pathogenesis of cardiac hypertrophy and failure. It is well established that renin-angiotensin-aldosterone system (RAAS) plays a critical role in the initiation and progression of cardiac hypertrophy and heart failure. However, even with the available treatment targeting RAAS and improving hemodynamics, the mortality rate associated with heart failure remains high. Inflammation has been shown to promote the development and progression of cardiac hypertrophy and heart failure. Proinflammatory responses mediated by activated macrophages are causally involved in the development of cardiac hypertrophy and heart failure.5,6 Meanwhile, the crosstalk between RAAS activation and inflammation has also been noted in the process of cardiac hypertrophy and heart failure. For example, it has been shown that cardiac hypertrophy induced by angiotensin II (Ang II), the principal functional mediator of RAAS, is in part mediated by macrophage expression of microRNA-155 (miR-155), a central regulator of macrophage activation in inflammatory processes.7 Therefore, inflammation may serve as a valuable drug target for the control of cardiac hypertrophy and heart failure.
Ginsenoside Rb1 (Rb1) is abundantly present in ginseng, a medicinal herb well recognized for its prophylactic and therapeutic cardiovascular and anti-inflammatory activities.8,9 It has been demonstrated that Rb1 is pharmacologically effective at attenuating heart failure induced by abdominal aorta constriction,10 monocrotaline,11 and prostaglandin F2 alpha.12 In addition, the activity of Rb1 at suppressing the inflammatory responses in activated macrophages has been reported.13 Our previous study has also shown that Rb1 decreases the expression of miR-155 in lipopolysaccharide (LPS)-stimulated macrophages.14 However, it is unknown whether Rb1 is effective at counteracting the pro-hypertrophic effect of Ang II. Meanwhile, whether Rb1 suppresses cardiac hypertrophy-associated inflammation remains to be addressed.
In the current study, Ang II infusion mouse model was adopted to investigate the pharmacological effects of Rb1 against RAAS activation-associated cardiac hypertrophy and inflammation in vivo. In addition, in vitro experiments addressing the direct impact of Rb1 on macrophage inflammation and cardiomyocyte hypertrophy were carried out to further elucidate the pharmacological implications of Rb1 in the treatment of cardiac hypertrophy.
Materials and Methods
Reagents
Rb1 (purity>98%) was purchased from Shanghai Yuanye Biotechnology Co., Ltd (China). Ang II and LPS were ordered from Sigma-Aldrich (USA). Phenylephrine (PE) was ordered from Tokyo Chemical Industry (Japan). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and penicillin/streptomycin were ordered from Thermo Fisher Scientific (USA). MiR-155 agomir (agomir-155) and negative control agomir (agomir-NC) were ordered from Guangzhou RiboBio Co., Ltd (China). Granulocyte-macrophage colony-stimulating factor was purchased from Sino Biological (China).
Animal Treatment
Seventy two C57BL/6J male mice (6–8 weeks of age weighing 24.1±1.6 g, Shanghai Laboratory Animal Research Center, China) were used in this study. The mice were maintained under controlled temperature (20±2°C), humidity (35–55%) and lighting (12 h light/dark cycle) conditions with access to food and water ad libitum. All animal handling procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Yueyang Hospital, Shanghai University of Traditional Chinese Medicine and performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Ang II was dissolved in sterile saline solution and infused at the dose of 1 μg/kg/min for 14 days via subcutaneously implanted micro-osmotic pumps (ALZET Model 1002, Durect Corporation, USA). For the gross assessment, morphological/immunohistochemical examinations, ultrastructural assessment and analysis of systemic inflammation, Ang II–infused mice received daily treatment of either vehicle (saline solution) or Rb1 at the doses of 6.25, 25 or 100 mg/kg body weight through intraperitoneal injection. For the experiments on cardiac gene expression, Ang II–infused mice received daily treatment of either vehicle or Rb1 at 100 mg/kg body weight through intraperitoneal injection. For sham controls, the vehicle-treated mice were implanted with subcutaneous micro-osmotic pumps loaded with saline solution. Rb1 or vehicle was delivered in a controlled volume of 100 μL for each intraperitoneal injection.
Gross Assessment and Histological Examination
The body weight was measured prior to euthanization and heart dissection. Following euthanization, the heart weight (HW) and the tibia length (TL) were measured. Subsequently, the dissected hearts were fixed in 4% paraformaldehyde, processed for paraffin embedding and cross-sectioned at the thickness of 5 μm. Heart sections were then stained with hematoxylin and eosin (H&E) and Picrosirius Red to evaluate the gross morphological changes and fibrosis, respectively. Micrographical images were acquired using a light microscope (DM2000, Leica, Germany). Image analysis was performed using Image J.
Immunohistochemistry (IHC)
Heart sections were antigen-retrieved, blocked and incubated with the indicated primary and secondary antibodies. The primary antibodies included Alexa Fluor 488-conjugated anti-wheat germ agglutinin (WGA) antibody (W11261, Thermo Fisher Scientific, USA), monoclonal mouse anti-CD68 antibody (ab31630, Abcam, USA) and monoclonal rat anti-CD11b antibody (14–0112-81, Thermo Fisher Scientific, USA). The primary antibody was incubated at 4°C overnight. FITC-conjugated goat anti-mouse or anti-rat secondary antibody (Sigma, USA) was then applied at room temperature for 2 h. The heart sections were also counterstained with 4-6-diamidino-2-phenylindole (DAPI) to help visualize the nuclei. The images were observed and recorded using a fluorescence microscope (DM6000B, Leica, Germany). The immunopositivity of the indicated staining was analyzed using Image J.
Transmission Electron Microscopy (TEM)
The heart specimens were fixed in 2.5% glutaraldehyde for 2 h before further processing. The ultrathin sections 50 nm-thick were then stained by uranyl acetate and lead citrate. The images were obtained using a transmission electron microscope (Tecnai G2 Spirit Bio TWIN, FEI Company, USA).
Cell Culture and Treatment
H9c2 cardiac cell line was purchased from Chinese Academy of Sciences Cell Bank of Type Culture Collection (Shanghai, China) and cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. H9c2 cells within 6 passages were seeded in 24-well plates at the number of 1×104 cells/well and subject to the indicated experiments. For the experiments involving Ang II stimulation, H9c2 cells were treated with Rb1 at the indicated concentrations for 30 min, followed by Ang II incubation at 1 μM for 48 h. To assess the effect of Rb1 on PE-induced hypertrophy, H9c2 cells were treated with Rb1 at the indicated concentrations for 30 min, followed by PE incubation at 100 μM for 48 or 72 h. To evaluate the effect of Rb1 on PE-induced changes in atrial natriuretic peptide (ANP) and mitochondrial function, H9c2 cells were treated with Rb1 at 100 μM for 30 min, followed by PE incubation at 100 μM for 72 h. RAW 264.7 macrophage cell line was obtained from Shanghai Institute of Biological Science (Shanghai, China) and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. RAW 264.7 cells within 6 passages were used for the indicated experiments. For the analysis of the levels of proinflammatory cytokines, RAW264.7 cells were seeded in 12-well plates at the number of 5×105 cells/well and incubated with Rb1 at the indicated concentrations or vehicle for 30 min, followed by LPS (5 ng/mL) incubation for 4 or 24 h. For the analysis of the expression of proinflammatory genes, RAW264.7 cells were seeded in 12-well plates at the number of 5×105 cells/well and incubated with Rb1 at the indicated concentrations or vehicle for 30 min, followed by LPS (5 ng/mL) incubation for 4 h. For Western blotting experiments, RAW264.7 cells were seeded in 6-well plates at the number of 1×106 cells/well and incubated with Rb1 at the indicated concentrations or vehicle for 30 min, followed by LPS (5 ng/mL) incubation for 30 min or 6 h. For the experiments involving miR-155 agomir transfection, RAW264.7 cells were seeded in 6-well plates at the number of 1×106 cells/well and transfected with either agomir-155 (100 nM) or agomir-NC (100 nM) using Lipofectamine™ RNAiMAX (Invitrogen, USA) following the manufacturer’s instructions. Twenty four h after the indicated transfections, cells were treated with LPS (5 ng/mL) in the presence or absence of Rb1 (100 μM) for 4 h, followed by the indicated analyses. The doses and the time points of the indicated treatments were determined by preliminary studies.
Isolation and Treatment of Bone Marrow-Derived Macrophages (BMDMs)
Bone marrow cells were isolated from the femurs and tibias of C57BL/6J mice and cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin and granulocyte-macrophage colony-stimulating factor (50 ng/mL). Cells were exposed to Ang II (1 μM) with or without Rb1 (250 μM) pretreatment from day 3 to 8. On day 9, BMDMs were harvested and subject to the indicated analyses. The doses of the indicated treatments were determined by preliminary studies.
Immunofluorescence
H9c2 cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% TritonX-100 in PBS for 15 min, and incubated with 1% BSA for 30 min at room temperature. The cells were then stained with rhodamine phalloidin (CA1610, Solarbio) or rabbit anti-ANP antibody (ab14348, Abcam) at 4°C overnight. Following anti-ANP antibody incubation, Cy3-conjugated anti-rabbit secondary antibody (C2306, Sigma-Aldrich) was applied at room temperature for 1 h. Counterstaining of DAPI was performed to visualize the nuclei. The immunofluorescence was observed and recorded using a fluorescence microscope (DMI6000, Leica, Germany). Quantification of immunofluorescence was performed using Image J.
Measurement of Mitochondrial Membrane Potential (MMP)
MMP was detected using a Mitochondrial Membrane Potential Assay kit (Beyotime, China) according to the manufacturer’s instructions. Briefly, after the indicated treatments, H9c2 cells were incubated in JC-1 detection solution at 37°C for 20 min. The red fluorescence of JC-1 aggregates and the green fluorescence of JC-1 monomers were observed and recorded using a fluorescence microscope (DMI6000, Leica, Germany). Quantification of immunofluorescence was performed using Image J.
Measurement of Mitochondrial Permeability Transition Pore (mPTP) Opening
The opening of mPTP was detected using a Mitochondrial Permeability Transition Pore Assay kit (Beyotime, China) following the manufacturer’s instructions. Briefly, after the indicated treatments, H9c2 cells were incubated in calcein acetoxymethyl ester staining solution and CoCl2 fluorescence quenching solution at 37°C for 50 min. The green fluorescent signal was visualized using a fluorescence microscope (DMI6000, Leica, Germany) and quantified by Image J.
Western Blotting
Cell lysate was extracted from RAW264.7 cells using RIPA lysis buffer (Beyotime, China) containing proteinase inhibitors and phosphatase inhibitors (Roche, Germany). The lysate was separated in 10% SDS-PAGE gels and transferred onto polyvinylidene fluoride membranes (Millipore, USA). Primary antibodies for Western blotting analysis were ordered from Cell Signaling Technology (USA), including monoclonal mouse anti-phospho-extracellular signal-regulated kinase 1/2 (Erk1/2) (#9106), polyclonal rabbit anti-Erk1/2 (#9102), monoclonal rabbit anti-phospho-p38 (#4511), monoclonal rabbit anti-p38 (#8690), monoclonal rabbit anti-phospho-c-Jun N-terminal kinase (JNK) (#4668), polyclonal rabbit anti-JNK (#9252), monoclonal rabbit anti-phospho-mitogen-activated protein kinase kinase 1/2 (MEK1/2) (#2338), monoclonal mouse anti-MEK1/2 (#4694), and monoclonal rabbit anti-β-tubulin antibody (#2128). After primary antibody incubation, horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibody (Promega, USA) was applied. WesternBright ECL reagent (Advansta, USA) was used to visualize the signal, which was then scanned using an UVP BioSpectrum imaging system (BioSpectrum 410, USA). Densitometry analysis was performed using Image J.
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum specimens from the experimental animals or culture media collected from RAW 264.7 cells were analyzed to quantify the level of interleukin (IL)-1 beta, IL-6, and tumor necrosis factor (TNF) using ELISA assay kits (Multi Sciences, China) following the manufacturer’s instructions. The absorbance at 450 nm and 630 nm were measured using a microplate reader (BioTek, USA).
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted from RAW 264.7 cells or homogenized heart samples using miRNeasy Mini Kit (Qiagen, Germany), followed by reverse transcription using PrimeScript RT Master Mix (TaKaRa, Japan) or miScript II RT Kit (Qiagen, Germany). The mRNA expression of Il1b, Il6, Tnf, natriuretic peptide type A (Nppa), natriuretic peptide type B (Nppb), myosin, heavy polypeptide 7, cardiac muscle, beta (Myh7) and miR-155 was analyzed using LightCycler 480 SYBR Green I Master (Roche, Germany) on a LightCycler 480 II system (Roche, USA). 18S ribosomal RNA (rRNA) or RNU6B was amplified as the internal control for the normalization purposes. The primer sequences are included in the Table 1 The fold change in the expression was calculated according to 2−[Ct (candidate)-Ct (internal control)].
 |
Table 1 Primer Sequences for Real-Time qPCR Analyses |
Statistical Analysis
The in vitro experiments were independently repeated at least 3 times. The data were expressed as mean±standard error of mean (S.E.M). The statistical analyses were performed by one-way ANOVA with the Turkey multiple-comparisons test. Statistically significance was defined as P<0.05.
Results
Rb1 Attenuates Ang II-Induced Cardiac Hypertrophy
To assess the pharmacological effects of Rb1 on Ang II–induced cardiac hypertrophy, Ang II infusion was delivered at 1 μg/kg/min for 14 days and the effects of Rb1 were assessed at the end of Ang II infusion. Increased heart weight indexes as indicated by higher HW/BW and HW/TL ratios were observed in Ang II–infused vehicle-treated mice compared to the sham controls. In contrast, significantly decreased HW/BW and HW/TL ratios were noted in Ang II–infused Rb1-treated mice at all of the doses examined (Figure 1A and B). At the histological level, WGA staining of the heart sections was performed to quantify the cross-sectional area (CSA) of cardiomyocytes. The results showed that the CSA of cardiomyocytes was increased in Ang II–infused vehicle-treated mice, whereas significantly decreased CSA of cardiomyocytes was observed in Ang II–infused Rb1-treated mice (Figure 1C and D). To validate the anti-hypertrophic effect of Rb1 at the molecular level, the experiment was independently repeated with Rb1 administered at 100 mg/kg. The gene expression analysis revealed that Ang II–induced upregulation of myh7 in the heart was significantly attenuated as a result of Rb1 treatment (Figure 1E). These results collectively indicate that Rb1 ameliorates Ang II–induced cardiac hypertrophy in vivo.
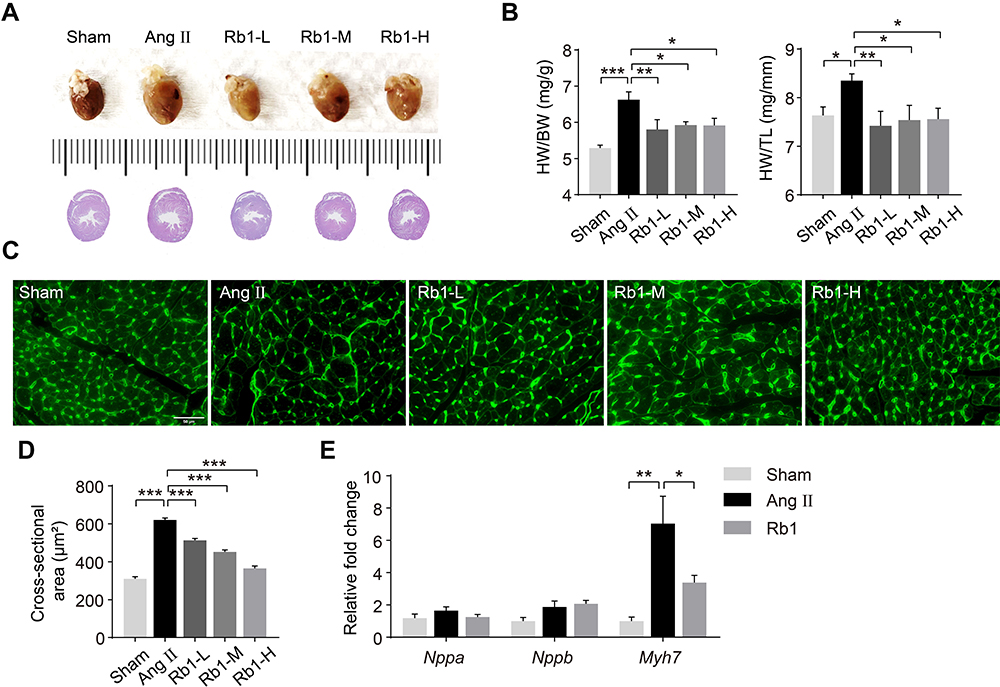 |
Figure 1 Rb1 attenuates cardiac hypertrophy in Ang II–infused mice. C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 6.25 mg/kg (Rb1-L), 25 mg/kg (Rb1-M) and 100 mg/kg (Rb1-H). (A) At the end of the indicated treatments, hearts were dissected and photographed. Additionally, H&E staining of the cardiac sections was performed to visualize the gross morphology of the heart. (B) The ratios of heart weight (HW) to body weight (BW) (left panel) and HW to tibia length (TL) (right panel) were measured (n=6-10 per group). (C) Heart sections were stained with wheat germ agglutinin (WGA) to visualize cardiomyocytes. (D) The cross-sectional area of cardiomyocytes was measured in WGA-stained heart sections (n=5 per group). Scale bar, 50 μm. (E). C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 100 mg/kg (Rb1). The mRNA expression of Nppa, Nppb and Myh7 in the heart was analyzed by real-time qPCR (n=6-7 per group). 18S rRNA was used as the internal reference. Relative fold change was plotted against that from the sham controls. Data were expressed as mean±S.E.M. * P<0.05, ** P<0.01, *** P<0.001. |
Rb1 Mitigates Ang II-Induced Myocardial Fibrosis
In addition to cardiac hypertrophy, Ang II infusion also results in myocardial fibrosis, which contributes to the structural and functional impairment of the heart. Therefore, the impact of Rb1 treatment on myocardial fibrosis was also assessed. H&E staining (Figure 2A) and Picrosirius Red staining (Figure 2B) revealed pronounced perivascular and interstitial fibrotic lesions in Ang II–infused vehicle-treated hearts. The perivascular and interstitial fibrosis appeared to be less pronounced in Ang II–infused Rb1-treated hearts. Consistent with the pathological impression, quantification of the Picrosirius Red positivity revealed significantly decreased perivascular (Figure 2C) and interstitial fibrosis areas (Figure 2D) in Ang II–infused Rb1-treated hearts compared to Ang II–infused vehicle-treated hearts. These observations indicate that Rb1 treatment alleviates Ang II–induced myocardial fibrosis in vivo.
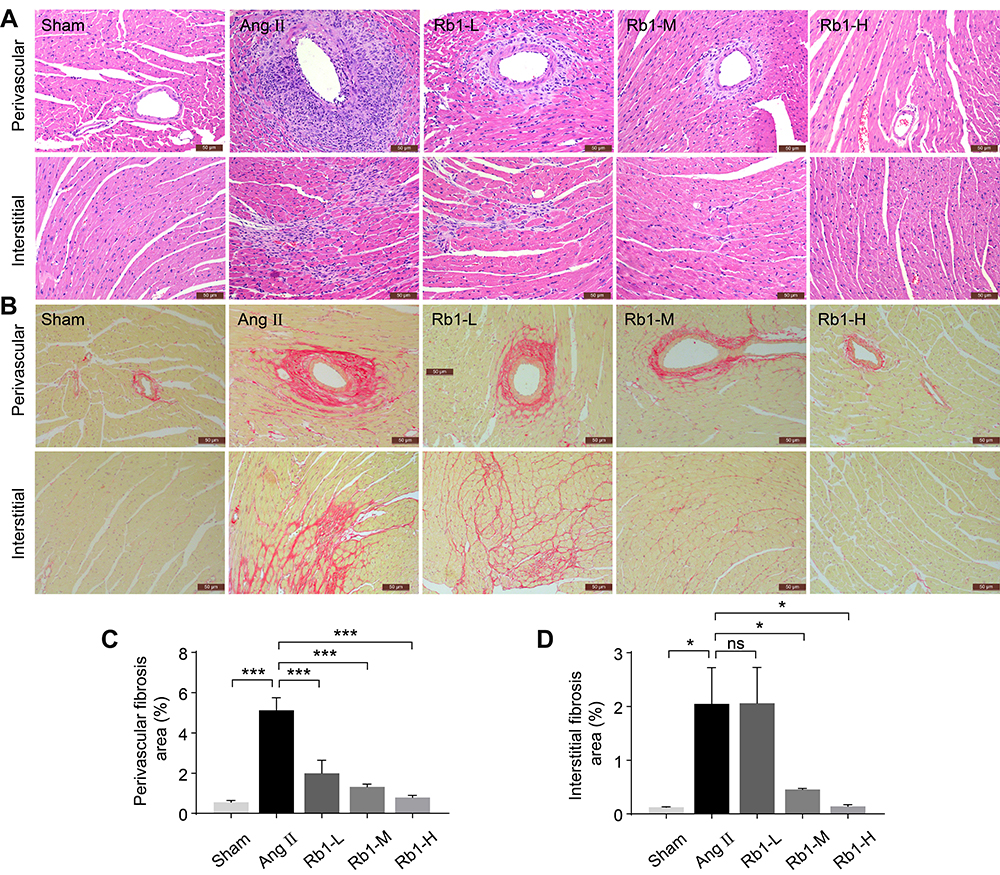 |
Figure 2 Rb1 alleviates cardiac fibrosis in Ang II–infused mice. C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 6.25 mg/kg (Rb1-L), 25 mg/kg (Rb1-M) and 100 mg/kg (Rb1-H). (A) Cardiac morphology was examined by H&E staining of paraffin-embedded heart sections. Scale bar, 50 μm. (B) Picrosirius Red staining was performed to visualize the collagen fibers in the heart sections. Scale bar, 50 μm. Perivascular (C) and interstitial fibrosis area (D) was measured in Picrosirius Red-stained heart sections (n=5 per group). Data were expressed as mean±S.E.M. * P<0.05, *** P<0.001. |
Rb1 Protects Against Ultrastructural Impairment of the Cardiomyocytes in Ang II-Infused Mice
To better understand the effect of Rb1 on the heart in Ang II–infused mice, TEM examination was further performed to assess the ultrastructural changes in the heart. As shown in Figure 3A and B, impaired mitochondrial morphology, specifically, disrupted cristae shape and loss of electron density were readily detected in the cardiomyocytes in Ang II–infused vehicle-treated hearts. In contrast, decreased number of structurally impaired mitochondria was found in Ang II–infused Rb1-treated hearts. Meanwhile, the sarcomeric Z-discs were disarrayed and the number of the Z-discs was reduced in Ang II–infused vehicle-treated hearts, whereas the pattern of the sarcomeric Z-discs was partially preserved and the number of the Z-discs was increased in Ang II–infused Rb1-treated hearts (Figure 3A and C). These observations provide ultrastructural evidence further supporting the protective effect of Rb1 against Ang II-triggered hypertrophic stress in the cardiomyocytes. Meanwhile, these results demonstrate that Rb1 protects against Ang II–induced cardiomyocyte mitochondrial impairment in vivo.
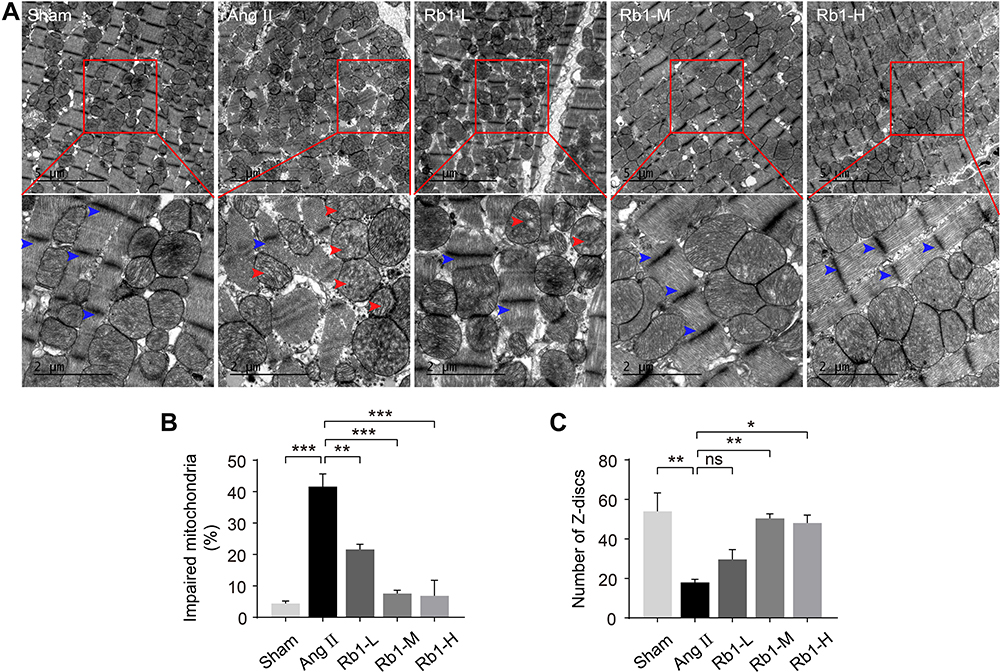 |
Figure 3 Rb1 protects against the ultrastructural impairment of cardiomyocytes in Ang II–infused mice. C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 6.25 mg/kg (Rb1-L), 25 mg/kg (Rb1-M) and 100 mg/kg (Rb1-H). Transmission electron microscopy (TEM) was performed to examine the ultrastructure of myocardium. (A) Representative TEM images showed impaired mitochondria (red arrowheads) and Z-discs (blue arrowheads). Top row: scale bar, 5 μm. Bottom row: scale bar, 2 μm. (B) The percentage of impaired mitochondria was quantified. (C) The number of Z-discs was quantified. Data were expressed as mean±S.E.M (n=3 per group). * P<0.05, ** P<0.01, *** P<0.001. |
Rb1 Alleviates Systemic and Cardiac Inflammation in Ang II-Infused Mice
It has been increasingly recognized that inflammation plays an important role in the pathogenesis of cardiac hypertrophy and failure.5 Thus, the impact of Rb1 on the inflammatory changes in Ang II–infused mice was further investigated. First, systemic inflammation was evaluated by measuring the plasma levels of IL-1 beta, IL-6 and TNF after the indicated treatments. The results showed that the plasma level of IL-6 was significantly increased in Ang II–infused vehicle-treated mice, whereas Rb1 treatment resulted in decreased plasma level of IL-6 in Ang II–infused mice (Figure 4A). IHC examination revealed that the number of CD68+ macrophages and that of CD11b+ leukocytes were significantly increased in Ang II–infused vehicle-treated hearts. In contrast, Rb1 treatment resulted in decreased number of CD68+ (Figure 4B and C) and CD11b+ cells (Figure 4D and E) in Ang II–infused hearts. In addition, real-time qPCR analysis of the expression of Il1b, Il6 and Tnf revealed that the cardiac expression of Il6 was increased in Ang II–infused vehicle-treated mice. On the contrary, significantly decreased cardiac expression of Il6 was observed in Ang II–infused Rb1-treated mice (Figure 4F). Taken together, these results indicate that Rb1 alleviates the systemic and cardiac inflammation in Ang II–infused mice.
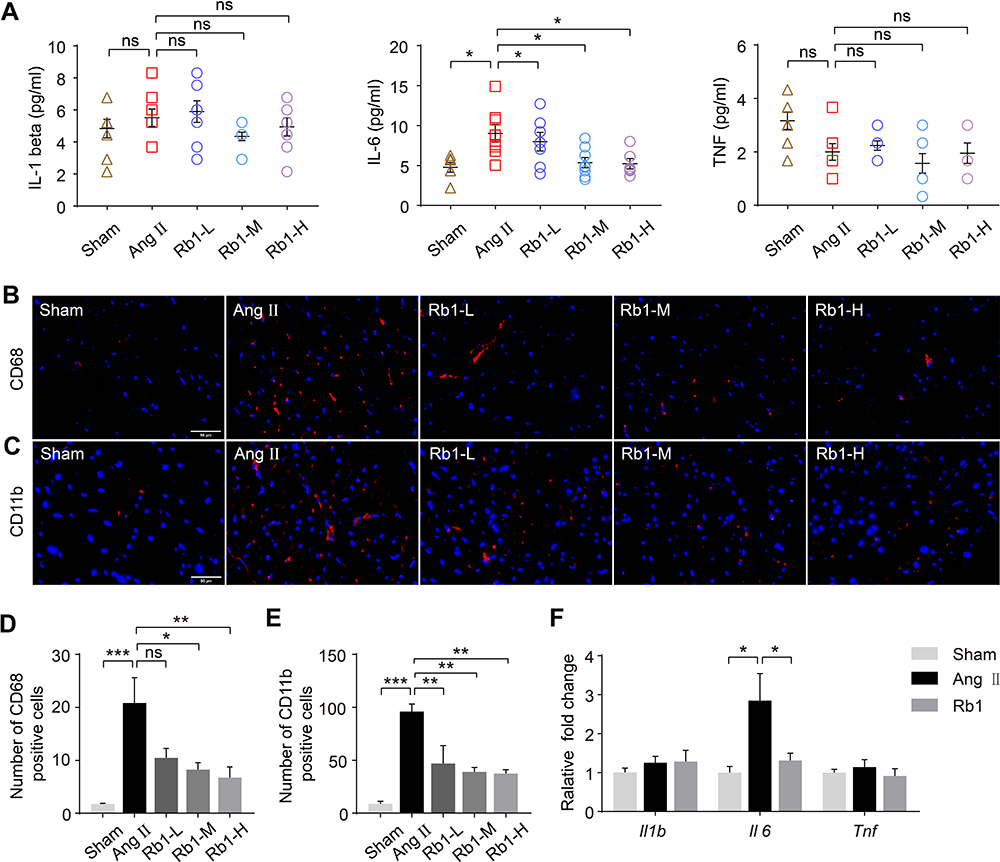 |
Figure 4 Rb1 mitigates inflammation in Ang II–infused mice. C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 6.25 mg/kg (Rb1-L), 25 mg/kg (Rb1-M) and 100 mg/kg (Rb1-H). (A) Plasma was isolated and analyze by ELISA for the level of IL-1 beta, IL-6 and TNF (n=6-8 per group). (B) Immunohistochemistry (IHC) was performed to examine the presence of CD68+ cells (in red) in the heart. DAPI (in blue) was counterstained to help visualize the nuclei. Scale bar, 50 μm. (C) The number of CD68+ cells in the heart was quantified (n=5 per group). (D) IHC was performed to reveal the presence of CD11b+ cells (in red) in the heart. DAPI (in blue) counterstaining was performed to visualize the nuclei. Scale bar, 50 μm. (E) The number of CD11b+ cells in the heart was quantified (n=5 per group). (F). C57BL/6J mice were subcutaneously infused with Ang II (1 μg/kg/min) or saline (sham) for 14 days. Ang II–infused mice received daily treatment of either vehicle (Ang II) or Rb1 at 100 mg/kg (Rb1). The cardiac expression of Il1b, II6, and Tnf was analyzed by real-time qPCR (n=6-7 per group). 18S rRNA was included as the internal control. Relative fold change was plotted against that from the sham controls. Data were expressed as mean±S.E.M. * P<0.05, ** P<0.01, *** P<0.001. |
Rb1 Exerts Direct Anti-Hypertrophic and Mitochondrial Protective Effects in Cardiomyocytes
To further address whether Rb1 exerts any direct impact on cardiac hypertrophy, the anti-hypertrophic effect of Rb1 was assessed in Ang II-stimulated H9c2 cardiomyocytes in vitro. As shown in Figure 5A and B, Ang II stimulation for 48 h resulted in a significant increase in the size of H9c2 cells. In contrast, decreased size of H9c2 cells was observed in Ang II-stimulated H9c2 cells treated with Rb1 at the concentration of 12.5, 50 or 100 μM. Rb1 also counteracted Ang II-augmented cellular level of ANP in H9c2 cells (Figure 5C and D). Similar results were obtained when H9c2 cells were subject to the stimulation of another pro-hypertrophic factor, PE. Rb1 mitigated the increases in the size of H9c2 cells that were stimulated by PE for 48 h (Supplementary Figure 1) or 72 h (Supplementary Figure 2). Taken together, these results support a direct anti-hypertrophic effect of Rb1 in cardiomyocytes.
 |
Figure 5 Rb1 antagonizes Ang II–induced hypertrophic responses in cardiomyocytes. H9c2 cells were pretreated with vehicle or Rb1 at the indicated concentrations for 30 min, followed by incubation in the presence or absence of Ang II (1 μM) for 48 h. (A) Rhodamine phalloidin staining (in red) was performed to label myofibril in the cells. DAPI counterstaining was performed to visualize the nuclei. Scale bar, 50 μm. (B) The size of H9c2 cells was measured after rhodamine phalloidin staining (n=6 per group). (C) H9c2 cells were pretreated with vehicle or Rb1 (50 μM) for 30 min, followed by incubation in the presence or absence of Ang II (1 μM) for 48 h. ANP (in red) and DAPI (in blue) staining was then performed to visualize intracellular ANP and nuclei, respectively. Scale bar, 50 μm. (D) The fluorescence intensity of ANP was measured and the relative fold change was plotted against that from the vehicle-treated cells (n=6 per group). Data were expressed as mean±S.E.M. *** P<0.001. |
As demonstrated in Figure 3, Rb1 treatment preserves the mitochondrial morphology in Ang II–infused mice in vivo, suggesting that Rb1 may protect against cardiac hypertrophy through preserving the mitochondrial function. To test this hypothesis, we further assessed the impact of Rb1 on MMP, an indicator of mitochondrial function, in Ang II-stimulated H9c2 cells using the membrane permeable JC1 dye. As shown in Figure 6A, Ang II stimulation for 48 h resulted in impaired MMP as reflected by decreased ratio of J-aggregates to J-monomers. In distinct contrast, Ang II–induced decrease in MMP was in part counteracted by Rb1 treatment, which resulted in increased ratio of J-aggregates to J-monomers. Similar observations were also made in PE-stimulated cells. Rb1 treatment increased the ratio of J-aggregates to J-monomers in H9c2 cells exposed to PE stimulation for 72 h (Supplementary Figure 3). Furthermore, the mitochondrial protective activity of Rb1 was also evaluated by the mPTP assay. As shown in Figure 6B, Ang II–induced opening of mPTP was in part attenuated by Rb1 treatment. These observations collectively indicate that Rb1 protects the mitochondrial function against pro-hypertrophic stress-induced impairment in cardiomyocytes.
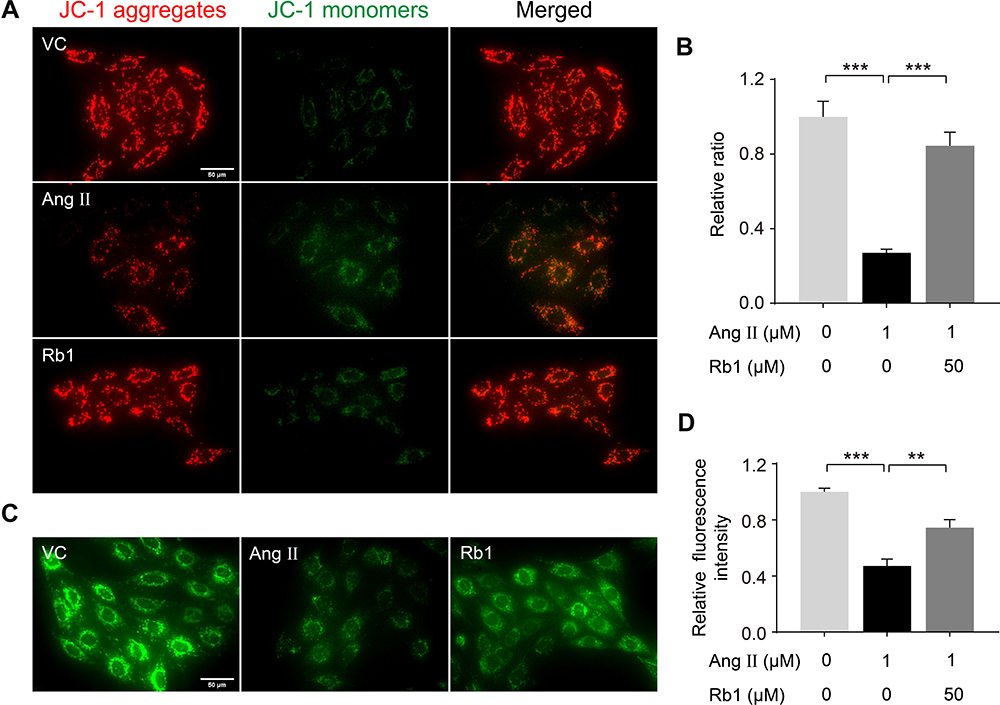 |
Figure 6 Rb1 protects against Ang II–induced impairment of mitochondrial function in cardiomyocytes. H9c2 cells pretreated with vehicle or Rb1 (50 μM) were incubated in the presence or absence of Ang II (1 μM) for 48 h. (A) MMP was analyzed by JC-1 staining, by which J-aggregates (in red) and J-monomers (in green) could be visualized by fluorescence microscopy. DAPI (in blue) was counterstained to visualize the nuclei. Scale bar, 50 μm. (B) The relative ratio of J-aggregates to J-monomers was plotted against that from the vehicle-treated cells (n=5 per group). (C) Calcein staining was performed to assess mPTP opening. Scale bar, 50 μm. (D) Relative fluorescence intensity of the calcein signal was plotted against that from the vehicle-treated cells (n=5 per group). Data were expressed as mean±S.E.M. ** P<0.01, *** P<0.001. |
Rb1 Mitigates Inflammatory Responses in Activated Macrophages in Part by Suppressing MAPK Signaling
To better understand the suppressive effect of Rb1 on inflammation, we started with confirming the anti-inflammatory activity of Rb1 in macrophages. The results showed that LPS triggered elevated production of IL-1 beta, IL-6 or TNF, whereas Rb1 treatment significantly decreased the production of IL-1 beta, IL-6 or TNF in RAW 264.7 cells 4 h (Figure 7A) or 24 h (Figure 7B) after LPS stimulation, validating the anti-inflammatory activity of Rb1 in macrophages. Meanwhile, Rb1 treatment resulted in decreased mRNA levels of Il1b, Il6 and Tnf in LPS-stimulated RAW 264.7 cells (Figure 7C), suggesting that the anti-inflammatory effects of Rb1 take place in part at the transcriptional level. Furthermore, the impact of Rb1 on the activation of MAPK signaling pathway was assessed given that it is the major intracellular signaling pathway activating the proinflammatory gene expression in macrophages.15 As shown in Figure 8, LPS-augmented phosphorylation of Erk1/2, JNK and p38 MAPKs was markedly attenuated in LPS-stimulated Rb1-treated RAW 264.7 cells. Meanwhile, LPS-augmented phosphorylation of MEK1/2, a kinase enzyme that activates Erk1/2, was significantly decreased as a result of Rb1 treatment. These results collectively validate the intrinsic anti-inflammatory activity of Rb1 in activated macrophages.
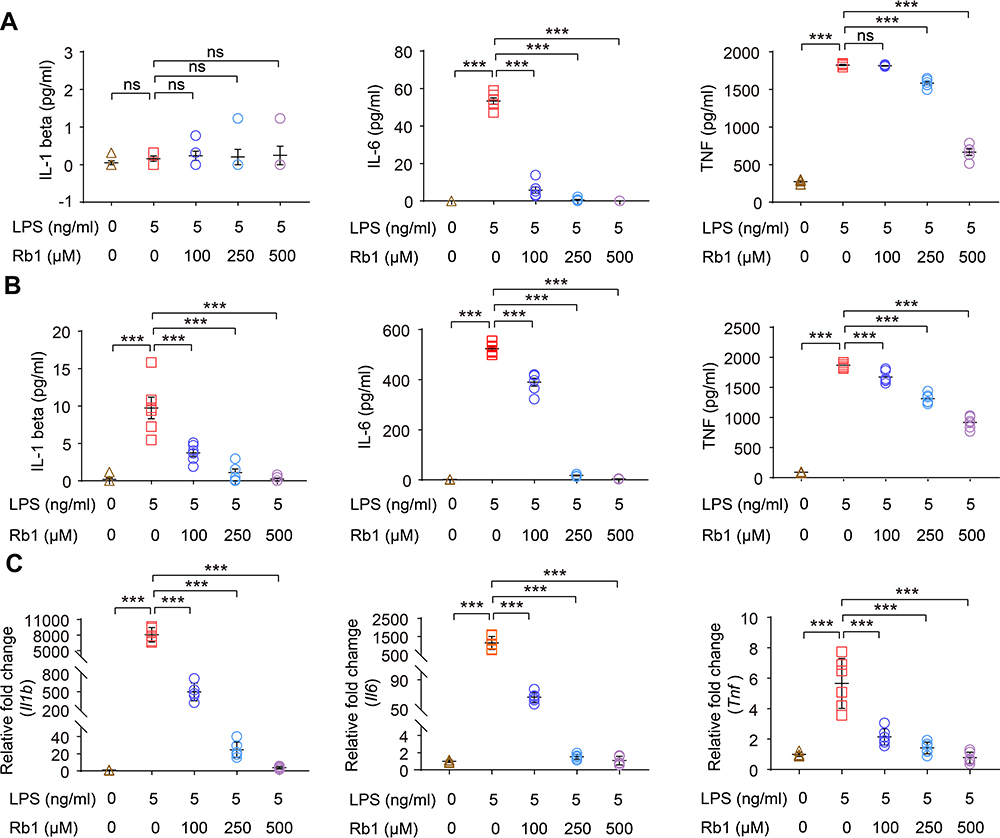 |
Figure 7 Rb1 suppresses the inflammatory responses in LPS-stimulated macrophages. RAW264.7 cells were treated with vehicle or Rb1 at the indicated concentrations, followed by incubation in the presence or absence or LPS (5 ng/mL) for 4 h or 24 h. Cell culture supernatants or cells were then collected for the indicated analyses. ELISA was performed to analyze the level of IL-1 beta, IL-6 and TNF 4 h (A) and 24 h (B) after LPS incubation (n=6 per group). (C) Real-time qPCR was performed to analyze the mRNA expression of Il1b, Il6, and Tnf 4 h after LPS incubation (n=6 per group). 18S rRNA was included as the internal reference. Relative fold change was plotted against that from the vehicle-treated cells. Data were expressed as mean±S.E.M. *** P<0.001. |
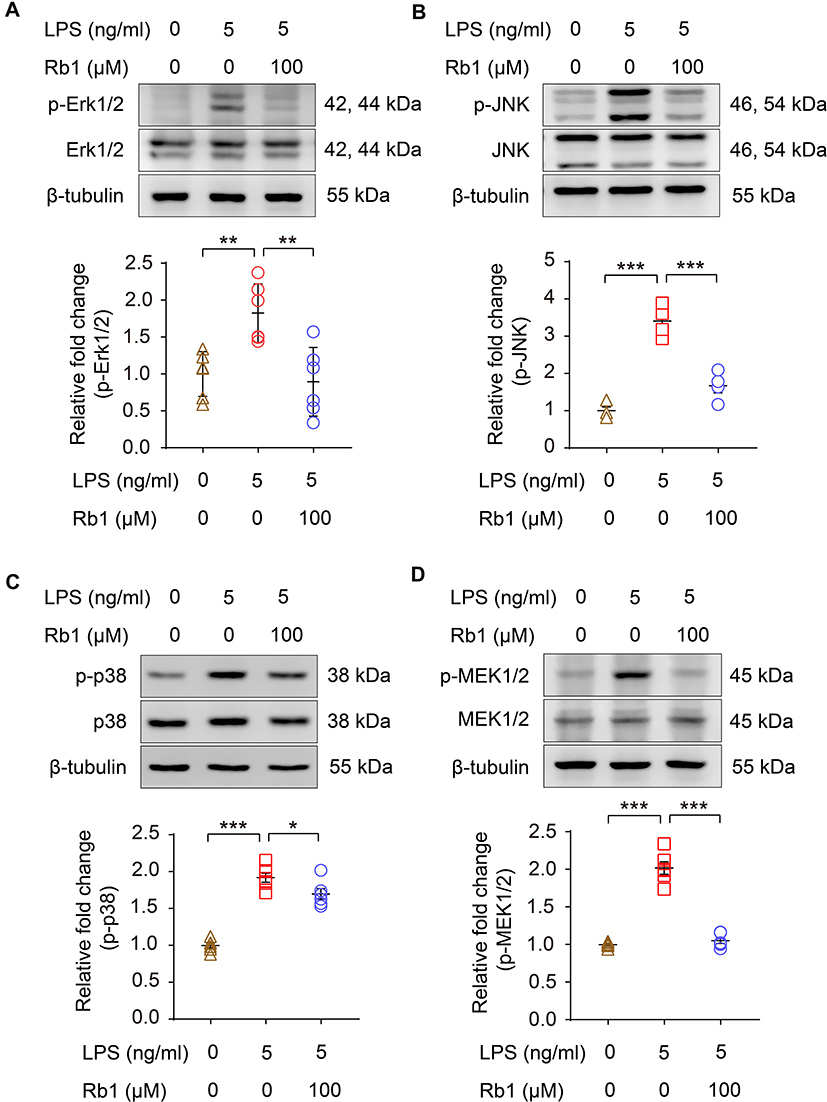 |
Figure 8 Rb1 attenuates the activation of MAPKs and MEK1/2 in LPS-stimulated macrophages RAW264.7 cells were treated with vehicle or Rb1 (100 μM), followed by incubation in the presence or absence of LPS (5 ng/mL) for 30 min or 6 h. Western blotting was performed to examine the level of p-Erk1/2, Erk1/2 (A), p-JNK, JNK (B), p-p38 and p38 (C) and p-MEK1/2, MEK1/2 (D) in the cells (n=4-6 per group). β-tubulin was probed for the normalization purposes. Densitometry was measured to quantify the protein signals. Relative fold change was plotted against that from the vehicle-treated cells. Data were expressed as mean±S.E.M. * P<0.05, ** P<0.01, *** P<0.001. |
Rb1 Suppresses the Expression of miR-155 in Macrophages
As one of the most potent proinflammatory miRNAs, macrophage miR-155 promotes the development of cardiac hypertrophy.7 Our previous study has shown that Rb1 suppresses LPS-stimulated expression of miR-155 in RAW 264.7 cells.14 Here, we confirmed that Rb1 treatment significantly counteracted LPS-induced increase in the expression of miR-155 in RAW 264.7 cells (Figure 9A). To further relate the anti-inflammatory activity of Rb1 to cardiac hypertrophy, the effects of Rb1 on Ang II–induced expression of miR-155 and proinflammatory genes were also examined in BMDMs. As shown in Figure 9B, increased expression of miR-505 was observed in response to Ang II stimulation, while significantly decreased expression of miR-155 was noted in Ang II-stimulated Rb1-treated BMDMs. Meanwhile, Ang II increased the expression of Il1b, Il6 and Tnf in BMDMs, supporting the proinflammatory action of Ang II in macrophages. In distinct contrast, Rb1 treatment resulted in remarkable reductions in the expression of Il1b, Il6 and Tnf in the presence of Ang II (Figure 9C). The results here indicate that Rb1 suppresses Ang II-triggered inflammatory gene expression and miR-155 upregulation in macrophages in a cell-autonomous manner. To further address the implication of downregulated miR-155 in the anti-inflammatory activity of Rb1 in activated macrophages, agomir-155 or agomir-NC was transfected in macrophages, followed by the analyses of the mRNA expression of Il1b, Il6 and Tnf as well as the production of IL-1 beta, IL-6 and TNF. The results showed that agomir-155 transfection partially abolished the inhibitory effect of Rb1 on the mRNA expression of Il6 (Figure 9D) and the production of IL-6 (Figure 9E) in LPS-stimulated RAW264.7 cells, while the levels of IL-1 beta and TNF were not affected by agomir-155 transfection (Supplementary Figure 4). Collectively, these results indicate that Rb1 therapeutically modulates the level of miR-155 in activated macrophages. Meanwhile, these results support that Rb1 suppresses the overproduction of IL-6 in activated macrophages in part through decreasing the expression of miR-155.
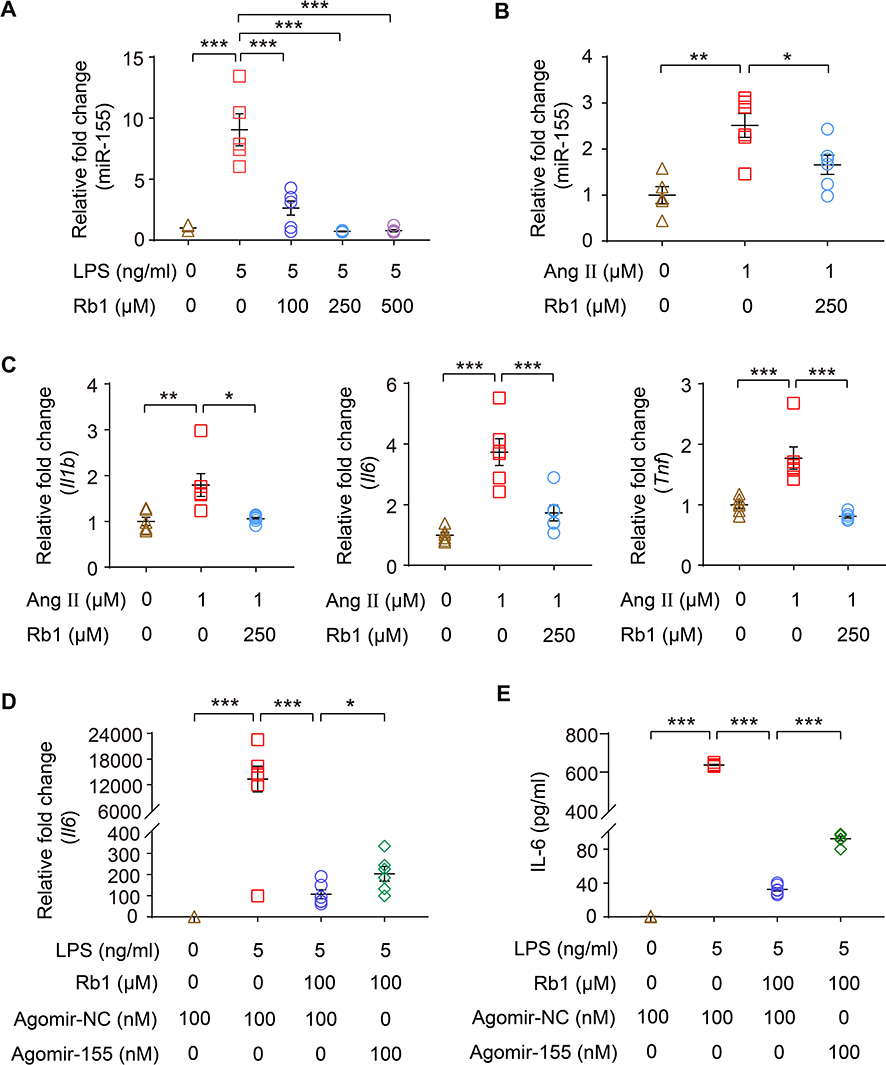 |
Figure 9 Rb1 suppresses the production of IL-6 in part through downregulating miR-155 in macrophages. RAW264.7 cells were treated with vehicle or Rb1 at the indicated concentrations, followed by LPS (5 ng/mL) incubation for 4 h. (A) Real-time qPCR was performed to analyze the expression of miR-155 in the cells (n=4-6 per group). RNU6B was analyzed in parallel for the normalization purposes. (B) Bone marrow-derived macrophages (BMDMs) were treated with vehicle or Rb1 (250 μM), followed by incubation in the presence or absence of Ang II (1 μM) starting from the day 3 in culture. Cells were collected after 6 days of continuous intervention. Real-time qPCR was performed to analyze the expression of miR-155 in BMDMs (n=6 per group). RNU6B was analyzed in parallel for the normalization purposes. (C) BMDMs were treated with vehicle or Rb1 (250 μM), followed by incubation in the presence or absence of Ang II (1 μM) starting from the day 3 in culture. Cells were collected after 6 days of continuous intervention. Real-time qPCR was performed to analyze the expression of Il1b, Il6 and Tnf in BMDMs (n=6 per group). 18S rRNA was analyzed in parallel for the normalization purposes. For the expression analyses in (A–C), relative fold change was plotted against that from the vehicle-treated cells. RAW264.7 cells were transfected with agomir-155 (100 nM) or agomir-NC (100 nM), treated with vehicle or Rb1 (100 μM) and exposed to LPS (5 ng/mL) for 4 h. The mRNA expression of Il6 (D) and the production of IL-6 in cell culture supernatant (E) were then analyzed by real-time qPCR and ELISA, respectively (n=6 per group). For real-time qPCR analysis of Il6 expression, 18S rRNA was analyzed in parallel for the normalization purposes. Relative fold change was plotted against that from vehicle-treated agomir-NC transfected cells. Data were expressed as mean±S.E.M. * P<0.05, ** P<0.01, *** P<0.001. |
Discussion
Complex mechanisms including the crosstalk of aberrantly elevated Ang II signaling and inflammation underlie the pathological development of cardiac hypertrophy and the ensuing heart failure. The findings here demonstrate that naturally occurring Rb1 protects against Ang II–induced cardiac hypertrophy, myocardial fibrosis and inflammation. At the cellular level, Rb1 suppresses cardiomyocyte hypertrophy and macrophage-mediated inflammation in a cell-autonomous manner. Furthermore, Rb1 protects the mitochondrial function in cardiomyocytes in the presence of pro-hypertrophic stimuli. Meanwhile, Rb1 suppresses the overproduction of IL-6 in part through decreasing the expression of miR-155 in activated macrophages, supporting that Rb1 is pharmacologically active at modulating the mechanisms involved in the crosstalk between macrophage inflammation and cardiomyocyte hypertrophy. The dual activity of Rb1 on cardiomyocyte hypertrophy and macrophage inflammation illustrates the potential therapeutic benefits of Rb1 in the treatment of cardiac hypertrophy (Figure 10).
 |
Figure 10 A schematic drawing summarizing the pharmacological mechanisms of Rb1 in the treatment of cardiac hypertrophy. In cardiomyocytes, Rb1 directly protects against pro-hypertrophic stimuli-induced cardiomyocyte hypertrophy in part through maintaining mitochondrial function. In macrophages, Rb1 mitigates inflammatory responses in activated macrophages in part through suppressing MAPK signaling and MEK1/2 activation. Furthermore, miR-155 is partially involved in the inhibitory effect of Rb1 on IL-6 production in activated macrophages. The suppressive effects of Rb1 on the expression of miR-155 and the production of IL-6 in activated macrophages suggest that Rb1 may modulate the crosstalk between activated macrophages and cardiomyocytes in cardiac hypertrophy. Dashed lines: published findings from reference 7 and 23–26. Solid lines: the results of the current work. |
First, our study provides in vivo evidence that directly supports the pharmacological activity of Rb1 in alleviating Ang II-associated myocardial remodeling (Figures 1 and 2). Cardiomyocyte hypertrophy and myocardial fibrosis, hallmark pathologies associated with elevated Ang II signaling, are attenuated as a result of Rb1 treatment. In vitro experiments further reveal that Rb1 exerts direct anti-hypertrophic effect in cardiomyocytes (Figure 5 and Supplementary Figure 1 and 2). Cardiomyocyte hypertrophy is fundamentally characterized by mitochondrial damage at the subcellular level.16 Both in vivo (Figure 3) and in vitro (Figure 6 and Supplementary Figure 3) findings here indicate that Rb1 protects against mitochondrial impairment and dysfunction associated with cardiomyocyte hypertrophy. Mitochondria are double-membraned organelles and primarily function to generate ATP molecules through oxidative phosphorylation that occurs in the inner membrane. During this process, reducing equivalents are transferred to the electron transport chain and protons are pumped to the intermembrane space. The functional mitochondria are therefore characterized by two interconnected features. One involves the innate impermeability of the inner membrane. This allows the proton pumping process to generate the membrane potential, another essential component of the mitochondrial function that is required for ATP production. Given that the heart obtains approximately 95% of ATP for consumption through oxidative phosphorylation, the structural and functional integrity of mitochondria is of critical importance to the cardiomyocytes. Regardless of the etiology of the cardiac disorders, mitochondrial dysfunction has been invariably noted in the failing heart, playing an important role in the pathogenesis of cardiac hypertrophy and heart failure.17,18 Therefore, the effect of Rb1 at protecting the cardiomyocytes against Ang II-triggered loss of MMP and opening of mPTP (Figure 6) not only corroborates the in vivo findings of Rb1-conferred mitochondrial protection (Figure 3), but also supports the possibility that mitochondrial protection may in part explain the anti-hypertrophic activity of Rb1 in the heart. Future studies are worth pursuing to further elucidate the molecular mechanisms underlying the mitochondrial protective effect of Rb1 in the cardiomyocytes.
Secondly, we show that Rb1 attenuates the inflammatory responses accompanying cardiac hypertrophy (Figure 4). Inflammation is a key process involved in the pathological cardiac remodeling and heart failure.19 Both low-grade systemic inflammation and local crosstalk between cardiomyocytes and activated macrophages contribute to the initiation and progression of cardiac hypertrophy and heart failure.20,21 Clinical analyses imply that increased level of proinflammatory cytokines is associated with pathological cardiac remodeling and poorer clinical outcomes in heart failure patients.19 Experimental studies also demonstrate that cardiac inflammation marked by increased number of macrophages in the heart and elevated cardiac expression of proinflammatory Il6 often accompanies cardiac hypertrophy and heart failure.22 Inflammation therefore has been considered as a promising druggable target for the treatment of cardiac hypertrophy and heart failure. Our work here presents multiple lines of evidence supporting the anti-inflammatory activities of Rb1 in the heart under pro-hypertrophic stress conditions. Decreased plasma IL-6, decreased cardiac expression of Il6 and reduced number of CD68+ macrophages and CD11b+ leukocytes in Ang II–infused Rb1-treated hearts support that Rb1 treatment is effective at suppressing cardiac hypertrophy-associated systemic and cardiac inflammation in vivo (Figure 4). The changes in IL-6 after Rb1 treatment are of particular pharmacological implications in the prevention and treatment of cardiac hypertrophy and heart failure. IL-6 infusion leads to the development of cardiac hypertrophy, fibrosis and cardiac dysfunction.23 Persistent activation of IL-6 signaling component gp130 results in cardiac hypertrophy.24 In a recently published analysis of a large cohort of heart failure patients, elevated level of IL-6 is found in over 50% of the patients and associated with decreased cardiac function and poorer prognosis.25 On the other hand, Il6 gene deletion protects against pressure overload-induced hypertrophic phenotypes and dysfunction in the heart.26 Therefore, our findings not only provide in vivo experimental evidence supporting that Rb1 suppresses the inflammatory responses in the pathological context of cardiac hypertrophy, but also highlight the pharmacological implications of Rb1-mediated regulation of IL-6 in the treatment of cardiac hypertrophy.
Thirdly, our work here further corroborates the pharmacological understanding of the direct anti-inflammatory activity of Rb1 in activated macrophages (Figure 7). It has been reported that the anti-inflammatory effects of Rb1 involve the inhibition of the activation of interleukin-1 receptor-associated kinase-1, IκB kinase β, nuclear transcription factor kB, and MAPKs (Erk1/2, JNK, and p38).13 Our results here consistently show that the anti-inflammatory activity of Rb1 in part involves downregulation of the expression of proinflammatory genes such as Il1b, Il6 and Tnf in activated macrophages (Figure 7C) and suppression of the phosphorylation of MAPKs (Figure 8). Meanwhile, Rb1 treatment also decreases the phosphorylation of MEK1/2 in activated macrophages (Figure 8). In inflammation, the highly conserved and ubiquitous MAPK signal transduction pathways are activated in response to various inflammatory stimuli, leading to the transcriptional activation of proinflammatory genes. MAPK pathways thus represent a novel class of therapeutic targets for anti-inflammatory treatment.15 It is worth noting that Erk1/2 is situated at the terminal of MAPK signaling and can translocate to the nucleus to regulate transcription programs. MEK1/2 directly interacts with Erk1/2 and catalyzes its phosphorylation. The suppressive effect of Rb1 on MEK1/2 phosphorylation suggests that Rb1 may act on the upstream components of MAPK signaling. This finding not only expands the molecular understanding of the anti-inflammatory activity of Rb1, but also warrants further investigations to identify the direct target of Rb1 in MAPK signaling. Nevertheless, the validated suppressive effects on MAPK activation further support the therapeutic value of Rb1 as an anti-inflammatory agent.
Most importantly, our study suggests that Rb1 may alleviate cardiac hypertrophy in part through downregulating the level of miR-155 in macrophages (Figure 9A and B). The remarkable effect of Rb1 on suppressing the expression of miR-155 in macrophages deserves special attention to understand its pharmacological implications in treating cardiac hypertrophy. MiR-155 is potently induced by multiple inflammatory stimuli and serves as a primary regulator of inflammation. It has been shown that miR-155 promotes cardiac inflammation in viral heart diseases.27 It has also been demonstrated that macrophages promote Ang II–induced cardiac hypertrophy and failure in a paracrine, miR-155-dependent manner.7 The macrophage expression of miR-155 is upregulated by Ang II. Genetic ablation of miR-155 in macrophages attenuates cardiac inflammation, hypertrophy and improves cardiac function in Ang II–infused mice, supporting the notion that macrophage miR-155 promotes the development of cardiac hypertrophy.7 Therefore, decreasing the level of miR-155 in macrophages is therapeutically significant in attenuating cardiac hypertrophy. In this study, we show that Rb1 suppresses the expression of miR-155 in both LPS and Ang II-stimulated macrophages (Figure 9A and B). These results collectively suggest that the anti-hypertrophic effect of Rb1 may in part be mediated by decreased expression of miR-155 in macrophages. Furthermore, our results reveal that miR-155 is in part involved in the suppressive effect of Rb1 on IL-6 production in activated macrophages (Figure 9C and D). As aforementioned, the level of IL-6 is of direct pathological significance in the development of cardiac hypertrophy.22–26 The inhibitory effect of Rb1 on the level of miR-155 in macrophages per se and the implication of miR-155 in Rb1-conferred suppression of IL-6 production shed new light on the anti-inflammatory mechanisms of Rb1, which possibly contribute in part to its therapeutic value in cardiac hypertrophy. Meanwhile, these findings support the potential modulatory effect of Rb1 on the crosstalk between macrophages and cardiomyocytes during the development of cardiac hypertrophy.
Conclusions
Effective mechanism-based intervention at the stage of cardiac hypertrophy may delay or alleviate the morphological and functional deterioration of the heart. The current study demonstrates that naturally occurring Rb1 is pharmacologically effective at directly modulating cardiomyocyte hypertrophic alterations and inhibiting macrophage inflammatory responses. Therefore, by elucidating the dual activity of Rb1 on hypertrophic cardiomyocytes and activated macrophages, the work here provides new mechanistic insights into the pharmacological implications of Rb1 in the treatment of cardiac hypertrophy.
Funding
This work was supported by the National Natural Science Foundation of China (82074049).
Disclosure
The authors declare no conflict of interest.
References
1. Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341.
2. Heinzel FR, Hohendanner F, Jin G, Sedej S, Edelmann F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J Appl Physiol. 2015;119:1233–1242.
3. Shah KS, Xu H, Matsouaka RA, et al. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol. 2017;70:2476–2486.
4. Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589.
5. Wrigley BJ, Lip GY, Shantsila E. The role of monocytes and inflammation in the pathophysiology of heart failure. Eur J Heart Fail. 2011;13:1161–1171.
6. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155.
7. Heymans S, Corsten MF, Verhesen W, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. 2013;128:1420–1432.
8. Kim JH. Cardiovascular diseases and panax ginseng: a review on molecular mechanisms and medical applications. J Ginseng Res. 2012;36:16–26.
9. Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol. 1999;58:1685–1693.
10. Zheng X, Wang S, Zou X, et al. Ginsenoside Rb1 improves cardiac function and remodeling in heart failure. Exp Anim. 2017;66:217–228.
11. Jiang QS, Huang XN, Dai ZK, et al. Inhibitory effect of ginsenoside Rb1 on cardiac hypertrophy induced by monocrotaline in rat. J Ethnopharmacol. 2007;111:567–572.
12. Jiang QS, Huang XN, Yang GZ, Jiang XY, Zhou QX. Inhibitory effect of ginsenoside Rb1 on calcineurin signal pathway in cardiomyocyte hypertrophy induced by prostaglandin F2alpha. Acta Pharmacol Sin. 2007;28:1149–1154.
13. Joh EH, Lee IA, Jung IH, Kim DH. Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1 activation–the key step of inflammation. Biochem Pharmacol. 2011;82:278–286.
14. Bian M, Du X, Wang P, et al. Combination of ginsenoside Rb1 and Rd protects the retina against bright light-induced degeneration. Sci Rep. 2017;7:6015.
15. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005;1754:253–262.
16. Dai DF, Johnson SC, Villarin JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846.
17. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–3726.
18. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139:1435–1450.
19. Shirazi LF, Bissett J, Romeo F, Mehta JL. Role of inflammation in heart failure. Curr Atheroscler Rep. 2017;19:27.
20. Sager HB, Hulsmans M, Lavine KJ, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res. 2016;119:853–864.
21. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282.
22. Plenz G, Song ZF, Tjan TD, et al. Activation of the cardiac interleukin-6 system in advanced heart failure. Eur J Heart Fail. 2001;3:415–421.
23. Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231.
24. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci U S A. 1995;92:4862–4866.
25. Markousis-Mavrogenis G, Tromp J, Ouwerkerk W, et al. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur J Heart Fail. 2019;21:965–973.
26. Zhao L, Cheng G, Jin R, et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118:1918–1929.
27. Corsten MF, Papageorgiou A, Verhesen W, et al. MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res. 2012;111:415–425.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.