")
Back to Journals » Infection and Drug Resistance » Volume 12
Drug–drug interaction study of imatinib and voriconazole in vitro and in vivo
Authors Lin QM , Xie S, Qiu X , Chen J, Xu RA
Received 26 December 2018
Accepted for publication 27 March 2019
Published 30 April 2019 Volume 2019:12 Pages 1021—1027
DOI https://doi.org/10.2147/IDR.S199526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Qianmeng Lin,1,2,* Saili Xie,1,* Xiangjun Qiu,3 Jingjing Chen,1 Ren-Ai Xu1
1The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, People’s Republic of China; 2School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325000, People’s Republic of China; 3Medical College of Henan University of Science and Technology, Luoyang 471003, People’s Republic of China
*These authors contributed equally to this work
Background: In clinical practice, common problem polypharmacy could result in the increased risks of drug–drug interactions (DDIs). Co-administered imatinib (IMA) and voriconazole (VOR) as one treatment protocol in cancer patients with fungal infections are common.
Purpose: The aim of the present study was to assess the potential DDIs associated with the concurrent use of IMA and VOR in rat liver microsomes (RLMs) and in rats.
Methods and results: The concentration levels of IMA, VOR, and their metabolites N-desmethyl IMA (CGP74588) and N-oxide voriconazole (N-oxide VOR) were determined by ultra performance liquid chromatography-tandem mass spectrometry. In vitro study of RLMs, VOR inhibited the IMA metabolism with the half-maximal inhibitory concentration (IC50) of 105.20 μM, while IC50 for IMA against VOR was 61.30 μM. After co-administered IMA and VOR in rats, the Cmax of IMA was increased significantly, while the AUC0→t, AUC0→∞, and Cmax of CGP74588 were decreased significantly. In addition, similar results were also found that the main pharmacokinetic parameters (AUC0→t, AUC0→∞, MRT0→∞, Tmax, and Cmax) of VOR were increased significantly, while the AUC0→t, AUC0→∞, and Cmax of N-oxide VOR were decreased significantly. Incorporation of all the results indicated that both drugs had a inhibitory effect on each other’s metabolism in vitro and in vivo.
Conclusion: Thus, it is of great value to monitor the concomitant use of IMA and VOR in the clinic to reduce the risks of unexpected clinical outcomes.
Keywords: drug–drug interaction, imatinib, voriconazole, rat liver microsome, metabolism
Introduction
Drug–drug interactions (DDIs) often occur, especially for patients with multiple underlying diseases who use various kinds of drugs.1 In clinical practice, polypharmacy is a common problem and results in the increased risks of DDIs.2,3 A primary reason of DDIs is the change of the cytochrome P450 (CYP450) isozyme activity by inducing or inhibiting.4,5 Therefore, it is very important to identify the potential DDIs in order to reduce the risks of unexpected outcomes.
In the clinic, invasive fungal disease is a serious problem with high mortality and morbidity rate, which could cause significant damage to human health, especially for hematological malignancies patients.6,7 As the first-line drug for the treatment of invasive aspergillosis, voriconazole (VOR) has a poor correlation between plasma levels and the drug dosing, because of its narrow therapeutic window (1.0–5.5 μg/mL) and variable pharmacokinetic profile.8,9 In addition, CYP2C19, CYP2C9, and CYP3A4 are the major enzymes responsible for VOR metabolized to N-oxide voriconazole (N-oxide VOR).10 Therefore, therapeutic drug monitoring (TDM) is proposed to ensure optimal VOR exposure for its wide variability and narrow therapeutic range.11
Imatinib (IMA) is the first synthetic tyrosine kinase inhibitor and is currently approved as standard care in patients with BCR-ABL-positive chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST).12,13 It is mainly metabolized by CYP3A4 enzyme to the primary circulating product N-desmethyl IMA (CGP74588), which was proved to be similar pharmacologic with the parent drug.14,15 Although IMA is considered to have an excellent pharmacokinetic profile, intra- and inter-individual variability of drug exposure is largely documented.16
In CML and GIST patients, the most serious fungal infection is invasive aspergillosis, which can lead to death.17 Co-administered IMA and VOR as one treatment protocol in those patients have been recognized, and the effect of VOR on the metabolism of IMA also has been studied in vivo18 and in vitro.19 However, to the best of our knowledge, there are currently no studies examining DDIs associated with the concurrent use of IMA and VOR, especially the effect of IMA on VOR metabolism. IMA was found to be a potent inhibitor of CYP3A4,20 which can interact with CYP3A4 substrates, like VOR. Thus, it is unclear what dosage of VOR to administer with IMA in the clinic is chosen.
In this study, we attempted to use an ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method for the determination of IMA, VOR, and their metabolites in plasma and investigate the DDIs of IMA and VOR in rats. Moreover, the potential DDIs in rat liver microsomes (RLMs) were also identified.
Materials and methods
Chemicals
IMA (purity >98%), CGP74588 (purity >98%), VOR (purity >98%), and N-oxide VOR (purity >98%) were bought from National Institutes for Food and Drug Control (Beijing, China). Diazepam was purchased from Sigma (St. Louis, MO, USA), and used as the internal standard (IS). The reduced nicotinamide adenine dinucleotide phosphate (NADPH) was obtained from Roche Pharmaceutical Ltd (Basel, Switzerland). High-performance liquid chrmotography grade acetonitrile and methanol were obtained from Merck Company (Darmstadt, Germany). Deionized water was produced using a Milli-Q academic reagent grade water purification system (Millipore, Bedford, USA).
Instrumentation and operation conditions
The LC and MS/MS condition were based on our report in the literature.21 An Acquity ultra-high performance liquid chromatography (UPLC) system (Waters Corp., Milford, MA, USA) with an Acquity BEH C18 column (2.1 mm×50 mm, 1.7 μm) at 45°C was used for the chromatographic separation. Acetonitrile (A) and water with 0.1% formic acid (B) were used as the gradient elution solvents, and were conducted with the 0.40 mL/mins flow rate as follow for the gradient program: 0–1.0 mins (20–90% A), 1.0–2.0 mins (90–90% A), 2.0–2.1 mins (90–20% A), and 2.1–4.0 mins (20–20% A).
In positive ionization mode, a XEVO TQ-S triple quadrupole mass spectrometer with electrospray ionization was employed for the quantitation. In the multiple reaction monitoring mode, the measurement was achieved with transitions of m/z 494.3→394.2 for IMA, m/z 480.3→394.2 for CGP74588, m/z 350.1→281.1 for VOR, m/z 366.1→224.1 for N-oxide VOR, and m/z 285.0→154.0 for IS, respectively. Masslynx 4.1 software (Waters Corp., Milford, MA, USA) was used to control the instrument and acquire the data.
Preparation of RLMs
The liver obtained from eight different rats were weighed and homogenized in a cold 0.01 mM PBS, which contains 0.25 mM sucrose. After centrifuged at 11,000 rpm for 15 mins, the supernatants separated from the homogenates were then transferred to a new tube and centrifuged at 11,000 rpm for 15 mins again. Moreover, the supernatants were then ultra-centrifuged at 100,000×g at 4°C for 1 hr. The microsomal pellets were reconstituted with 0.01 mM cold PBS and stored at −80°C.22 The protein concentrations of the liver microsomes were assayed by Bradford Protein Assay Kit (Thermo Scientific, Waltham, MA, USA).
VOR–IMA interaction studies in RLMs
When VOR was used as the inhibitor to determine the half-maximal inhibitory concentration (IC50), the incubation mixture (total volume, 200 μL) contained 0.5 mg/mL RLMs, 1 M potassium phosphate buffer (pH 7.4), IMA (5.0 μΜ, approximately the Km of RLMs, which was determined as reported19), VOR (0, 0.01, 0.1, 1, 10, 50, 100, and 1,000 μM) and 1 mM NADPH. In a shaking water bath at 37°C, NADPH was added to initiate the reaction after pre-incubation for 5 mins. The final volume of the mixture was 200 μL, and was performed for 40 mins, stopped by cooling to −80°C immediately. Then, 400 μL IS working solution (5 ng/mL IS in acetonitrile) was added. After vortex mixing for 1 min, the tubes were centrifuged at 15,000×g for 15 mins. The supernatant mixture (2 μL) was injected for analysis.
When IMA was used as the inhibitor, the incubation mixture and sample preparation were as the same as the above mentioned, except for VOR (2.0 μΜ, approximately the Km of RLMs) and IMA (0, 0.01, 0.1, 1, 10, 20, 50, and 100 μM). All incubations were performed in triplicate and data are presented as means ± SD.
In vivo pharmacokinetic interaction studies
Male Sprague–Dawley rats (200±20 g) obtained from the Laboratory Animal Center of Henan University of Science and Technology (Luoyang, China) were used to study the pharmacokinetic interaction of IMA and VOR. All experimental procedures and protocols were reviewed and approved by the Animal Care and Use Committee of Henan University of Science and Technology and were in accordance with the Guide for the Care and Use of Laboratory Animals.
Twenty-four SD rats were divided randomly into three groups: the IMA group (Group A, n=8), the VOR group (Group B, n=8), and the co-administered IMA and VOR group (Group C, n=8). All animals were fasted overnight and allowed free to water within the period of the experiment. IMA and VOR were dissolved in 0.5% sodium carboxymethyl cellulose (CMC-Na) solution alone or together. The Group A and B were treated with 20 mg/kg IMA and 20 mg/kg VOR, respectively, while the Group C with an equivalent amount of 20 mg/kg IMA and 20 mg/kg VOR together. Blood samples (300 µL) were collected via tail vein to 1.5 mL centrifuge tube at the time points of 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 36 hrs after oral administration. The obtained samples were immediately centrifuged at 4,000×g for 8 mins, and the supernatant (100 µL plasma samples) were collected and stored at −80°C until analysis. As for sample preparation, 200 μL IS working solution (5 ng/mL IS in acetonitrile) was added to each plasma sample for the extraction. After vortex-mixed for 1 min, the mixture was centrifuged at 15,000×g for 15 mins, and the supernatant (2 µL) was injected into the UPLC-MS/MS system for analysis.
Statistical analysis
The non-compartmental analysis was used to calculate the pharmacokinetic parameters by DAS version 2.0 (Shanghai University of Traditional Chinese Medicine, China). The mean plasma concentration-time curve was plotted by Origin 8.0 (Originlab Company, Northampton, MA, USA), and the IC50 was calculated by GraphPad (Version 6.0; Graphpad Software Inc., San Diego, CA, USA). Statistical comparisons of the main pharmacokinetic parameters within each group were carried out with the Statistical Package for the Social Sciences (version 17.0; SPSS Inc., Chicago, IL, USA) by one-way analysis of variance for repeated measures coupled with the Dunnett’s test. In all cases, P<0.05 was considered to be of statistical significance.
Results
Effects of VOR on the metabolism of IMA in RLMs
As shown in Figure 1, when the concentration of VOR (100 μM) was used, VOR inhibited the IMA metabolism rate in RLMs to 54.4%. Furthermore, the IC50 for inhibition activity in RLMs was 105.20 μM (Figure 1).
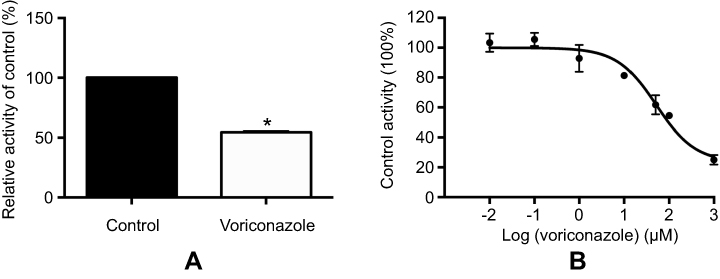 | Figure 1 The effect of 100 μM VOR on IMA metabolite (CGP74588) formation (A) and various concentrations (0, 0.01, 0.1, 1, 10, 50, 100, and 1,000 μM) of VOR for IC50 (B) in RLMs. Values are Mean ± SD, n=3. |
Effects of IMA on the metabolism of VOR in RLMs
Figure 2 exhibits the inhibitory effect of IMA on VOR metabolism. It indicated that 100 μM IMA inhibited the VOR metabolism rate in RLMs to 38.9%. In addition, the IC50 for inhibition activity in RLMs was 61.30 μM (Figure 2).
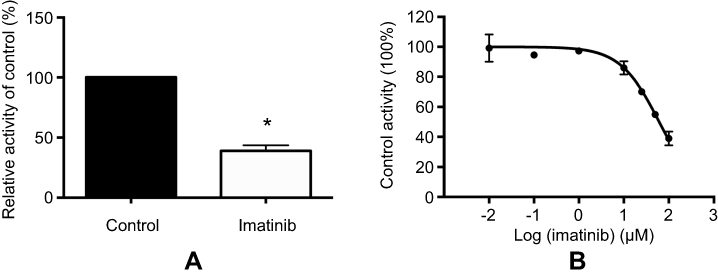 | Figure 2 The effect of 100 μM IMA on VOR metabolite (N-oxide VOR) formation (A) and various concentrations (0, 0.01, 0.1, 1, 10, 20, 50, and 100 μM) of IMA for IC50 (B) in RLMs. Values are Mean ± SD, n=3. *P<0.05. |
Pharmacokinetic interaction studies in rats
Table 1 shows the statistical analysis results for the mean pharmacokinetic parameters of IMA (Group A), VOR (Group B) administered alone, and in combination (Group C), which were analyzed by DAS 2.0. Mean plasma concentration–time curves of IMA, VOR, and their metabolites in three groups are presented in Figure 3.
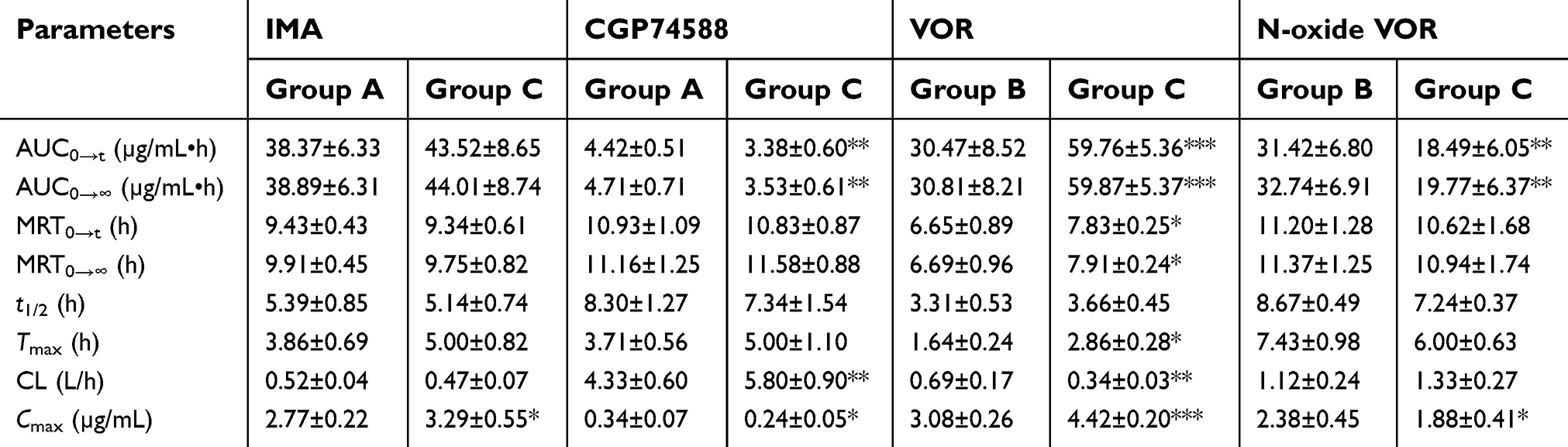 | Table 1 Pharmacokinetic parameters of IMA, VOR, and their metabolites in three different groups (n=8) |
 | Figure 3 Mean plasma concentration versus time of IMA (A), CGP74588 (B), VOR (C), N-oxide VOR (D) in three group rats (Mean ± SD, n=8). |
The mean plasma concentration–time curve and mean pharmacokinetic parameters showed that the Cmax of IMA in Group C was increased significantly by 18.8%, when compared to Group A (P<0.05). In addition, the AUC0→t, AUC0→∞, and Cmax of CGP74588 were decreased significantly by 23.5%, 25.1%, and 29.4%, respectively, compared to those of the Group A, while CL of CGP74588 was increased significantly by 33.9%. These results indicated that VOR had the inhibitory effect on the IMA metabolism in rats, which were consistent with the results of DDIs in RLMs.
As for VOR, the main pharmacokinetic parameters (AUC0→t, AUC0→∞, MRT0→∞, Tmax, and Cmax) of VOR in Group C were increased significantly by 96.1%, 94.3%, 18.2%, 74.4%, and 43.5%, respectively, compared to those of the Group A, while CL of VOR was decreased significantly by 50.7%. Moreover, the AUC0→t, AUC0→∞, and Cmax of N-oxide VOR in Group C were decreased significantly by 41.2%, 39.6%, and 21.0%, respectively. Our results indicated that IMA had the inhibitory effect on the VOR metabolism in rats, which were also consistent with the results of DDIs in RLMs.
Discussion
IMA has been considered as the first-line treatment for CML, as well as for GIST and other hematological disorders. It is primarily metabolized by CYP3A4, and its bioavailability is thus expected to be drastically affected by the co-administration of drug known to alter the CYP activity in vitro19,23 and in vivo.24 In cancer patients with low immune function, especially under chemotherapy, opportunistic fungal infections are common problems and result in a high mortality rate. Because of individual variation and narrow therapeutic index in plasma concentration levels, VOR requires TDM as the first-line drug for the treatment of fungal infection. VOR is a CYP3A4 inhibitor, and can lead to high plasma levels of IMA, which result in a severe adverse drug event.25 Although IMA and VOR are co-administered to cancer patients, DDIs associated with the concurrent use have not been fully explored.
In this study, we investigated the effect of VOR on the metabolism of IMA via the formation of CGP74588 in RLMs. The results of our study showed that VOR had a slight inhibition on the IMA metabolism based on IC50 value (105.20 μM) in our vitro assay, which was consistent with the reported findings.19 In pharmacokinetic interaction studies, the results indicated that VOR had no impact on the clearance or bioavailability of IMA except an 18.8% increase of the Cmax in rats. In addition, the main pharmacokinetic parameters (AUC0→t, AUC0→∞, and Cmax) of CGP74588 were decreased significantly. Our results of the pharmacokinetic interaction studies in rats were in agreement with the reported article by Lin et al18. These results in a combination of in vitro and in vivo demonstrated that VOR would increase the IMA exposure. Although a previous study showed that strong CYP3A4 inhibitor ritonavir had no significant effect on the pharmacokinetic parameters of IMA,26 there was sufficient evidence to confirm that VOR would increase the IMA exposure by inhibiting CYP3A4 activity.
To the best of our knowledge, this is the first report about the inhibitory effect of IMA on the metabolism of VOR via the formation of N-oxide VOR in RLMs. Our vitro study indicated that IMA had a moderate inhibition on the VOR metabolism based on IC50 value (61.30 μM). Moreover, the results of pharmacokinetic interaction studies in rats exhibited that the concentration of VOR increased and the formation of N-oxide VOR decreased, which showed IMA inhibited the metabolism of VOR in rats. Therefore, the results in vitro and in vivo were consistent. VOR is extensively metabolized in the liver, primarily through CYP2C19 and, to a lesser extent, through CYP3A4 and CYP2C9.10 The relative importance of the pathways in conjunction with the relative higher abundance of the CYP3A4 compared with low contents of CYP2C19 in human livers may still translate to higher overall contribution of CYP3A4, even having lower CYP3A activities compared with CYP2C19.27 It was suggested that CYP3A4 is important in the metabolism of VOR, which occurred DDI with ritonavir in individuals with poor CYP2C19 catalytic function.28 It was also validated that coadministration of drugs that modulate CYP3A4 activity could effect VOR plasma levels.10,29 IMA was found to be a potent inhibitor of CYP3A4 from the finding of the IMA-simvastatin interaction in patients with CML.20 It was reported that IMA could increase the exposure of a single dose of simvastatin when given concomitantly, and the reason most likely to be the inhibition of CYP3A4-mediated metabolism of simvastatin in the liver. Potent mechanism-based inhibition of CYP3A4 by IMA in vitro was confirmed and suggested that IMA could markedly increase plasma concentrations of CYP3A4 substrates by reducing the clearance rate of the latters.30 This may explain why IMA inhibits the metabolism of VOR. Therefore, caution is required when administering IMA with CYP3A4 substrates with a narrow therapeutic window, especially VOR.
Conclusions
In conclusion, the present study exhibited IMA and VOR inhibited the metabolism of each other and confirmed the importance of CYP3A4 in DDI of IMA and VOR. For the first time, we found that IMA can inhibit the metabolism of VOR in vitro and alter the pharmacokinetic profiles of both VOR and N-oxide VOR in vivo. Given a big chance of IMA co-formulated with VOR, our study gives a novel direction to the guidance of clinical medication and treatment. Clearly, further studies to elucidate the DDI of IMA and VOR are warranted.
Acknowledgments
This study was supported by a grant from the Medical and Health Science and Technology Program of Zhejiang Province in China (Grant No. 2017KY461).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. van Leeuwen RW, van Gelder T, Mathijssen RH, Jansman FG. Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15(8):e315–e326. doi:10.1016/S1470-2045(13)70579-5
2. Dookeeram D, Bidaisee S, Paul JF, et al. Polypharmacy and potential drug-drug interactions in emergency department patients in the Caribbean. Int J Clin Pharm. 2017;39(5):1119–1127. doi:10.1007/s11096-017-0520-9
3. Rodrigues MCS, de Oliveira C. Drug-drug interactions and adverse drug reactions in polypharmacy among older adults: an integrative review. Rev Lat Am Enfermagem. 2016;24: e2800.
4. Gu H, Dutreix C, Rebello S, et al. Simultaneous physiologically based pharmacokinetic (PBPK) modeling of parent and active metabolites to investigate complex CYP3A4 drug-drug interaction potential: a case example of midostaurin. Drug Metab Dispos. 2018;46(2):109–121. doi:10.1124/dmd.117.078006
5. Luo M, Dai MY, Lin HT, et al. Species-related exposure of phase II metabolite gemfibrozil 1-O–glucuronide between human and mice: a net induction of mouse P450 activity was revealed. Biopharm Drug Dispos. 2017;38(9):535–542. doi:10.1002/bdd.2105
6. Groll AH, Castagnola E, Cesaro S, et al. Fourth European Conference on Infections in Leukaemia (ECIL-4): guidelines for diagnosis, prevention, and treatment of invasive fungal diseases in paediatric patients with cancer or allogeneic haemopoietic stem-cell transplantation. Lancet Oncol. 2014;15(8):e327–e340. doi:10.1016/S1470-2045(13)70510-2
7. Solano C, Slavin M, Shaul AJ, et al. Economic evaluation of azoles as primary prophylaxis for the prevention of invasive fungal infections in Spanish patients undergoing allogeneic haematopoietic stem cell transplant. Mycoses. 2017;60(2):79–88. doi:10.1111/myc.12552
8. Alanio A, Denis B, Hamane S, et al. Azole resistance of aspergillus fumigatus in immunocompromised patients with invasive aspergillosis. Emerg Infect Dis. 2016;22(1):157–158. doi:10.3201/eid2201.150848
9. Troke PF, Hockey HP, Hope WW. Observational study of the clinical efficacy of voriconazole and its relationship to plasma concentrations in patients. Antimicrob Agents Chemother. 2011;55(10):4782–4788. doi:10.1128/AAC.01083-10
10. Hyland R, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metab Dispos. 2003;31(5):540–547.
11. Pascual A, Calandra T, Bolay S, Buclin T, Bille J, Marchetti O. Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin Infect Dis. 2008;46(2):201–211. doi:10.1086/524669
12. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2012 Update on diagnosis, monitoring, and management. Am J Hematol. 2012;87(11):1038–1045. doi:10.1002/ajh.23282
13. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. doi:10.1182/blood-2013-05-501569
14. Manley PW, Blasco F, Mestan J, Aichholz R. The kinetic deuterium isotope effect as applied to metabolic deactivation of imatinib to the des-methyl metabolite, CGP74588. Bioorg Med Chem. 2013;21(11):3231–3239. doi:10.1016/j.bmc.2013.03.038
15. Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44(9):879–894. doi:10.2165/00003088-200544090-00001
16. Herbrink M, Nuijen B, Schellens JHM, Beijnen JH. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat Rev. 2015;41(5):412–422. doi:10.1016/j.ctrv.2015.03.005
17. Hachem R, Gomes MZ, El Helou G, et al. Invasive aspergillosis caused by Aspergillus terreus: an emerging opportunistic infection with poor outcome independent of azole therapy. J Antimicrob Chemother. 2014;69(11):3148–3155. doi:10.1093/jac/dku241
18. Lin G, Wang C, Qiu X, et al. Differential effects of ketoconazole, itraconazole and voriconazole on the pharmacokinetics of imatinib and its main metabolite GCP74588 in rat. Drug Dev Ind Pharm. 2014;40(12):1616–1622.
19. Luo X, Li T, Yu Z, et al. The impact of azole antifungal drugs on imatinib metabolism in human liver microsomes. Xenobiotica. 2018;1–18.
20. O‘Brien SG, Meinhardt P, Bond E, et al. Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer. 2003;89(10):1855–1859. doi:10.1038/sj.bjc.6601152
21. Xu RA, Lin Q, Qiu X, et al. UPLC-MS/MS method for the simultaneous determination of imatinib, voriconazole and their metabolites concentrations in rat plasma. J Pharm Biomed Anal. 2019;166:6–12. doi:10.1016/j.jpba.2018.12.036
22. Wang Z, Sun W, Huang CK, et al. Inhibitory effects of curcumin on activity of cytochrome P450 2C9 enzyme in human and 2C11 in rat liver microsomes. Drug Dev Ind Pharm. 2015;41(4):613–616. doi:10.3109/03639045.2014.886697
23. Beumer JH, Pillai VC, Parise RA, et al. Human hepatocyte assessment of imatinib drug-drug interactions – complexities in clinical translation. Br J Clin Pharmacol. 2015;80(5):1097–1108. doi:10.1111/bcp.12723
24. Dutreix C, Peng B, Mehring G, et al. Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother Pharmacol. 2004;54(4):290–294. doi:10.1007/s00280-004-0832-z
25. Gambillara E, Laffitte E, Widmer N, et al. Severe pustular eruption associated with imatinib and voriconazole in a patient with chronic myeloid leukemia. Dermatology. 2005;211(4):363–365. doi:10.1159/000088510
26. van Erp NP, Gelderblom H, Karlsson MO, et al. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin Cancer Res. 2007;13(24):7394–7400. doi:10.1158/1078-0432.CCR-07-0346
27. Yamazaki H, Inoue K, Shaw PM, Checovich WJ, Guengerich FP, Shimada T. Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeprazole by human liver microsomes: effects of contents of these two forms in individual human samples. J Pharmacol Exp Ther. 1997;283(2):434–442.
28. Mikus G, Schowel V, Drzewinska M, et al. Potent cytochrome P4502C19 genotype-related interaction between voriconazole and the cytochrome P450 3A4 inhibitor ritonavir. Clin Pharmacol Ther. 2006;80(2):126–135. doi:10.1016/j.clpt.2006.04.004
29. Murayama N, Imai N, Nakane T, Shimizu M, Yamazaki H. Roles of CYP3A4 and CYP2C19 in methyl hydroxylated and N-oxidized metabolite formation from voriconazole, a new anti-fungal agent, in human liver microsomes. Biochem Pharmacol. 2007;73(12):2020–2026. doi:10.1016/j.bcp.2007.03.012
30. Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787–2798. doi:10.1111/j.1476-5381.2011.01732.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.