")
Back to Journals » ImmunoTargets and Therapy » Volume 10
Drug Treatment of Neuromyelitis Optica Spectrum Disorders: Out with the Old, in with the New?
Authors Held F, Klein AK, Berthele A
Received 5 January 2021
Accepted for publication 16 February 2021
Published 19 March 2021 Volume 2021:10 Pages 87—101
DOI https://doi.org/10.2147/ITT.S287652
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Friederike Held, Ana-Katharina Klein, Achim Berthele
Department of Neurology, School of Medicine, Klinikum Rechts der Isar, Technical University of Munich, Munich, Germany
Correspondence: Achim Berthele
Department of Neurology, School of Medicine, Klinikum Rechts der Isar, Technical University of Munich, Ismaninger Str. 22, Munich, 81675, Germany
Tel +49 89 4140 4673
Fax +49 89 4140 4867
Email [email protected]
Introduction: Neuromyelitis optica spectrum disorders (NMOSD) are rare neuroinflammatory demyelinating diseases of the CNS, mainly affecting optic nerves, spinal cord and brainstem regions. The diagnosis depends on clinical symptoms, MRI findings and the detection of autoantibodies against the water channel aquaporin 4 (AQP4-Ab). This autoantibody is particularly important for diagnostic sensitivity and specificity and further sets the course for major therapeutic decisions. Due to a relapsing course with the accumulation of disability, relapse prevention by immunotherapy is crucial in NMOSD. Until recently, disease-modifying agents specific to NMOSD were not available, and patients were treated with various immunosuppressive drugs and regimens - with variable success. Fortunately, since 2019, three new therapeutic antibodies have entered the market.
Areas Covered: We aim to shortly summarise the pathogenesis and biological targets for acute and preventive therapy of adult NMOSD. We will focus on conventional immunotherapies and the recently approved novel biological drugs satralizumab, eculizumab and inebilizumab, and conclude with a brief outlook on future therapeutic approaches.
Expert Opinion: Although satralizumab, eculizumab and inebilizumab are a breakthrough concerning short-term efficacy, important questions on their future use remain open. There is no data from head-to-head comparisons, and data on long-term safety and efficacy of the new medicines are pending. Whether any of the biologics are efficacious in AQP4-Ab negative NMOSD patients is not yet known – as is how they will succeed in non-responders to conventional immunotherapies. Further, (autoimmune) comorbidities, affordability, and market availability of drugs may be decisive factors for choosing treatments in the near future. We are fortunate to have these new drugs available now, but they will not immediately supersede established off-label drugs in this indication. It is still too early to definitively revise the treatment algorithms for NMOSD - although we are probably on the way.
Keywords: NMO, NMOSD, AQP4, MOG, monoclonal antibody
Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are rare demyelinating diseases of the CNS, affecting 0.52–10/100,000 persons worldwide.1 Female gender, age over 35 years and Asian or African descent constitute risk factors for NMOSD.2 Most NMOSD patients show a relapsing course, and full recovery from relapses is less likely than in multiple sclerosis (MS).3,4 Thus, accrual of disability is generally relapse-dependent, and attack-prevention with immunomodulatory drugs is currently considered the best available treatment in NMOSD.5,6 However, the response to conventional immunosuppressive therapy (IST) is variable, and there is a strong medical need for new therapeutic options or regimens. Fortunately, since NMOSD is a rare disease, drug development may benefit from an orphan drug designation, and NMOSD is increasingly recognised by drug companies as an emerging market.
The Classification of NMOSD
The concept of a “Neuromyelitis optica spectrum” was initially suggested by Wingerchuk et al in 2007 to account for the clinical manifestations beyond optic neuritis or myelitis known from classical Devic disease.7 This definition originated after the discovery of the aquaporin 4 autoantibody (AQP4-Ab), which allowed the description of NMO spectrum manifestations that formerly remained unrecognised.
Currently, NMOSD can be categorised into two pathophysiological entities depending on the presence or absence of AQP4-Ab. In approx. 80% of cases, AQP4-Ab can be detected, which cause a primarily astrocytopathic disease.8,9 Interestingly, in about 50% of the AQP4-Ab seronegative NMOSD patients autoantibodies against myelin-oligodendrocyte-glycoprotein (MOG-Ab) can be seen, whose pathomorphological correlate is primarily oligodendrocytopathic.10–12 Here, NMOSD overlaps with the spectrum of MOG-AD (MOG Antibody disease).13,14 Lastly, double-negative NMOSD cases remain, and whether these are driven by as yet unknown autoantibodies or are rather MS variants is an open question. Irrespective of the primary target, inflammatory damage of axons and myelin are the consequence, which determines the clinical phenotype with opticospinal cerebral lesions.3,12,15,16
The term “Neuromyelitis optica spectrum disorders (NMOSD)” was coined by the International Panel for NMO Diagnosis (IPND), which convened in 2015 to consent on new diagnostic criteria. To date, NMOSD is diagnosed using these IPND diagnostic criteria, which differentiate into AQP4-Ab positive or negative cases.17 A separate diagnostic category for MOG-Ab positive NMOSD is as yet missing. At least, this argues for sticking to the term “Neuromyelitis optica spectrum disorders” rather than lumping all variants into a “Neuromyelitis optica spectrum disorder”.
Pathophysiology of AQP4-Ab Positive NMOSD
Central and peripheral tolerance defects of the innate immune system lead to the development of autoreactive CD20-positive B-Lymphocytes.18–20 The differentiation of lymphocytes and maintenance of antibody production by CD19-CD27-CD138 positive-plasmablasts are triggered by interleukin-6 (IL-6). Both aspects determine the importance of IL-6 as a marker for NMOSD disease severity.21,22 Next to these humoral immune system processes, cellular immune responses aggravate inflammation. Migration of antibodies from the periphery into the CNS and binding to their epitope activates the complement cascade and effector cells.23,24 Complement-dependent cytotoxicity (CDC) and complement-dependent cellular cytotoxicity (CDCC) cause necrotic lesions with complement deposition and infiltration of neutrophils. The latter is preceded by antibody-driven cellular toxicity (ADCC) due to binding of AQP4-Ab to its target.3 The lack of co-expressed complement regulator proteins such as CD59 in the brain is assumed to be the reason for the preferential auto-inflammatory response to antibody binding within the CNS.3,25 Aggregation and internalisation of AQP4 and its accompanied glutamate transporter (excitatory amino acid transporter 2 (EAAT2)) creates an inflammatory milieu due to glutamate-dependent cytotoxicity by extracellular accumulation.26,27 Thus, complement and antibody-mediated cellular toxicity drive progressive destruction of astrocytes and disrupt the blood-brain barrier with consecutive parenchymal and vascular damage to the brain (see Figure 1).3,16,24,27
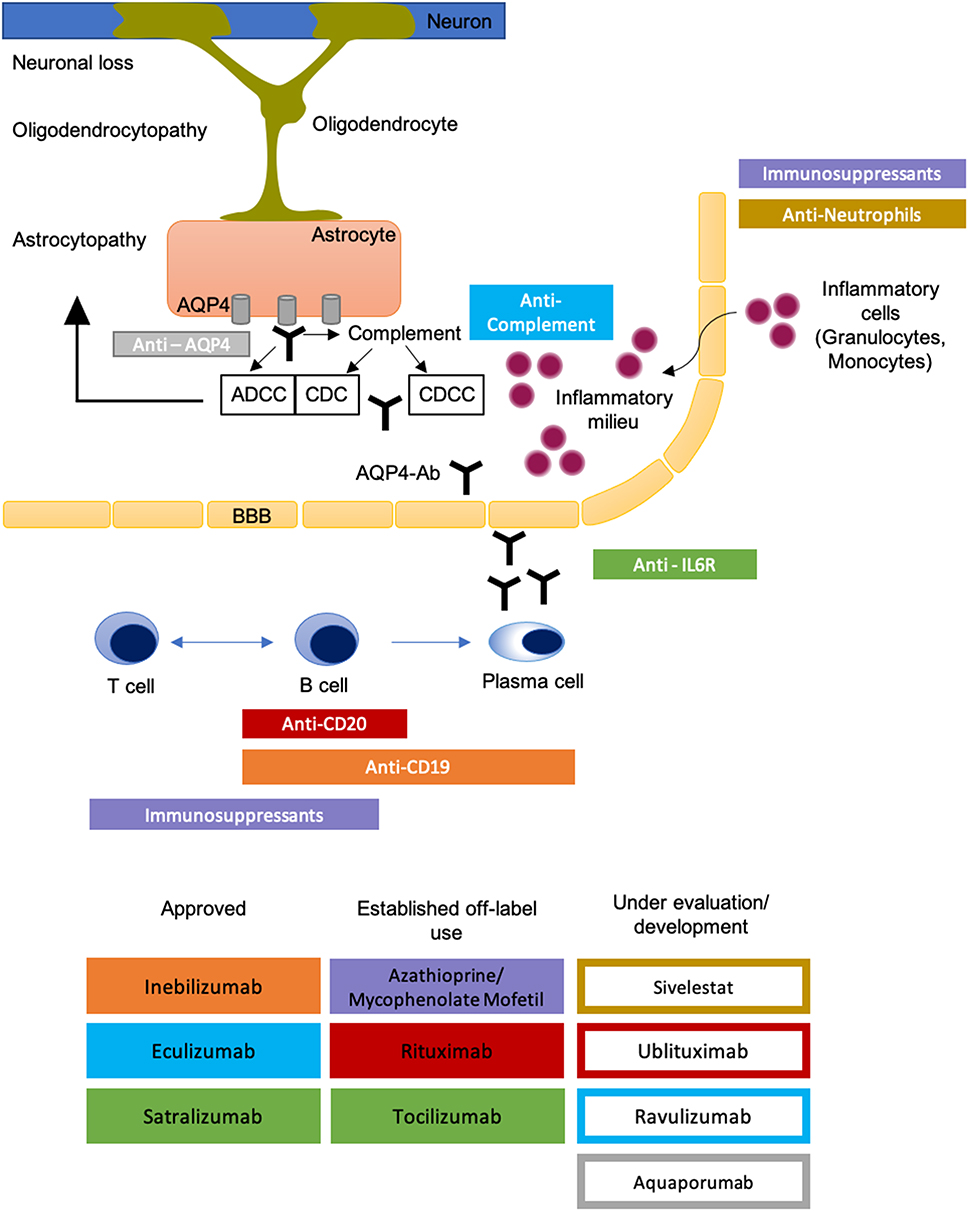 |
Figure 1 Drugs for relapse prevention in NMOSD. Approved therapeutic agents (filled rectangles) and agents under clinical evaluation or development (framed rectangles) and their pharmacological targets. Colour of rectangles represents epitopes. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, Nature Reviews Neurology. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies., COPYRIGHT 2014.24 Abbreviations: ADCC, antibody-dependent cellular cytotoxicity; AQP4-Ab, aquaporin 4 antibody; BBB, blood-brain barrier; CDC, complement-dependent cytotoxicity; CDCC, complement-dependent cellular cytotoxicity. |
Therapy of Acute Attacks
Accumulation of physical impairment in NMOSD is relapse-dependent.28,29 For this reason, acute relapse therapy is crucial to minimise residual deficits. As in multiple sclerosis, high-dose intravenous (i.v.) methylprednisolone (1g/d for five days) is the accepted standard therapy. Retrospective data from cohort studies indicate that early initiation of treatment seems to be associated with a better clinical outcome.28,30,31 A small retrospective study on 27 patients showed that the outcome in patients suffering from optic neuritis was likely to be better when relapse therapy was initiated within four days after onset of symptoms - regardless of antibody serostatus.32 Atrophy of the retinal ganglion layer within a few days after relapse was observed as the pathophysiological correlate.32 For relapses refractory to steroids, escalated therapy with plasma separation/immunoadsorption is warranted. For patients refractory to steroids in previous relapses, apheresis techniques may even be used as first-line therapy.28,30
Prevention of Relapses
Traditional Disease-Modifying Drugs
Predominantly retrospective studies or uncontrolled case series published before 2019 led to the recommendation of azathioprine (AZA), mycophenolate mofetil (MMF) or Rituximab (RTX) as effective first-line agents. All of these drugs lead to a reduction of relapse rates and may stabilise or even improve neurological disability (reflected by the Expanded Disability Status Scale (EDSS)).33–40 Low-dose corticosteroids have not been systematically studied. Nevertheless, they are frequently used, either as maintenance therapy, mono41 or add-on to conventional immunosuppressants, or very slowly tapered after a relapse has occurred.
RTX is a chimeric monoclonal CD 20 antibody inducing B-cell depletion. The clinical efficacy of RTX has recently been replicated in prospective cohorts, most interestingly not only in AQP4-Ab positive patients but also in AQP4-Ab negative patients.42–44 Moreover, in AQP4-Ab positive patients, there is no clear association between the clinical efficacy of RTX and AQP4-Ab levels.33
Compared to RTX, the immunomodulatory effects of AZA and MMF are conveyed by rather unselective suppression of fast-dividing immune cells. The efficacy of AZA and MMF has been shown repeatedly in retrospective studies, reducing the annualised relapse rate (ARR) to a variable extent in each study.34,35,37,38,45 In 2018, Yang et al prospectively compared efficacy and safety of RTX, AZA and MMF: RTX and MMF were reported to be superior to AZA in terms of efficacy and safety profile.46 Adverse events occur mainly under therapy with AZA.43,46
In line with this, Mealy et al revealed the superiority of RTX over MMF or AZA when retrospectively analysing a cohort of 90 NMOSD patients. Moreover, the study exemplifies two current clinical problems: firstly, AQP4-Ab negative patients are more likely to be treated with AZA or MMF than with RTX.47 This may reflect some sort of a “cautionary rule” concerning the use of highly potent and selective B-cell depleting agents in AQP4-Ab negative NMOSD patients. However, the AQP4-Ab serostatus per se did not predict the treatment efficacy of conventional disease-modifying drugs in another cohort study.48
Secondly, when relapses continue to occur under one of these drugs, the success of switching to another drug is somewhat unpredictable – both unexpectedly, switching from AZA/MMF to RTX may fail as switching from RTX to AZA may be successful.47 To this end, unfortunately, studies comparing treatment sequences are entirely missing.
Another therapeutic antibody which is re-purposed as rescue therapy in NMOSD is tocilizumab. Tocilizumab is a humanised monoclonal antibody which binds to the interleukin-6 receptor (IL6-R) and blocks IL6 signalling. Tocilizumab is administered intravenously (i.v.) or subcutaneously (s.c.) as monotherapy or as an add-on. Retrospective case series have shown a reduction in relapse rate and disability, also in highly active NMOSD.49–51
RCTs Evaluating Traditional Biologics
A first randomised controlled clinical trial was published in 2017 by Nikoo et al (see Table 1), which was open-label.43 Comparing treatment with either RTX or AZA revealed a significant reduction of the ARR in both groups compared to baseline, but RTX was more effective than AZA. 78% of participants allocated to RTX, and 54.3% of patients receiving AZA became relapse-free. Notably, only approx. 50% of patients were AQP4-Ab positive.
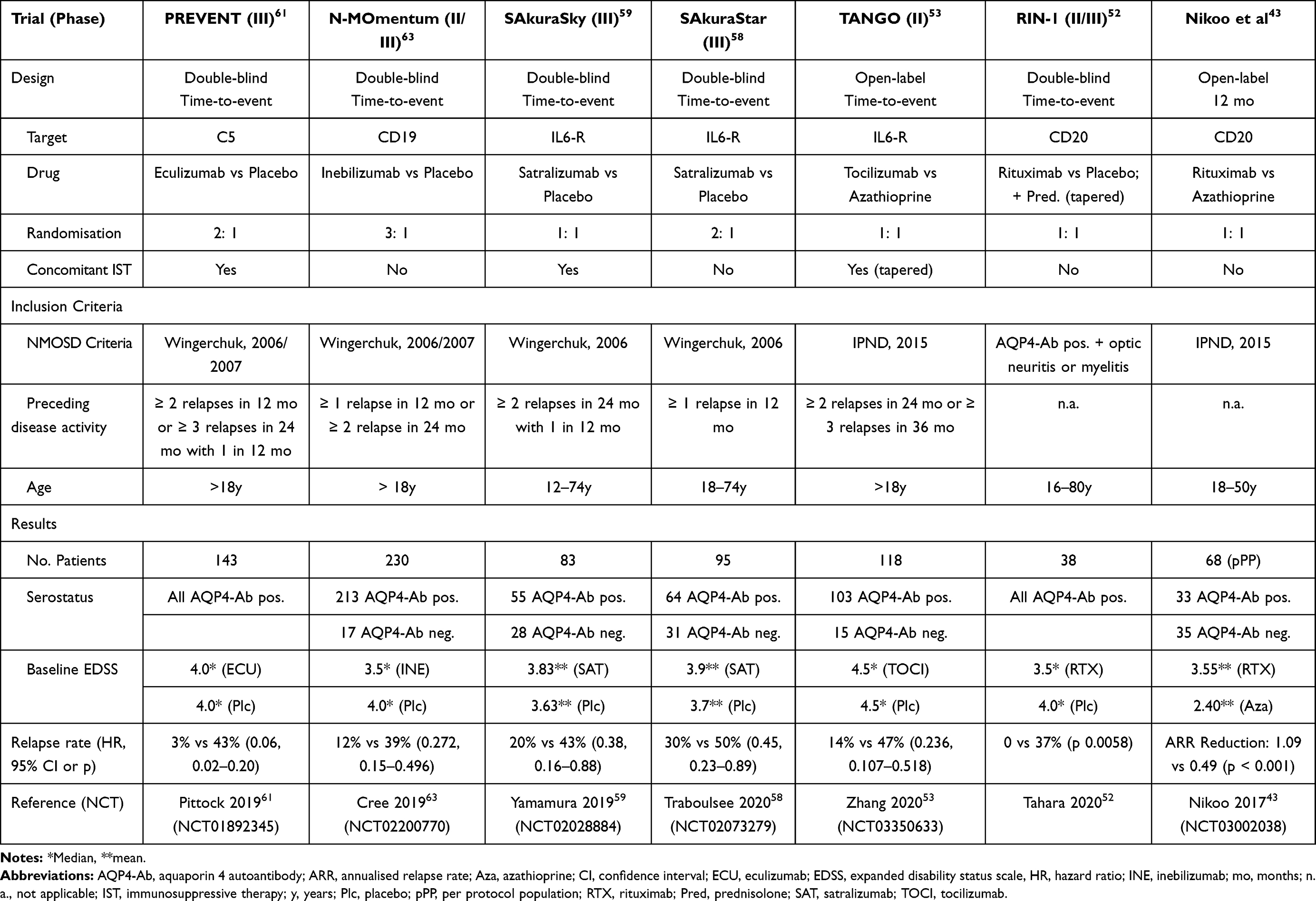 |
Table 1 Overview of Clinical Trials |
In 2020 two additional clinical trials on classical biologics were published, RIN-1 and TANGO (see Table 1).
Rituximab: RIN-1 Trial
Although RTX is a well-established first-line treatment, it is still an off-label use in this indication, and reimbursement often remains a challenging issue. For this reason, in many countries RTX is not available for treating NMOSD patients in a routine setting. In Japan, this led to the RIN-1 study, which was the first multicenter randomised double-blind placebo-controlled Phase III time-to-event clinical trial on RTX in NMOSD.52
38 AQP4-Ab positive patients with an EDSS of 7.0 or less were randomised 1:1 to receive either rituximab i.v. (375 mg/m2 body surface each week 1 to 4, then 1000 mg i.v. at week 24, 26, 48 and 50) or matching placebo and concomitant oral prednisolone, which was tapered over the study’s duration of 72 weeks. A historical AQP4-Ab finding was sufficient; 11 out of 38 patients were AQP4-Ab negative at inclusion. No other immunosuppressants were allowed.
The primary endpoint was set as the time to first relapse. Seven out of 19 patients (37%) treated with placebo relapsed vs none (0 out of 19) on RTX. Regarding secondary endpoints, changes in EDSS and the degree of oral steroid reduction was not significantly different between both groups. There were no unexpected safety issues.
Notably, the positive results of the RIN-1 study may lead to a conditional approval of RTX for the treatment of NMOSD in Japan.
Tocilizumab: TANGO Trial
The TANGO trial was an open-label, multicenter, randomised Phase II clinical trial conducted in China.53 118 patients suffering from an AQP4-Ab seropositive (n=103) or negative (n=15) NMOSD with an EDSS of 7.5 or less were randomised 1:1 to receive either tocilizumab 8mg/kg i.v. every four weeks or AZA 2–3mg/kg p.o. per day. Concomitant IST to AZA was permitted up to 24 weeks after study initiation. The primary endpoint was the time to first relapse, and patients were treated at least 60 weeks. The primary endpoint was the time to first relapse, and patients were treated at least 60 weeks.
Patients allocated to tocilizumab showed a significantly lower risk of relapse: 14% (8 of 59) of patients receiving tocilizumab vs 47% (28 of 59) allocated to AZA suffered a relapse (hazard ratio 0.236, 95% confidence interval 0.107 to 0.518), and the median time to relapse was significantly longer in the tocilizumab group (67.2 weeks vs 38 weeks). A subgroup analysis revealed that tocilizumab worked both in patients with and without concomitant autoimmune diseases (e.g. Sjögren’s syndrome, rheumatoid arthritis). Three patients were tested positive for MOG-Ab (one received tocilizumab, two placebo). Of those, one placebo-treated patient relapsed. Concerning secondary endpoints, a significant reduction of AQP4-Ab titers in 50% of patients treated with tocilizumab was reported, whereas treatment with AZA did not show any effect. The rate of confirmed disease progression at 12 and 24 weeks (increase in EDSS score of at least 1.0 from baseline) was significantly different between both groups.
The incidence of total adverse events was similar in both groups. However, treatment-related adverse events occurred in 61% of patients receiving tocilizumab compared to 83% of patients with AZA. Three people died, two of them receiving tocilizumab. The authors declared that none of the deaths were treatment-related.
Novel Biologics
Recently, the FDA approved three monoclonal antibodies for the treatment of AQP4-Ab positive NMOSD, namely eculizumab (Soliris®, in June 2019), inebilizumab (Uplizna®, in June 2020) and satralizumab (Enspryng®, in August 2020). Eculizumab was also approved in Australia, Canada, Europe, Japan and Switzerland, and satralizumab in Canada, Switzerland and Japan. For inebilizumab, approval in other countries is still pending.
These three new drugs all utilise a unique mode of action: satralizumab inhibits interleukin-6 signalling, eculizumab targets the terminal complement system, and inebilizumab depletes CD19 positive B-cells.
Interleukin-6 Inhibition
IL6 is known to promote differentiation of lymphocytes with a predominance of TH17-cells. IL6-dependent maintenance of AQP4-Ab production by plasmablasts and a proinflammatory milieu increase blood-brain barrier permeability with infiltration of proinflammatory cytokines and antibodies into the CNS. In NMOSD, CSF IL6-levels are higher in relapse than in remission, and IL6-levels during relapse correlate with serum AQP4-Ab titers. Thus, IL-6 is considered a marker of disease activity.54–56
Satralizumab is a s.c. administered monoclonal IgG2 antibody, which binds membrane-bound or soluble interleukin 6-Receptor (IL6-R) (see Table 2). Thereby, it inhibits IL6 signal transduction. Compared to its predecessor tocilizumab, pharmacokinetics have been further optimised by enhanced “Ab-recycling”: After ph-dependent dissociation of the satralizumab-IL6-R complex within the endosome, satralizumab is recycled to bind IL6-R again.57,58 This allows extending the interval of re-dosing.
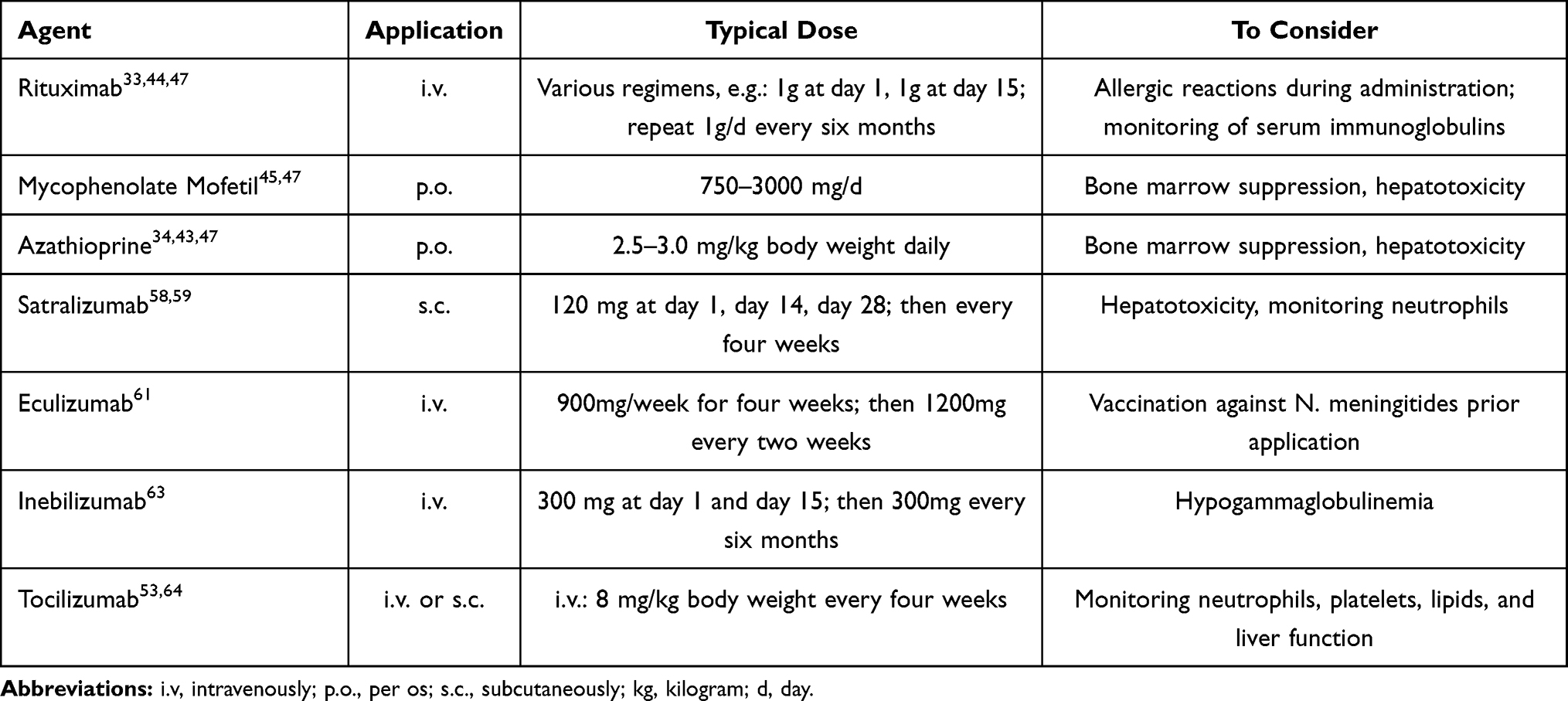 |
Table 2 Dosing Regimens of Therapeutic Agents for Relapse Prevention in NMOSD |
Satralizumab: SAkuraSky Trial
A phase III clinical trial SAkuraSky (see Table 1) evaluated the efficacy and safety of satralizumab when added to baseline immunosuppressants in NMOSD patients.59 83 patients with stable doses of either AZA, MMF or oral glucocorticoids were 1:1 randomised to receive add-on satralizumab or placebo. 55 AQP4-Ab positive and 28 AQP4-Ab negative patients were enrolled (see Table 1). As a time-to-event trial, the primary endpoint was the time to the first adjudicated relapse. The core study ended after 26 protocol-defined relapses had occurred, and participants were eligible to enter an open-label extension.
The study met its primary endpoint: 20% of patients treated with satralizumab vs 43% of patients receiving placebo had a protocol-defined relapse (hazard ratio 0.38, 95% confidence interval 0.16 to 0.88) and a significantly longer time to relapse. However, in AQP4-Ab negative patients, time to relapse and number of relapses (hazard ratio 0.66, 95% confidence interval 0.20 to 2.24) were not significantly different.
Regarding the main secondary endpoints, no significant between-group differences in pain and fatigue scores were observed. Changes in the EDSS at 24 weeks were not significantly different, too.
The frequency of adverse events or severe adverse advents was similar in both groups. There were no deaths and no anaphylactic reactions in both groups.
Satralizumab: SAkuraStar Trial
SAkuraStar was a phase III double-blind, placebo-controlled trial comparing satralizumab monotherapy vs placebo (see Table 1).58 The study enrolled NMOSD patients with or without AQP4-Ab (64 vs 31) and an EDSS of 6.5 or less in 13 countries. Patients were randomised 2:1. The primary endpoint was the time to relapse. The double-blind phase was planned to end when 44 protocol-defined relapses had occurred or 1.5 years after the last patient had been randomised (see Table 1). An open-label extension followed the placebo-controlled double-blind phase.
The double-blind phase of the study ended 1.5 years after the last patient has entered. Only 35 relapses had occurred at this time point, leaving the study less powered than initially planned. Compared to placebo, satralizumab significantly reduced the relapse rate and prolonged the time to relapse. Overall, the study showed a 55% reduction of relapses under therapy with satralizumab than placebo (hazard ratio 0.45, 95% confidence interval 0.23 to 0.89). Relapses occurred in 30% of patients receiving satralizumab versus 50% receiving placebo.
In AQP4-Ab positive patients, only 22% of patients treated with satralizumab had a relapse compared to 57% of those receiving placebo (hazard ratio 0.26, 95% confidence interval 0.11 to 0.63). In contrast, AQP4-Ab negative patients had an even higher relapse rate under therapy with satralizumab than placebo (46% vs 33% respectively). Thus, as in SAkuraSky, AQP4-Ab negative patients seemingly did not profit from satralizumab; however, both studies were most probably underpowered to address this reliably.
Moreover, key secondary endpoints like fatigue and pain did not significantly improve in patients treated with satralizumab. Changes in the EDSS score at 24 weeks were not significantly different between groups.
The rate of serious adverse events was similar in both groups. Only one patient receiving satralizumab discontinued the trial due to severe pneumonia.
Complement Inhibition
AQP4-Ab belong to the IgG1 subclass, and the activation of the complement system is a major feature of inflammatory lesions in NMOSD. The terminal complement cascade comprises C5a, which takes effect as a proinflammatory mediator, and C5b, which is directly involved in forming the membrane attack complex C5b-9.
Eculizumab is a humanised IgG2/IgG4-hybrid monoclonal antibody against complement factor 5 (see Table 2). Its binding prevents the cleavage of C5 to C5a and C5b, thereby inhibiting both final downstream effector mechanisms of the complement system.
In an open-label study published in 2013, eculizumab was highly efficacious in 14 AQP4-Ab positive female NMOSD patients by reducing the median annual relapse number from 3 to 0 and improving the EDSS.60 Unfortunately, one patient developed meningococcal sepsis. Nevertheless, the promising results on efficacy gave rise to a large placebo-controlled trial PREVENT, and a trial on pediatric patients is underway (NCT04155424).
Eculizumab: PREVENT Trial
PREVENT was a Phase III, time-to-event, double-blind, randomised controlled trial assessing the efficacy and safety of eculizumab as add-on or monotherapy compared to placebo.61 The results were published in 2019; an open-label extension is still ongoing for a small number of patients. In the core trial, 143 patients from 18 countries suffering from AQP4-Ab positive NMOSD with an EDSS of 7.0 or less received either eculizumab or placebo (1:1) (see Table 2). Continuation of a pre-existing stable immunosuppressive therapy was permitted and occurred in 76% of the cases (see Table 1).
The trial had a time-to-event design and stopped after 23 of 24 prespecified adjusted relapses. Overall, there was a 94% reduction of relapse risk under therapy with eculizumab. Relapses occurred in 3% of patients (3 out of 96) receiving eculizumab vs 43% of patients (20 out of 47) receiving placebo (hazard ratio 0.06, 95% confidence interval 0.02 to 0.20). Eculizumab showed consistent efficacy in patients both with or without concomitant immunosuppressive therapy in AQP4-Ab positive NMOSD. Remarkably, none of the patients treated with eculizumab as a monotherapy suffered from a relapse - in contrast to 4% of patients treated with eculizumab and concomitant IST.
Concerning secondary endpoints, EDSS, modified Ranking scale, Hauser Ambulation index and European Quality of Life 5-Dimension 3-Level (EQ-5D-3L) showed no significant difference between both groups.
All patients received vaccination against Neisseria meningitides before receiving any study drug. There was no meningococcal infection, and the rates of adverse events or serious adverse events were similar in both groups. There was one death in the eculizumab group due to pulmonary empyema. Two patients discontinued the study due to adverse events - both of them receiving placebo.
CD19 B-Cell Depletion
Besides the pivotal role of AQP4-Ab, B-cells contribute to NMOSD pathology by B-cell T-cell collaboration.
Inebilizumab (formerly MEDI-551) is an i.v. administered humanised IgG1 kappa monoclonal antibody targeting the CD19 surface antigen on B-Lymphocytes (see Table 2). In contrast to CD20-directed antibodies like RTX, inebilizumab depletes a broader range of B-cells, including plasmablasts and some plasma cells. Thus, inebilizumab may especially be advantageous concerning the inhibition of AQP4-Ab production.
A phase Ib dose-escalating trial in relapsing MS patients showed an acceptable safety profile of inebilizumab up to 2x 600 mg administered i.v. With all doses investigated, depletion of peripheral B-cells was complete and sustained over 24 weeks.62
Inebilizumab: N-MOmentum Trial
Up to now, N-MOmentum is the largest placebo-controlled phase II/III clinical trial in NMOSD (see Table 1). The core study results were published in 2019, the open-label extension ist ongoing.63 231 patients from 25 countries suffering from an AQP4-Ab positive (n=212) or negative (n=18) NMOSD with an EDSS of 8.0 or less were randomised 3:1 to receive either i.v. inebilizumab monotherapy (see Table 2) or placebo. All participants started with a short course of oral prednisolone as comedication to prevent early relapses after B-cell therapy initiation. After that, no concomitant immunosuppressants were permitted. The study had a time-to-event design and was planned to last until 67 relapses had occurred. For each participant, the double-blind phase ended after a relapse or after day 197 (see Table 1). These alternative criteria were introduced to limit the period patients in the placebo arm remained without immunomodulatory treatment to an ethically acceptable length of time.
At the instigation of the data safety monitoring board, the N-MOmentum trial recruitment was halted early due to better than expected efficacy. With only 43 relapses observed, the primary endpoint was met: Under treatment with inebilizumab relapses occurred in 12% (21/174) vs 39% (22/56) in the placebo group (hazard ratio 0.272, 95% confidence interval 0.15 to 0.496). When analysing AQP4-Ab positive patients separately, relapses occurred in 11% under treatment with inebilizumab vs 42% in the placebo group. Interestingly, three out of 13 AQP4-Ab negative patients suffered a relapse under therapy with inebilizumab, but none under placebo (0/4).
As a secondary endpoint, the EDSS was analysed by assessing the number of patients with EDSS worsening. In this regard, inebilizumab patients did significantly better (odds ratio 0.371, 95% confidence interval 0.181 to 0.763). Changes in the mean EDSS were not reported. Additionally, inebilizumab treated patients were less likely to develop new active MRI lesions in the CNS.44 Data on AQP4-Ab levels are not yet published.
The rate of adverse or serious adverse events was similar in both groups of the core study; however, treatment interruptions or discontinuations due to adverse effects only occurred in the inebilizumab-treated group (in three resp. two out of 174 patients). After six months, serum immunoglobulin levels decreased by approx 10% (most pronounced for IgM) in inebilizumab treated patients, and this decrease was seemingly ongoing with treatment duration. Therefore, the FDA issued the requirement to monitor immunoglobulin levels during therapy.
Moreover, there were two fatalities in the open-label extension. One was most likely due to an NMOSD relapse-related respiratory insufficiency, and the second resulted from an encephalopathy of unknown origin.
Although all Phase 3 studies had a time-to-event design, trial settings differed in various aspects (see Table 1), making comparisons difficult.65 AQP4-Ab negative patients were not allowed in the eculizumab trial; concomitant IST or prednisolone was permitted in some, but not all clinical trials. Inclusion criteria differed concerning preceding disease activity and former immunomodulatory treatments. Last but not least, relapses as the primary outcome were identified with differing scrutiny: in the eculizumab PREVENT trial, a relapse adjudication committee was installed amid the ongoing study, whereas in both the satralizumab and inebilizumab trials, relapse adjudication committees were in place right from the beginning. Moreover, in the inebilizumab trial, this committee followed the most rigorous criteria to confirm these clinical events.
Drugs Under Development
Aquaporumab
Aquaporumab is a monoclonal antibody that binds AQP4 with high affinity (see Figure 1, Table 3) and may displace AQP4-Ab from binding. The Fc portion of aquaporumab is engineered to incapacitate AQP4-Ab from triggering CDC or ADCC downstream mechanisms. Thus, binding of aquaporumab is not pathogenic, but is expected to block the cell- and complement-mediated cytotoxicity of pathogenic AQP4-Ab in NMO patients. Aquaporumab has been shown to be effective in a mouse model of NMO.66,67
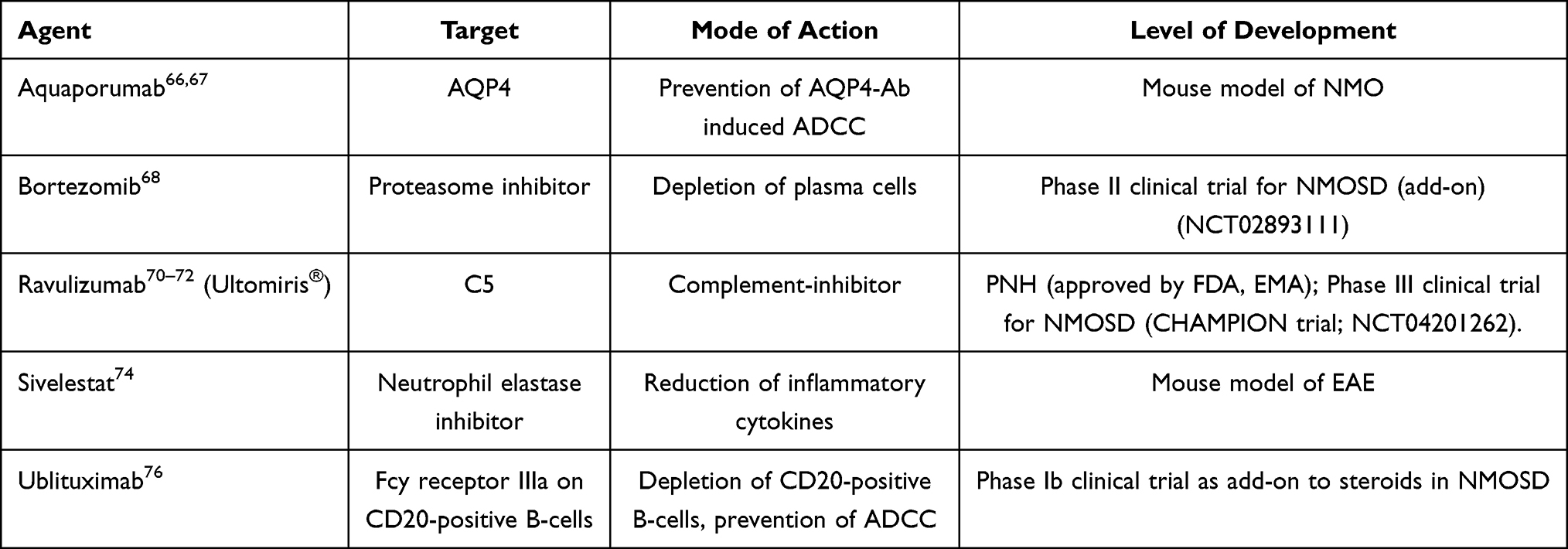 |
Table 3 Drugs Under Development |
Bortezomib
Bortezomib (see Table 3) is a proteasome inhibitor, developed and approved for the treatment of multiple myeloma. As such, it depletes plasma cells, and its use is evaluated in various autoantibody-driven neurologic autoimmune diseases, including myasthenia gravis or Anti-NMDA-Receptor encephalitis. Next to the induction of peripheral neuropathy, which is a typical side effect of the drug, a rebound of plasma cell activity with an overshooting production of autoantibodies after cessation of the drug is a concern. In NMOSD, bortezomib was investigated as an add-on medication in a small open-label study of five AQP4-Ab positive Chinese patients who were refractory to other drugs (NCT02893111). Four out of five patients were relapse-free during a one-year follow-up. Observed side effects were mild, but long-term outcome and safety profiles were not reported.68 It remains to be determined whether bortezomib, with its potentially unfavourable side effect profile, will be further developed in the treatment of NMOSD.
Ravulizumab (Ultomiris®)
Ravulizumab (see Figure 1, Table 3) is a next-generation antibody targeting C5, which was designed to provide prolonged therapy intervals by utilising the “Ab-recycling” approach.69 Ravulizumab is applied only every eight weeks. Non-inferiority to eculizumab has been proven by two large phase III trials in patients with paroxysmal nocturnal hemoglobinuria (PNH).70,71 Based on these findings, ravulizumab was approved by the FDA and EMA for the use in adult PNH in 2018 and 2019.72 In December 2019, a phase 3 open-label study of ravulizumab in AQP4-Ab positive NMOSD patients had started (CHAMPION trial; NCT04201262).
Sivelestat
Sivelestat (see Figure 1, Table 3) is a neutrophil elastase inhibitor. Neutrophil elastase is a proteolytic, highly destructive enzyme, that triggers the production of inflammatory cytokines.73 In a mouse model of experimental autoimmune encephalomyelitis sivelestat reduced ADCC.74 Although phase I/II clinical trials were discontinued for various reasons, a granulocyte-targeting therapy remains a promising therapeutic approach.75
Ublituximab
A pilot safety study of Ublituximab as a novel add-on therapy in acute relapses of NMOSD was published by Mealy et al.76 Ublituximab is a third-generation chimeric IgG1 monoclonal antibody with high affinity to the Fcy receptor IIIa (FCyRIIIA), an epitope on CD20-positive B-cells which is not targeted by rituximab (see Figure 1, Table 3). Ublituximab has enhanced ADCC activity while CDC is retained.44,77 In the phase Ib clinical trial with five patients, ublituximab was applied as an add-on to high dose i.v. steroids within five days of relapse onset. Ublituximab was well tolerated. In three subjects EDSS improved at 90d follow-up. Two of the five patients exhibited relapses within three months due to insufficient depletion of B-cells.76 Further clinical development in NMOSD is pending.
What Comes Next?
Refractory courses of autoimmunological diseases of the CNS under immunomodulatory therapy are common. In these cases, hematopoietic stem cell therapy (HSCT) is increasingly gaining relevance. The objective is the reconstruction of a new, self-tolerant immune system. Due to the lower occurrence of graft versus host disease (GvHD) and transplant-related mortality, autologous stem cell therapy is currently preferred to allogenic stem cell therapy.78–80
The first case report of an autologous stem cell transplantation in a 23-year-old severely affected patient with refractory NMOSD was published in 2010.81 In a 12-month follow-up, the patient remained blind, but paraparesis and dysesthesia remitted. Stem cell therapy also aims at preventing further relapses. A Europe-wide retrospective multicenter study with 16 patients diagnosed with NMO or NMOSD had a mixed outcome with recurrent relapses and AQP4-Ab persistence in some patients.82 HSCT conducted in a cohort of 12 patients suffering from an NMOSD at Northwestern University achieved resounding success with prolonged drug-free remission and suppression of AQP4-Ab.83 The choice of drugs for the conditioning regimen of autologous stem cells was discussed as the underlying cause for the contrasting results of both studies. The use of rituximab seems to be promising. In terms of safety, no grade 4 toxicity has been described.83 The promising therapeutic effects of autologous HSCT with an acceptable safety profile led to the recently published level 2 recommendation of the European Society for Blood and Marrow Transplantation for refractory cases of NMOSD.84,85
MOG-Ab Associated Disease with NMOSD Phenotype
The prognosis, therapy and outcome of patients with MOG-Ab and NMOSD phenotype correspond to MOG-associated disease (MOG-AD) in general.
Treatment of MOG-AD is highly variable, and recommendations remain scarce.86 MOG-AD shows a relapsing course in around 50%. Monophasic courses can occur especially in low-titer MOG-Ab positive patients.87 Thus, after the first relapse of MOG-AD, low-dose steroid therapy alone for three to six months is often sufficient and is frequently recommended as such.87 Other immunosuppressive drugs are regularly started when a second relapse occurred. Alternatively, persisting high MOG-Ab titers after relapse therapy may indicate a relapsing course, which may warrant the initiation of long-term immunosuppressive therapy.13,87
Prospective clinical trials on MOG-AD are still lacking, and treatment recommendations rely on observational studies. A prolonged steroid therapy following a relapse therapy in patients with persistent MOG-Ab reduces the risk of early relapses.87 For maintenance therapy, AZA, MMF and methotrexate, as well as i.v. immunoglobulins (IvIg) or rituximab, have been described.87–90 In 70 patients suffering relapsing MOG-AD the long-term use of IvIg showed the most effective reduction of relapse rate during a study period of 18 months: only 20% of patients treated with IvIg experienced further relapses compared to 74% under therapy with MMF, 61% under RTX and 59% under AZA.91 Previous small retrospective studies support these results, especially in children.88,92,93
The comparably low efficacy of RTX in MOG-AD was further supported by a retrospective study of 121 patients of whom 78.9% developed relapses under RTX despite sufficient B-cell depletion.90 The underlying pathophysiology is still unknown. Nevertheless, for MOG-AD intravenous immunoglobulins seem to be the most effective therapy for relapse prevention.
Problems Solved?
The approval of three new biologicals is a hallmark in the therapy of NMOSD. However, the benefits are only evident in the large subset of AQP4-Ab positive patients, and, so far, data on long-term efficacy and safety of these new biologicals are not available. In AQP4-Ab seronegative NMOSD, inebilizumab and satralizumab do not seem to have a relevant impact on the disease course. Eculizumab was only tested in AQP4-Ab seropositive NMOSD. Moreover, the benefits for MOG-AD with NMOSD phenotype remains uncertain, and therapy-refractory patients still pose a further challenge.
High costs for treatment with novel monoclonal antibodies lead to an exclusive availability of these agents in high-income countries. For low-income countries, the approval of such drugs does not impact the treatment of NMO. The use of conventional immunosuppressive drugs is also unevenly distributed: while RTX and MMF are preferred drugs in high-income countries, AZA is most often used in low-income countries due to low therapy costs despite the higher percentage of adverse events.94
Conclusion/Expert Opinion
Whereas, until two years ago, only off-label therapies were available, there are currently three drugs FDA-approved for treating AQP4-Ab positive NMOSD patients, as well as a level 2 recommendation for HSCT in refractory courses. Monoclonal antibodies with class I evidence inhibiting complement activation (eculizumab), IL6-R signalling (satralizumab) or promoting B-cell-depletion (inebilizumab) were consistently more effective than placebo. Head-to-head studies of these three drugs would be of interest but are admittedly challenging. Moreover, a true comparison with RTX would be helpful, since small head-to-head studies have already revealed RTX’s and Tocilizumab’s superiority compared to AZA.
Regarding their safety profiles, data on long-term effects do not exist for all three novel agents. RTX has shown superiority regarding adverse events compared to AZA over the last years. Eculizumab and tocilizumab both have a well-known safety profile in other inflammatory diseases.
Next, although approved for the use in many countries, the allowed indications may still differ. For example, eculizumab has an unrestricted first-line label in the US, but, in the EU, it can be utilised only after the second relapse. In Australia, authorities favour the use of eculizumab as an add-on therapy.
Thus, recommending a treatment algorithm is still far from straight forward. Apart from the lack of approval, available data keep supporting RTX as a first-line option in AQP4-Ab positive and -negative NMOSD patients. Eculizumab might be superior in AQP4-Ab positive patients. It can be recommended in highly-active patients or even RTX non-responders, although the latter assumption cannot be derived with certainty from the available data. In contrast, eculizumab is not an immunosuppressant in the strict sense, so that the autoimmune cause of NMOSD remains largely unaffected. In the long term, satralizumab, inebilizumab or RTX, may therefore be the more effective therapeutic agents. Finally, costs, the immediacy of immunosuppression, the patient’s preferences and burden of therapy (e.g. route and frequency of administration) have to be taken into account.
Overall, therapy for NMOSD must be tailored to each patient. Fortunately, the drug armamentarium has grown and continues to grow, which helps to achieve this.
Funding
There was no funding in support of the preparation of this manuscript.
Disclosure
F. Held reports no financial or non-financial interests. A. Klein reports no financial or non-financial interests. A. Berthele reports speaker and consulting honoraria from Alexion. His institution has received compensations for the participation in the eculizumab in NMOSD (PREVENT) and ravulizumab in NMOSD (CHAMPION) trials. Further, AB reports speaker and consulting honoraria from Biogen, Bayer Healthcare, Celgene, Merck, Novartis Pharma and Roche; all outside the submitted work.
References
1. Papp V, Illes Z, Magyari M, et al. Nationwide prevalence and incidence study of neuromyelitis optica spectrum disorder in Denmark. Neurology. 2018;91(24):E2265–E2275. doi:10.1212/WNL.0000000000006645
2. Jonsson DI, Sveinsson O, Hakim R, et al. Epidemiology of NMOSD in Sweden from 1987 to 2013: a nationwide population-based study. Neurology. 2019;93(2):E181–E189. doi:10.1212/WNL.0000000000007746
3. Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci. 2016;1366(1):20–39. doi:10.1111/nyas.12794
4. Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. 2012;135(6):1834–1849. doi:10.1093/brain/aws109
5. Stellmann J-P, Krumbholz M, Friede T, et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry. 2017;88(8):639–647. doi:10.1136/jnnp-2017-315603
6. Kawachi I, Lassmann H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2017;88(2):137–145. doi:10.1136/jnnp-2016-313300
7. Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6(9):805–815. doi:10.1016/S1474-4422(07)70216-8
8. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9(1):503. doi:10.1186/1742-2094-9-14
9. Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78(9):665–671. doi:10.1212/WNL.0b013e318248dec1
10. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088–2094. doi:10.1007/s00415-017-8596-7
11. Sepúlveda M, Aldea M, Escudero D, et al. Epidemiology of NMOSD in Catalonia: influence of the new 2015 criteria in incidence and prevalence estimates. Mult Scler J. 2018;24(14):1843–1851. doi:10.1177/1352458517735191
12. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15(2):89–102. doi:10.1038/s41582-018-0112-x
13. Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol. 2019;266(5):1280–1286. doi:10.1007/s00415-018-9122-2
14. Kunchok A, Chen JJ, Saadeh RS, et al. Application of 2015 seronegative neuromyelitis optica spectrum disorder diagnostic criteria for patients with myelin oligodendrocyte glycoprotein IgG–associated disorders. JAMA Neurol. 2020;77(12):1572. doi:10.1001/jamaneurol.2020.2743
15. Wu Y, Zhong L, Geng J. Neuromyelitis optica spectrum disorder: pathogenesis, treatment, and experimental models. Mult Scler Relat Disord. 2019;27:412–418. doi:10.1016/j.msard.2018.12.002
16. Höftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139(5):875–892. doi:10.1007/s00401-020-02132-y
17. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–189. doi:10.1212/WNL.0000000000001729
18. Bennett JL, O’Connor KC, Bar-Or A, et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2015;2:e104. doi:10.1212/NXI.0000000000000104
19. Kowarik MC, Astling D, Gasperi C, et al. CNS aquaporin-4-specific B cells connect with multiple B-cell compartments in neuromyelitis optica spectrum disorder. Ann Clin Transl Neurol. 2017;4(6):369–380. doi:10.1002/acn3.418
20. Wilson R, Makuch M, Kienzler A-K, et al. Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain. 2018;141(4):1063–1074. doi:10.1093/brain/awy010
21. Fujihara K, Bennett JL, de Seze J, et al. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2020;7:1–12. doi:10.1212/NXI.0000000000000841
22. Barros PO, Cassano T, Hygino J, et al. Prediction of disease severity in neuromyelitis optica by the levels of interleukin (IL)-6 produced during remission phase. Clin Exp Immunol. 2016;183(3):480–489. doi:10.1111/cei.12733
23. Duan T, Smith AJ, Verkman AS. Complement-independent bystander injury in AQP4-IgG seropositive neuromyelitis optica produced by antibody-dependent cellular cytotoxicity. Acta Neuropathol Commun. 2019;7(1):112. doi:10.1186/s40478-019-0766-7
24. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10(9):493–506. doi:10.1038/nrneurol.2014.141
25. Yao X, Verkman AS. Marked central nervous system pathology in CD59 knockout rats following passive transfer of Neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun. 2017;5(1):15. doi:10.1186/s40478-017-0417-9
26. Hinson SR, Romero MF, Popescu BFG, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci U S A. 2012;109(4):1245–1250. doi:10.1073/pnas.1109980108
27. Hinson SR, Clift IC, Luo N, et al. Autoantibody-induced internalization of CNS AQP4 water channel and EAAT2 glutamate transporter requires astrocytic Fc receptor. Proc Natl Acad Sci U S A. 2017;114(21):5491–5496. doi:10.1073/pnas.1701960114
28. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1153 treatment courses. Ann Neurol. 2016;79(2):206–216. doi:10.1002/ana.24554
29. Mader S, Kümpfel T, Meinl E. Novel insights into pathophysiology and therapeutic possibilities reveal further differences between AQP4-IgG- and MOG-IgG-associated diseases. Curr Opin Neurol. 2020;33(3):362–371. doi:10.1097/WCO.0000000000000813
30. Kleiter I, Gahlen A, Borisow N, et al. Apheresis therapies for NMOSD attacks. Neurol Neuroimmunol Neuroinflamm. 2018;5:e504. doi:10.1212/NXI.0000000000000504
31. Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346–351. doi:10.1136/jnnp-2017-316286
32. Stiebel-Kalish H, Hellmann MA, Mimouni M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm. 2019;6:1–8. doi:10.1212/NXI.0000000000000572
33. Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology. 2011;76(15):1310–1315. doi:10.1212/WNL.0b013e3182152881
34. Costanzi C, Matiello M, Lucchinetti CF, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77(7):659–666. doi:10.1212/WNL.0b013e31822a2780
35. Elsone L, Kitley J, Luppe S, et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: a multicentre retrospective observational study from the UK. Mult Scler J. 2014;20(11):1533–1540. doi:10.1177/1352458514525870
36. Lau LW, Cua R, Keough MB, et al. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci. 2013;14(10):722–729. doi:10.1038/nrn3550
37. Huh SY, Kim SH, Hyun JW, et al. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol. 2014;71:1372–1378. doi:10.1001/jamaneurol.2014.2057
38. Chen H, Qiu W, Zhang Q, et al. Comparisons of the efficacy and tolerability of mycophenolate mofetil and azathioprine as treatments for neuromyelitis optica and neuromyelitis optica spectrum disorder. Eur J Neurol. 2017;24(1):219–226. doi:10.1111/ene.13186
39. Torres J, Pruitt A, Balcer L, et al. Analysis of the treatment of neuromyelitis optica. J Neurol Sci. 2015;351(1–2):31–35. doi:10.1016/j.jns.2015.02.012
40. Jeong IH, Park B, Kim S-H, et al. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler J. 2016;22(3):329–339. doi:10.1177/1352458515587752
41. Watanabe S, Misu T, Miyazawa I, et al. Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler J. 2007;13(8):968–974. doi:10.1177/1352458507077189
42. Cree BAC, Lamb S, Morgan K, et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64(7):1270–1272. doi:10.1212/01.WNL.0000159399.81861.D5
43. Nikoo Z, Badihian S, Shaygannejad V, et al. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264(9):2003–2009. doi:10.1007/s00415-017-8590-0
44. Kim W, Kim HJ. Monoclonal antibody therapies for multiple sclerosis and neuromyelitis optica spectrum disorder. J Clin Neurol. 2020;16(3):355–368. doi:10.3988/jcn.2020.16.3.355
45. Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. 2009;66(9):1128–1133. doi:10.1001/archneurol.2009.175
46. Yang Y, Wang C-J, Wang B-J, et al. Comparison of efficacy and tolerability of azathioprine, mycophenolate mofetil, and lower dosages of rituximab among patients with neuromyelitis optica spectrum disorder. J Neurol Sci. 2018;385:192–197. doi:10.1016/j.jns.2017.12.034
47. Mealy MA, Wingerchuk DM, Palace J, et al. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71(3):324–330. doi:10.1001/jamaneurol.2013.5699
48. Mealy MA, Kim S-H, Schmidt F, et al. Aquaporin-4 serostatus does not predict response to immunotherapy in neuromyelitis optica spectrum disorders. Mult Scler J. 2018;24(13):1737–1742. doi:10.1177/1352458517730131
49. Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82(15):1302–1306. doi:10.1212/WNL.0000000000000317
50. Ringelstein M, Ayzenberg I, Harmel J, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72(7):756. doi:10.1001/jamaneurol.2015.0533
51. Ayzenberg I, Kleiter I, Schröder A, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-cd20 therapy. JAMA Neurol. 2013;70(3):394–397. doi:10.1001/jamaneurol.2013.1246
52. Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19(4):298–306. doi:10.1016/S1474-4422(20)30066-1
53. Zhang C, Zhang M, Qiu W, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, Phase 2 trial. Lancet Neurol. 2020;19(5):391–401. doi:10.1016/S1474-4422(20)30070-3
54. Uzawa A, Mori M, Arai K, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler. 2010;16(12):1443–1452. doi:10.1177/1352458510379247
55. Içöz S, Tüzün E, Kürtüncü M, et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010;120(1):71–75. doi:10.3109/00207450903428970
56. Matsushita T, Tateishi T, Isobe N, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One. 2013;8(4):2–9. doi:10.1371/journal.pone.0061835
57. Igawa T, Ishii S, Tachibana T, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28(11):1203–1207. doi:10.1038/nbt.1691
58. Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402–412. doi:10.1016/S1474-4422(20)30078-8
59. Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114–2124. doi:10.1056/NEJMoa1901747
60. Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12(6):554–562. doi:10.1016/S1474-4422(13)70076-0
61. Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(7):614–625. doi:10.1056/NEJMoa1900866
62. Agius MA, Klodowska-Duda G, Maciejowski M, et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: results from a Phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler J. 2019;25(2):235–245. doi:10.1177/1352458517740641
63. Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352–1363. doi:10.1016/S0140-6736(19)31817-3
64. Akaishi T, Nakashima I. Efficiency of antibody therapy in demyelinating diseases. Int Immunol. 2017;29(7):327–335. doi:10.1093/intimm/dxx037
65. Xue T, Yang Y, Lu Q, et al. Efficacy and safety of monoclonal antibody therapy in neuromyelitis optica spectrum disorders: evidence from randomized controlled trials. Mult Scler Relat Disord. 2020;43:102166. doi:10.1016/j.msard.2020.102166
66. Tradtrantip L, Zhang H, Saadoun S, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71(3):314–322. doi:10.1002/ana.22657
67. Duan T, Tradtrantip L, Phuan P-W, et al. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology. 2020;162:107827. doi:10.1016/j.neuropharm.2019.107827
68. Zhang C, Tian D-C, Yang C-S, et al. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 2017;74(8):1010. doi:10.1001/jamaneurol.2017.1336
69. Sheridan D, Yu Z-X, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. doi:10.1371/journal.pone.0195909
70. Lee JW, de Fontbrune FS, Lee LWL, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
71. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor–experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
72. McKeage K. Ravulizumab: first global approval. Drugs. 2019;79(3):347–352. doi:10.1007/s40265-019-01068-2
73. Aikawa N, Kawasaki Y. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther Clin Risk Manag. 2014;10:621–629. doi:10.2147/TCRM.S65066
74. Saadoun S, Waters P, MacDonald C, et al. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012;71(3):323–333. doi:10.1002/ana.22686
75. Araki M, Yamamura T. Neuromyelitis optica spectrum disorders: emerging therapies. Clin Exp Neuroimmunol. 2017;8(2):107–116. doi:10.1111/cen3.12394
76. Mealy MA, Levy M. A pilot safety study of ublituximab, a monoclonal antibody against CD20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine. 2019;98(25):e15944. doi:10.1097/MD.0000000000015944
77. Le Garff-tavernier M, Herbi L, De Romeuf C, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28(1):230–233. doi:10.1038/leu.2013.240
78. Sykes M, Nikolic B. Treatment of severe autoimmune disease by stem-cell transplantation. Nature. 2005;435(7042):620–627. doi:10.1038/nature03728
79. Dominietto A, Lamparelli T, Raiola AM, et al. Transplant-related mortality and long-term graft function are significantly influenced by cell dose in patients undergoing allogeneic marrow transplantation. Blood. 2002;100(12):3930–3934. doi:10.1182/blood-2002-01-0339
80. Burman J, Tolf A, Hägglund H, et al. Autologous haematopoietic stem cell transplantation for neurological diseases. J Neurol Neurosurg Psychiatry. 2018;89(2):147–155. doi:10.1136/jnnp-2017-316271
81. Peng F, Qiu W, Li J, et al. A preliminary result of treatment of neuromyelitis optica with autologous peripheral hematopoietic stem cell transplantation. Neurologist. 2010;16(6):375–378. doi:10.1097/NRL.0b013e3181b126e3
82. Greco R, Bondanza A, Vago L, et al. Allogeneic hematopoietic stem cell transplantation for neuromyelitis optica. Ann Neurol. 2014;75(3):447–453. doi:10.1002/ana.24079
83. Burt RK, Balabanov R, Han X, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology. 2019;93(18):E1732–E1741. doi:10.1212/WNL.0000000000008394
84. Sharrack B, Saccardi R, Alexander T, et al. Autologous haematopoietic stem cell transplantation and other cellular therapy in multiple sclerosis and immune-mediated neurological diseases: updated guidelines and recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transplant. 2020;55(2):283–306. doi:10.1038/s41409-019-0684-0
85. Ceglie G, Papetti L, Valeriani M, et al. Hematopoietic stem cell transplantation in neuromyelitis optica-spectrum disorders (NMO-SD): state-of-the-art and future perspectives. Int J Mol Sci. 2020;21(15):1–17. doi:10.3390/ijms21155304
86. Whittam DH, Karthikeayan V, Gibbons E, et al. Treatment of MOG antibody associated disorders: results of an international survey. J Neurol. 2020;267(12):3565–3577. doi:10.1007/s00415-020-10026-y
87. Juryńczyk M, Jacob A, Fujihara K, et al. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol. 2019;19(3):187–195. doi:10.1136/practneurol-2017-001787
88. Ramanathan S, Mohammad S, Tantsis E, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127–137. doi:10.1136/jnnp-2017-316880
89. Hacohen Y, Mankad K, Chong WK, et al. Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology. 2017;89(3):269–278. doi:10.1212/WNL.0000000000004117
90. Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, et al. Treatment of MOG-IgG-associated disorder with rituximab: an international study of 121 patients. Mult Scler Relat Disord. 2020;44:102251. doi:10.1016/j.msard.2020.102251
91. Chen JJ, Flanagan EP, Bhatti MT, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95(2):e111–e120. doi:10.1212/WNL.0000000000009758
92. Tsantes E, Curti E, Siena E, et al. Successful intravenous immunoglobulin treatment in relapsing MOG-antibody-associated disease. Mult Scler Relat Disord. 2019;32:27–29. doi:10.1016/j.msard.2019.04.021
93. Hacohen Y, Banwell B. Treatment approaches for MOG-Ab-associated demyelination in children. Curr Treat Options Neurol. 2019;21(1):2. doi:10.1007/s11940-019-0541-x
94. Holroyd K, Vogel A, Lynch K, et al. Neuromyelitis optica testing and treatment: availability and affordability in 60 countries. Mult Scler Relat Disord. 2019;33:44–50. doi:10.1016/j.msard.2019.05.013
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.