")
Back to Journals » Infection and Drug Resistance » Volume 11
Drug-resistance profiling and transmission dynamics of multidrug-resistant Mycobacterium tuberculosis in Saudi Arabia revealed by whole genome sequencing
Authors Al-Ghafli H, Kohl TA, Merker M, Varghese B, Halees A, Niemann S, Al-Hajoj S
Received 23 July 2018
Accepted for publication 20 August 2018
Published 12 November 2018 Volume 2018:11 Pages 2219—2229
DOI https://doi.org/10.2147/IDR.S181124
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Hawra Al-Ghafli,1,* Thomas A Kohl,2,3,* Matthias Merker,2,3,* Bright Varghese,1,* Anason Halees,4 Stefan Niemann,2,3 Sahal Al-Hajoj1
1Mycobacteriology Research Section, Department of Infection and Immunity, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia; 2Molecular and Experimental Mycobacteriology, Forschungszentrum Borstel, Leibniz-Zentrum für Medizin und Biowissenschaften, Borstel 23845, Germany; 3German Centre for Infection Research (DZIF), Partner site Borstel, Borstel 38124, Germany; 4Data and Information Office, Anfas Medical Centre, Hittin District, Riyadh, Saudi Arabia
*These authors contributed equally to this work
Background: In Saudi Arabia, cross-border transmission of multidrug-resistant (MDR) Mycobacterium tuberculosis complex (MTBC) strains might be particularly fostered by high immigration rates. Herein, we aimed to elucidate the transmission dynamics of MDR-MTBC strains and reveal a detailed prediction of all resistance-conferring mutations for the first- and second-line drugs.
Methods: We investigated all MDR-MTBC strains collected between 2015 and 2017 from provincial mycobacteria referral laboratories and compared demographic and clinical parameters to a cohort of non-MDR-TB patients using a whole genome sequencing approach. Clusters were defined based on a maximum strain-to-strain genetic distance of five single-nucleotide polymorphisms (SNPs) as surrogate marker for recent transmission, and then investigated molecular drug-resistance markers (37 genes).
Results: Forty-eight (67.6%) MDR-MTBC strains were grouped in 14 different clusters, ranging in size from two to six strains; 22.5% (16/71) of all MDR-MTBC isolates were predicted to be fully resistant to all five first-line drugs, and five strains (7.0%) exhibited fluoroquinolone resistance. Moreover, we revealed the presence of 12 compensatory mutations as well as 26 non-synonymous SNPs in the rpoC gene and non-hotspot region in rpoB, respectively
Conclusion: Optimized TB molecular surveillance, diagnosis, and patient management are urgently needed to contain MDR-MTBC transmission and prevent the development of additional drug resistance.
Keywords: molecular profiling, drug resistance, genotyping, detailed prediction, compensatory mutations, drug-resistance mutations
Introduction
The global figures on tuberculosis (TB) urge for optimized control strategies and efficient allocation of resources to reduce TB mortality and morbidity rates. In 2016, the WHO reported 10.4 million new cases and approximately 1.8 million deaths caused by TB.1 A particular concern in TB control is the worldwide emergence of resistant, multidrug-resistant (MDR), and extensively resistant (XDR) Mycobacterium tuberculosis complex (MTBC) strains.1 M/XDR-TB is considerably more difficult to treat (longer duration, second-line drugs with severe side effects) and has much poorer treatment outcomes.2
Saudi Arabia is classified by the WHO as a moderate TB-burden country;1 however, several factors may contribute to TB transmission and the emergence of drug resistance among new cases. The country has a unique geographical location, situated in the heart of three different continents: Africa, Asia, and Europe. As such, it serves as a major migration hub. In addition, the country hosts millions of Islamic pilgrims annually, most of whom arrive from highly TB-endemic regions, and only 60% of the total population are Saudis.3 The resulting mobility increases the risk of the introduction and transmission of “imported” M/XDR strains from patients who have not been registered or identified by local TB-control measures.
The first molecular epidemiology study in the country, based on spoligotyping, revealed the presence of the geographically dominating Delhi/CAS genotype among others, such as Latin American Mediterranean (LAM), East African Indian (EAI), Uganda, Ghana, Cameroon, Haarlem, and Beijing, which may reflect transmission from immigrated TB patients.4 Recently, Varghese et al applied 24 loci MIRU-VNTR typing for the analysis of drug resistant (mono, poly, and multidrug resistant) MTBC strains, and showed the predominance of Delhi/CAS, EAI, Beijing, Ghana, and Haarlem genotypes. In addition, the study revealed a high cluster rate between local and immigrant population with no association between patient origin and strain genotype.5 However, both, spoligotyping and MIRU-VNTR typing have a low discriminatory power to precisely analyze the genetic relationship of MTBC strains as a surrogate marker for the extent of recent patient-to-patient MTBC transmission, especially in high-incidence populations.2,6,7 Here, whole genome sequencing (WGS)-based approaches have a clear advantage by allowing the discrimination of clinical MTBC strains at the highest resolution and simultaneously enabling detailed resistance predictions for, virtually, all drugs.2,8 More recently, the first national surveillance study conducted on 1,902 MTBC strains revealed an alarming proportion of 4.5% MDR-TB cases in the country.9 However, high-resolution data on transmission patterns or molecular drug-resistance profiles using WGS in Saudi Arabia is lacking, which is a critical step to define the underlying epidemiology of MDR-TB and its transmission dynamics with high precision.
To address these questions, we utilized a WGS approach to characterize all MDR-MTBC strains sampled from different provinces of the country between 2015 and 2017. In addition, we included a cohort of non-MDR MTBC strains to point out putative associations between patients infected with MDR and non-MDR MTBC strains, which were also stratified on the basis of their demographic and clinical parameters.
Materials and methods
Study design
The Mycobacteriology Research Section of King Faisal Specialist Hospital and Research Center maintains a national-level collection of clinical isolates as part of the National TB Surveillance Study during August 2015 to July 2017. The collection of isolates applied the WHO’s 100% collection policy during the execution time; thus, a fair and extensive coverage rate was acquired. During the period, 2,156 isolates were collected; 394/2156 were either not successfully cultured or recognized as non-MTBC strains. The remaining 1,762 isolates were subjected to phenotypic drug-susceptibility testing (DST) and mutation-detection via Line Probe Assay (LPA) as an initial screening method. A total of 71 isolates were phenotypically defined as MDR strains and were accordingly considered in the study. Another 134 non-MDR-TB strains were included as background samples to obtain a final sample size of 205 isolates.
Demographic and clinical data were gathered using data collection forms from the provincial laboratories on the basis of national TB codes, and the data were verified at the National TB Registry. The data collection was strictly anonymous and no patient identifiers were used throughout the collection and analysis period. The study has been reviewed and approved by the Office of Research Affairs at King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
Laboratory methods and data analysis
All the isolates were subjected to DST of four first-line drugs (isoniazid, rifampicin, streptomycin, and ethambutol) using the MGIT SIRE kit (Becton Dickinson, USA) as an initial screening method. Genomic DNA was extracted with a standard spin-column technique as per the manufacturer’s recommendations (QIAamp DNA mini Kit, Qiagen, Germany). Primary rapid screening of mutations conferring resistance to rifampicin and isoniazid was carried out using the LPA Genotype MTBDR Plus (Hain Life Science, Nehren, Germany).
From the extracted genomic DNA, WGS was carried out with Illumina next-generation sequencing technology, using the Nextera XT library preparation kit and the NextSeq-500 sequencing platform as instructed by the manufacturer (Illumina, San Diego, CA, USA). Reads were mapped to the genome of M. tuberculosis H37Rv (GenBank ID: NC_000962·3) with the alignment program SARUMAN.10 All datasets reached a minimum average coverage depth of 54.8-fold, with at least 98.6% of the reference genome covered by at least one read for all isolates. For variant detection in the mapped reads, we employed minimum thresholds of 10× coverage and 75% allele frequency. Variant positions were then combined, supplementing the joint list with the respective information from the original mappings where necessary. SNP positions with a reliable base call (10× coverage and 75% allele frequency) in at least 95% of the isolates were concatenated to a sequence alignment, excluding SNPs occurring within a window of 12 bp from each other in the same isolate. Genome clusters were defined by grouping all isolates within a maximum distance of five SNPs to the nearest group member – an empirically defined cutoff for epidemiologically related cases (ie, household contacts).11 Polymorphisms in 37 drug-resistance and bacterial fitness-associated target genes were investigated and used to construct molecular drug-resistance profiles of all MDR-MTBC strains (Table S1). SNPs in 37 resistance-related target genes that were found in at least two isolates of the same MTBC genotype but were not associated to phenotypic resistance were excluded as putative phylogenetic variants (Table S1).
Data analysis was conducted using the software package SPSS version 19.0 (IBM Corporation, Armonk, NY, USA). Pearson’s χ2 statistics or Fisher’s two-tailed test was used to compare categorical variables. All variables were studied in a univariate logistic regression analysis. In all analyses, 95% CIs and P-values were corrected for the definite population of genomic clusters.
Results
Overall, 71 genotypically confirmed MDR-TB and 134 non-MDR-TB cases were enrolled for final data analysis, including the mainly pulmonary specimens (88.3%) and extra-pulmonary specimens (12%; Table 1). All enrolled isolates were culture-positive and 72% were smear-positive (Table 1). MTBC strains were derived from 62.9% male and 37.1% female patients; the dominating age groups were 16–29 years (32.2%) and 30–45 years (37.1%). Similar proportions of Saudi and non-Saudi patients were included, mainly originating from the Central (27.3%) and Western provinces (34.1%). On comparing MDR and non-MDR-TB cases in a univariate analysis, we found no differences in the general patient characteristics described earlier. However, MDR-TB patients were more often observed with smear-positive acid-fast bacilli specimens (P=0.001), higher treatment-failure rates (P=0.002), deportation (P=0.006), and patient death (P=0.017; Table 1).
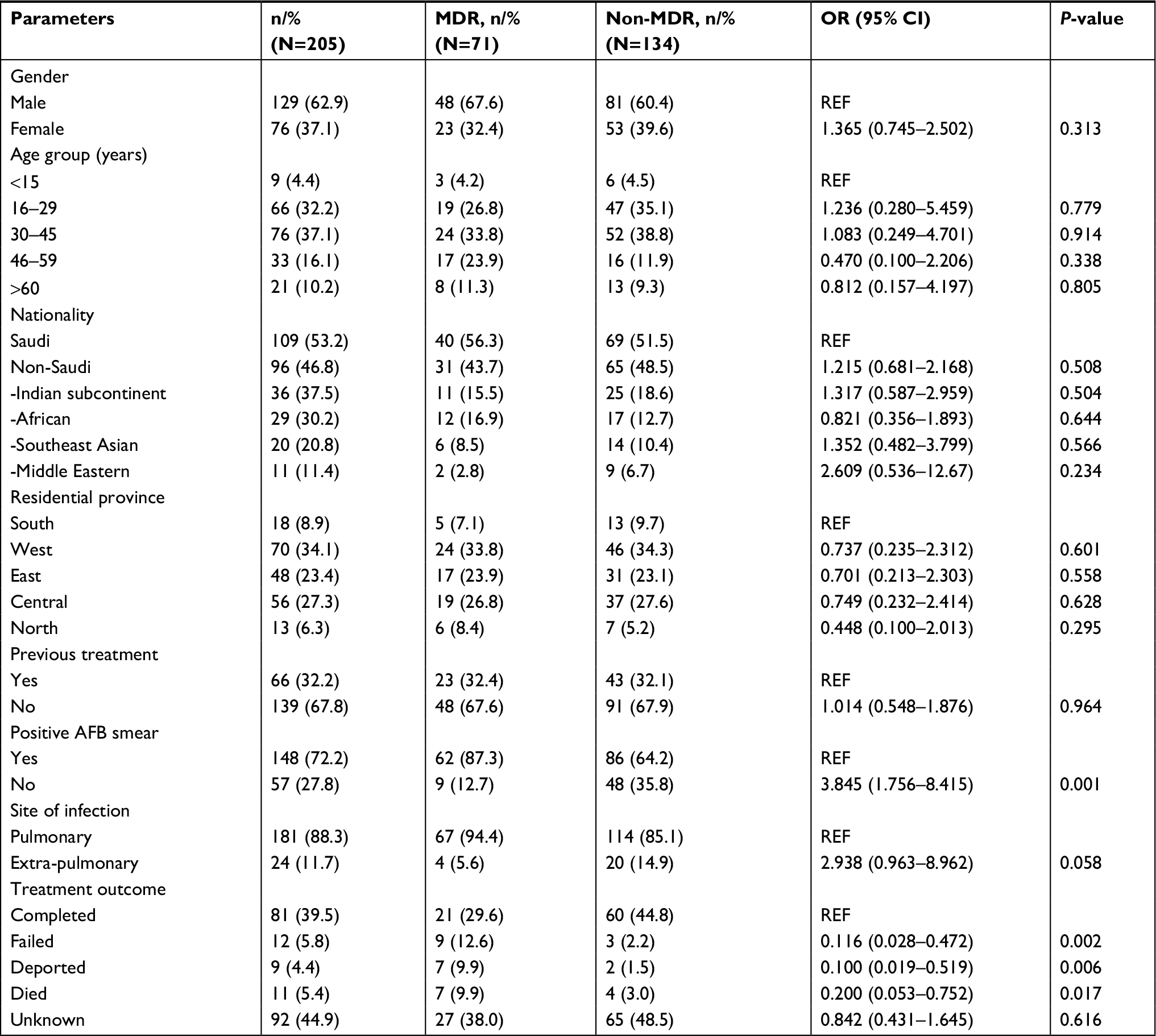 | Table 1 Demographic and clinical summary of study subjects Notes: Indian subcontinent – Afghanistan, India, Pakistan, Bangladesh, Nepal, and Myanmar; African – Somalia, Sudan, Nigeria, Eritrea, Ethiopia, and Egypt; Middle Eastern – Yemen and Syria; South East Asian – Philippines and Indonesia. Provincial cities: South – Jizan and Asir; West –Jeddah, Baha, Taif, and Medina; East – Dammam; Central – Riyadh; North – Hail. Unknown: After diagnosis, the patient’s follow-up was lost from the records. Abbreviations: AFB, acid-fast bacilli; MDR, multidrug-resistant; M. bovis, Mycobacterium bovis; REF, reference. |
All strains were phylogenetically classified in accordance with a recently reported SNP barcode nomenclature.12 Overall, four major MTBC lineages were identified: 32/205 (15.6%) of isolates were assigned to Lineage 1(EAI), 16/205 (7.8%) to Lineage 2 (Beijing), 43/205 (21.0%) to Lineage 3 (Delhi/CAS), 113/205 (55.1%) to Lineage 4 (Euro-American super-lineage), and one isolate was classified as Mycobacterium bovis (Figure 1, Figure S1). With regard to the MDR-MTBC population structure in particular, we assigned 11/71 (15.5%) isolates to Lineage 1, 11/71 (15.5%) to Lineage 2, 17/71 (23.9%) to Lineage 3, and 32/71 (45.0%) to Lineage 4 (Table 2).
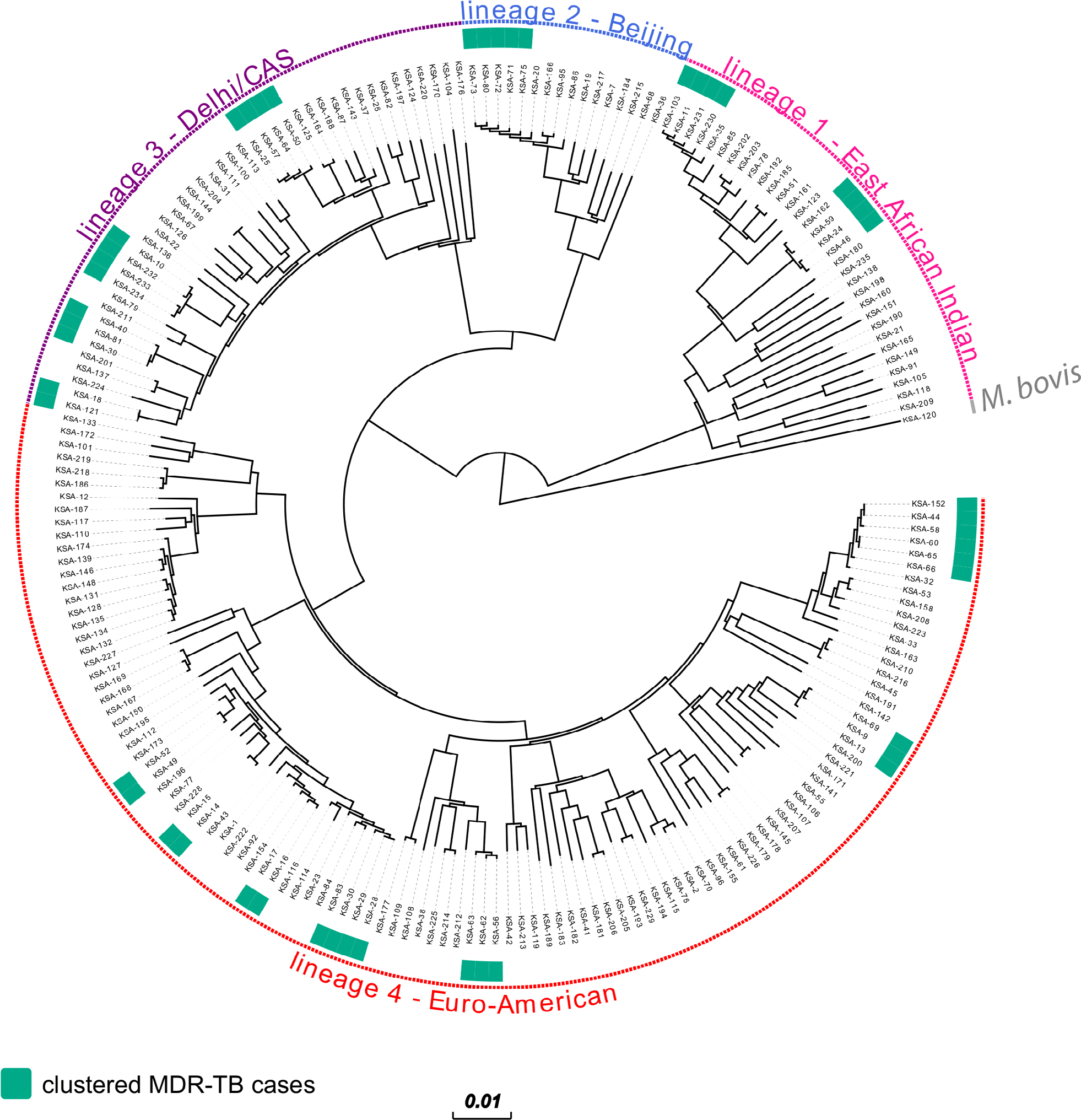 | Figure 1 Phylogeny of 71 MDR and 134 non-MDR-MTBC strains from Saudi Arabia. Notes: Maximum likelihood phylogeny of 205 isolates with annotated genomic MDR-MTBC clusters using 21,745 variable genome positions and 1,000 bootstrap resamples. Main MTBC lineages are annotated. Abbreviations: MDR, multidrug-resistant; MTBC, Mycobacterium tuberculosis complex; M. bovis, Mycobacterium bovis; CAS, Central Asian. |
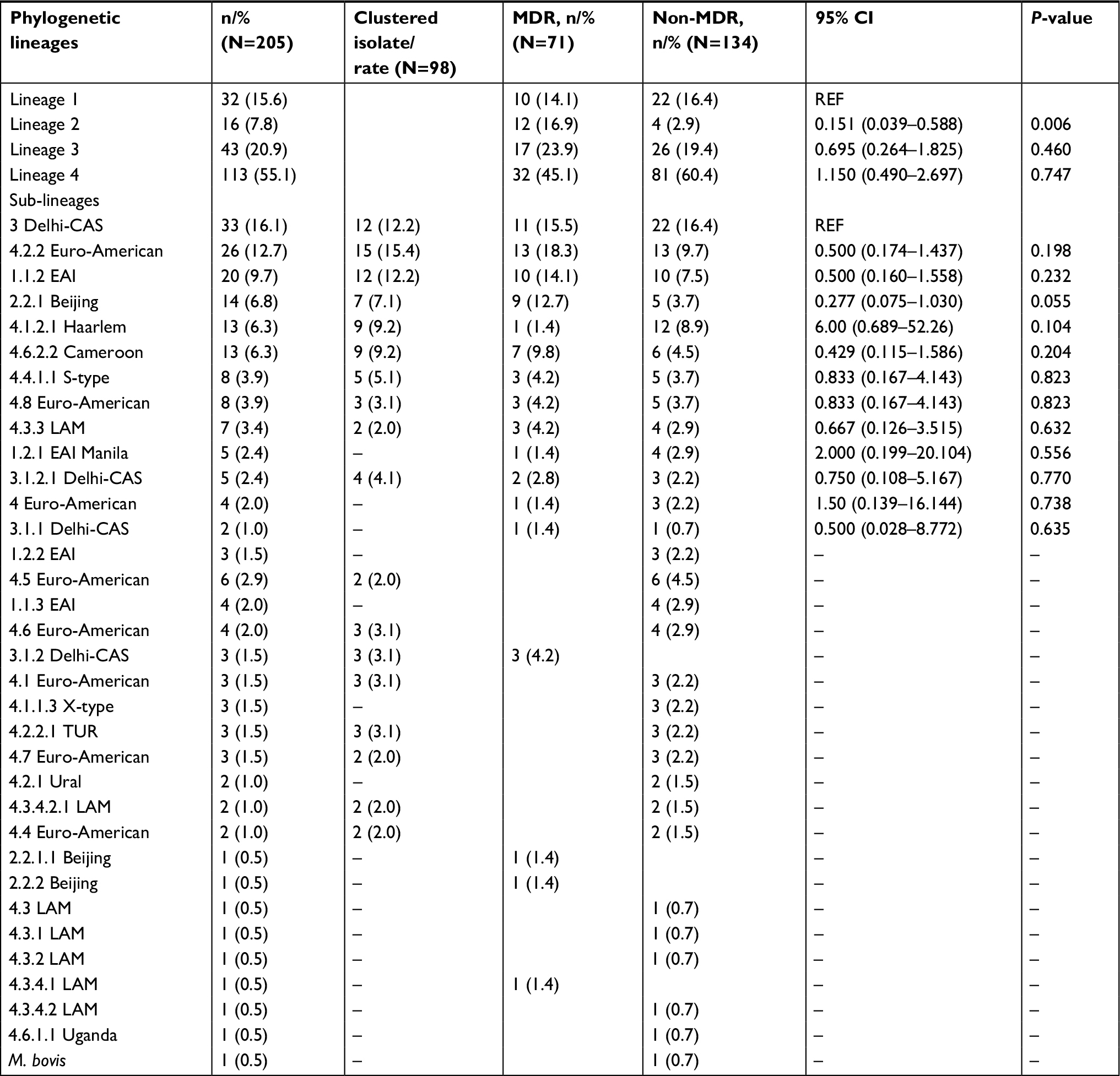 | Table 2 Major phylogenetic lineages and sub-lineages classified by whole genome sequencing approach Abbreviations: MDR, multidrug-resistant; M. bovis, Mycobacterium bovis; EAI, East African Indian; LAM, Latin American Mediterranean; TUR, Turkish. |
On a sub-lineage level, the most prevalent MDR-MTBC groups that could be further differentiated by the applied SNP barcode were Lineage 4.2.2 (associated with the TUR genotype; 13/71, 18.3%), Lineage 1.1.2 (associated with the EAI genotype; 10/71, 14.1%), Lineage 2.2.1 (associated with the Beijing genotype; 10/71, 14.1%), and Lineage 4.6.2.2 (associated with the Cameroon genotype; 7/71, 9.9%; Table 2). Compared to the non-MDR-MTBC strains (n=134), strains belonging to Lineage 2 were enriched in the MDR-TB cohort (P=0.006) whereas all other identified lineages and sub-lineages were observed with similar proportions in both datasets (Table 2).
Primary screening of phenotypically and genotypically confirmed MDR-MTBC strains with LPA identified 43/71 (60.6%) S531L (ie, S450L in the M. tuberculosis reference genome, see “Methods” section), 4/71 (5.6%) H526Y (ie, H445Y) and 4/71 (5.6%) D516V (ie, D435V) mutations in the rpoB gene, and 20/71 (28.2%) had a missing wild-type band that indicated another rare mutation in the rpoB hotspot region (Tables S1 and S2). With regard to isoniazid-resistance-mediating mutations, 59/71 (83.8%) of the MDR-MTBC isolates showed the mutation katG S315T, (8/71, 10.3%) inhA mutation in Position –15 (C>T) and four missing wild-type bands for the katG gene (Table S2).
Using WGS, we analyzed insertions, deletions, and SNPs in 37 target genes associated with bacterial fitness and antibiotic resistance. Isoniazid resistance was dominant with the mutation (S315T, 61/71, 85.9%) in katG gene. The second common mutation was found in the inhA gene (–15 C>T, 8/71 (11.3%; Table S2). Rifampicin resistance was associated with S450L mutation (43/71, 60.6%) followed by H445Y (9/71, 12.7%) of the rpoB gene. All strains with undefined mutations in the rpoB gene on LPA have mutations at S450W, L452P, L430P, H445R, H544N, and S428G codon positions (Tables S1 and S2).
We analyzed the rpsL and rrs genes for the mutations conferring streptomycin resistance. The K43R mutation in the rpsL gene was found to be the most common mutation (16/71, 22.5%) followed by the 514 A>C mutation in the rrs gene (12/71, 16.9%). Ethambutol-resistant isolates were mainly associated with the mutation embB M306V (28/71, 39.4%; Table S2). Furthermore, we identified variants in the upstream region of the gene pncA, associated with pyrazinamide resistance, as well as non-synonymous and frame-shift mutations in 52.1% (37/71) of isolates. Overall, 22.5% (16/71) of MDR-MTBC isolates were identified with molecular resistance markers to all five first-line drugs; 5/71 (7.0%) of the MDR-MTBC strains were found with a fluoroquinolone-resistance-mediating mutation (ie, gyrA A90V and gyrA D94G, respectively) and were defined accordingly as pre-XDR (Table S2). Overall 18/205 isolates (including 13 MDR-MTBC isolates) were observed with prothionamide- and ethionamide-resistance-mediating mutations – that is, fabG1/inhA –15 c/t, ethA 672 insG, and ethA 377 delT (Tables S1 and S2). No resistance marker was identified for second-line injectable drugs or WHO Group Five anti-TB drugs (ie, bedaquiline, delamanid, clofazimine, linezolid, and paraminosalicylic acid). Among 47/71 (66.2%) MDR-MTBC isolates, we identified putative compensatory mutations in the RNA-polymerase genes rpoA (1/71; 1.4%), rpoB (32/71; 45.1%), and rpoC (23/71; 32.4%). The mutation in the rpoC gene was found to dominate (17/23, 73.9%) among isolates as was the S450L mutation of the rpoB gene (Table S2).
In order to determine molecular strain clusters indicative of recent chains of MDR-MTBC transmission (within the last 3 years), we defined groups of at least two strains that had a maximum distance of five SNPs to at least one other group member. This threshold was recently shown to be a reliable indicator to identify transmission among household contacts in the UK.11 Overall, 48/71 (67.6%) MDR-MTBC isolates were assigned into one of 14 molecular clusters, each comprising two to six patients (Figure 1, Table 3). In contrast, non-MDR strains showed a clustering rate of only 30.6%. Individual MDR-MTBC strains within genomic clusters each presented with identical resistance-conferring mutations in the genes katG or inhA, and rpoB mediating a MDR phenotype (Table 3 and S1). In addition, 29 MDR-TB patients in eight different transmission networks were infected with strains showing cluster-specific pncA mutations associated with pyrazinamide resistance – again, pointing toward ongoing patient-to-patient transmission. However, molecular markers associated with second-line injectable drug resistance were identical only in one cluster.
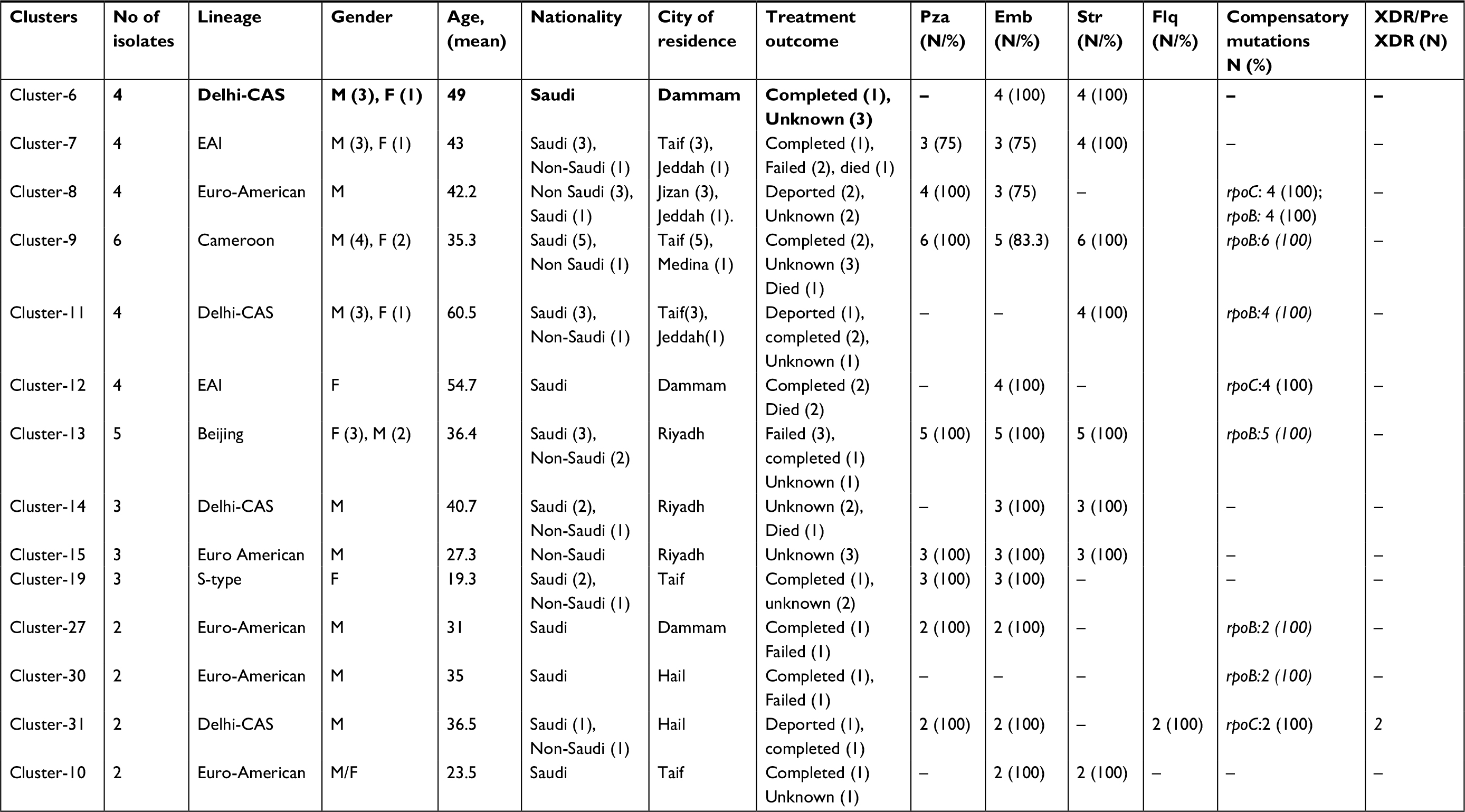 | Table 3 Characteristics of 14 MDR-TB genomic clusters Notes: Treatment regiments have been given to patients; no culture conversion was documented (failed), with confirmed culture conversion (completed). Abbreviation: EMB, ethambutol; Flq, fluoroquinolone; PZA, pyrazinamide; Str, streptomycin; XDR, extensively resistant. |
Demographic data from patients involved in the 14 suggested transmission networks revealed five clusters with only Saudi nationals and one cluster with only non-Saudi nationals. The remaining eight clusters comprised both Saudi and non-Saudi nationals. Ten out of the 14 clusters were linked to a single city, with the remaining four clusters linked to two cities. There was no significant difference in the MDR-MTBC cluster rate on comparing patient origin – Saudi nationals (57.1%) and non-Saudi nationals (41.2%; P=0.629; Table 4). However, lack of proper social information restricts further analysis of epidemiological associations among the MDR-TB patients (Table 4). The two major clusters comprised MTBC strains belonging to the Cameroon sub-lineage (4.6.2.2; six cases) and the Beijing lineage (2.2.1; five cases), and included both Saudi and non-Saudi nationals (Table 3).The following mutations were shared among all strains from the Cameroon genotype cluster (katG S315T, rpoB S450L, Rvnr01 514A>C, embB M306V, and pncA T76P). Strains from the Beijing genotype cluster shared the mutations katG S315T, rpoB S450L, rpsL K43R, embB M306V, and pncA T47I. Overall, 12/14 (85.7%) of the clusters were observed with identical resistance-conferring mutations (Table 3).
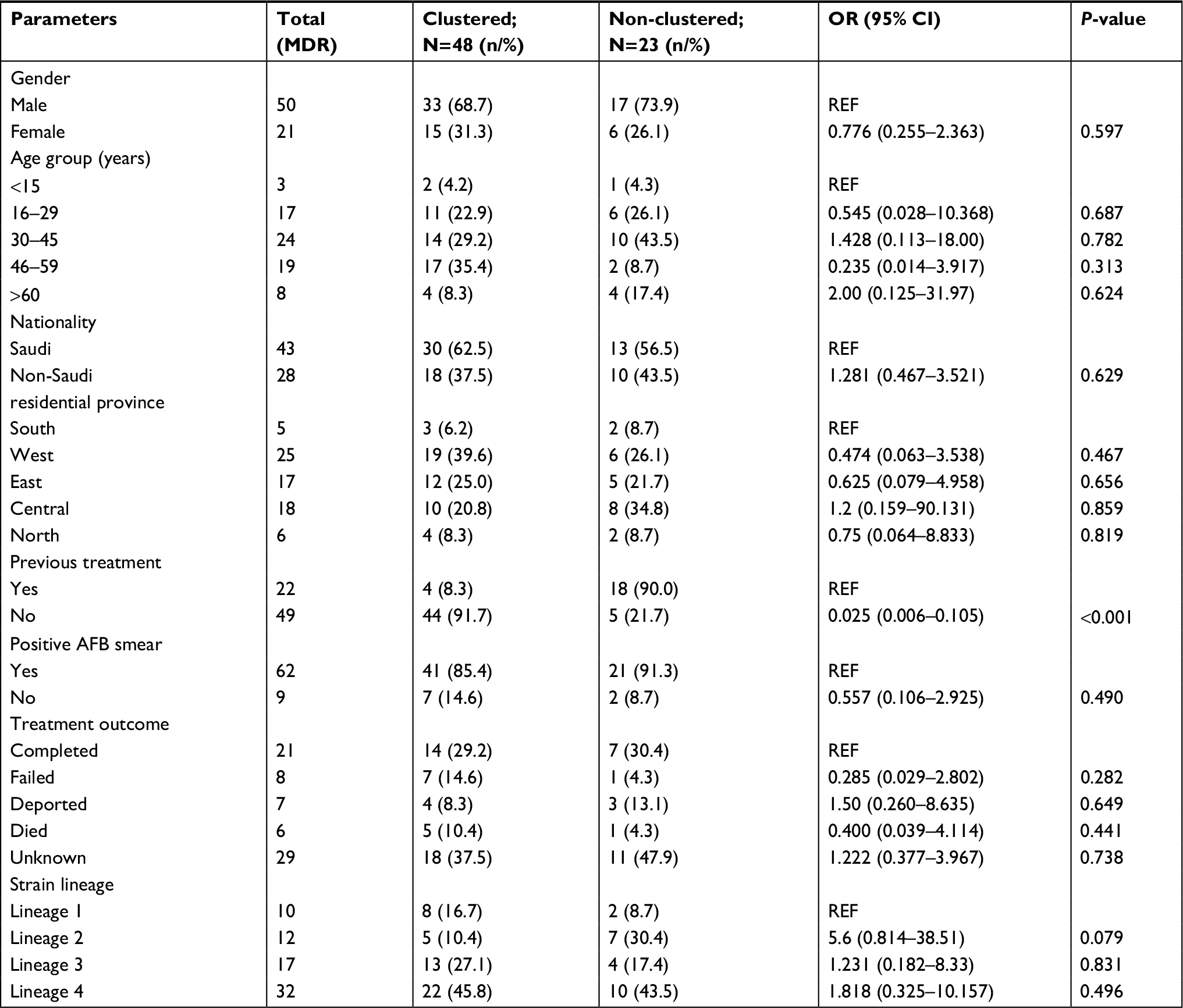 | Table 4 Demographic and clinical risk Abbreviations: AFB, acid-fast bacilli; MDR, multidrug-resistant. |
Analysis on confounding factors for MDR-TB clustering showed no statistically significant association with other variables tested, except for the treatment history. Clustered MDR-TB cases were more likely to be new cases (P<0·001), confirming the cluster-definition threshold of five SNPs as an indicator for patient-to-patient transmission (Table 4).
Discussion
To the best of our knowledge, this is the first study elucidating the transmission dynamics of MDR-MTBC strains using a WGS approach in Saudi Arabia – a country challenged by high immigration rates. Alarmingly, high cluster rates (67.6%) were determined among the MDR-MTBC strains, leading to a total of 14 putative recent transmission chains. Together with the fact that MDR-TB was not associated with patient origin, this points to ongoing, endemic transmission of MDR-TB within Saudi Arabia. This is particularly important as several clustered strains have already acquired resistance-conferring mutations against all first-line drugs. At the same time, genomic markers for second-line drug resistance are still rare among our study cohort, urging for improved TB-control measures for complete resistance detection and tailored therapy.
An association of MDR-TB with recent transmission has been collectively reported from different geographical settings.13–15 Moreover, our findings are in concordance with high rates of MDR-MTBC transmission (45.1%) in countries such as Ethiopia.15 Other intermediate TB-burden countries, such as Iran, Iraq, and Turkey reported lower rates of transmission, ranging from 24% to 35.9%, based on classical genotyping methods.14,16,17 Nevertheless, the different methodological approaches employed herein – compared with studies conducted in nearby regions – make the process of contextualizing transmission dynamics very challenging. However, strikingly, our data suggest that, transmission of full first-line-resistant MTBC strains leading to extended primary MDR-TB cases is a major factor for the emergence of MDR-TB in Saudi Arabia. This has been further evidenced by identical resistance-conferring mutations in katG, inhA, rpoB, and pncA in clustered strains. Thus, transmission chains must be effectively interrupted by proper case finding (beyond conventional contact-tracing), isolation of MDR-TB cases until they are negative, and comprehensive diagnosis to allow for prompt and effective treatment. The latter are critical to significantly decrease the incidence of MDR-TB and avert the emergence of XDR-TB in Saudi Arabia.
A slightly increased cluster rate was evident among Saudi nationals (62.5%) compared to non-Saudi cases. Of the total clusters, 13 had Saudi cases whereas non-Saudis were represented in only nine clusters. There were four clusters exclusively composed of Saudi nationals, whereas only one cluster was exclusively composed of non-Saudi cases. Interestingly, eight clusters contained both local and foreign patients, which corroborate findings from previous studies;5 71.4% of the clusters were found limited to a single city, which supports the possibility of an active ongoing transmission in the local area. However, no statistical evidence has been observed in the geographical origin (province of residence) of patients and strain clusters.
Among the MDR-TB cases found as part of any cluster, only 29.2% of the patients received a full course of therapy, whereas 30.4% patients received therapy among non-clustered cases. The majority of the MDR-TB cases found in clusters had an “unknown” status without follow-up visits, according to the medical records. Thus, Saudi Arabia is still far (62%) from the WHO’s MDR-TB treatment-success target (85%). In addition, several previous studies showed a large scale non-compliance of treatment among Saudi nationals in particular.18,19 Unsuccessful treatment is, therefore, another likely factor for the ongoing transmission of MDR-TB strains.
As expected from the high immigration rate reported in Saudi Arabia, a high phylogenetic diversity of MTBC strains was identified, unlike what has been reported elsewhere.20 Besides Lineage 3, MTBC strains that are associated with a South Asian evolutionary origin, and which are also frequently isolated from TB patients in East Africa and the Middle East, we found 55% of all patients being infected with Lineage 4 strains. Strains of the TUR/4.2.2 sub-lineage are most prevalent, followed by strains of the Haarlem/4.1.2.1, and Cameroon/4.6.2.2 lineages. Although Haarlem is one of the globally widespread sub-lineages, the sub-lineages Cameroon and TUR are geographically restricted and have been identified mainly in Asia and Africa, highlighting the impact of high immigration rates from these regions into Saudi Arabia.21 In fact, the large clusters of Cameroon and TUR strains identified in our study included non-Saudi nationals, particularly of African (Nigeria) and Middle Eastern (Syria) origin. Similarly, Lineage 3 is frequently identified in Asia and Africa and has been previously reported in Saudi Arabia and Oman with dominating rates.22,23 The high influx of immigrants coming from Asia and Africa is well reflected by the highly diverse patient population in the study (Figure S1). This might explain the predominance of these sub-lineages in our settings. Strains of lineages 1 and 2, on the other hand, accounted for less than 10% of the investigated strains in this study. This is in contrast to studies in Russia and China, which reported a dominance of Beijing strains and have elucidated an association between Lineage 2 strains and drug resistance.24,25 In addition, four strains from Saudi patients belonging to Lineage 4 failed to be identified to the sub-lineage level in this study. This might be attributed to a small “blind spot” in the employed SNP-based algorithm,12 potentially representing minor novel sub-lineages. Analysis on mutations conferring resistance to isoniazid and rifampicin showed a clear domination of well-established mutations in the katG codon 315 and the rpoB hotspot region in concordance with previous Saudi Arabian studies by Varghese et al.13,26 However, there were six other mutations in the rpoB hotspot region and one mutation in katG which have never been reported in the country before. Likewise, we have identified variants in the upstream region of the gene pncA as well as non-synonymous and frame-shift mutations that are associated with pyrazinamide resistance in 22.4% of the isolates. Similar to our results, many studies have reported a variety of scattered and variable set of pyrazinamide resistance-conferring mutations.27,28 Of note, pyrazinamide is not part of the routine first-line therapy in Saudi Arabia and phenotypic DST is not applied. Thus, our WGS results suggest that mutations in the pncA gene have been acquired elsewhere or that individual patients had non-programmatic access to this drug.
Compensatory mutations can be a major factor in restoring the fitness reduction of strains due to drug-resistance-conferring mutations. Compensation for rifampicin resistance has been shown to be associated with mutations in rpoA, rpoC, and the non-hotspot region of the rpoB gene.29 Our analysis detected mutations in the rpoC gene and outside the rifampicin-resistance-determining region, with four mutations (one in MDR-MTBC strain and three in non-MDR-MTBC strains) found in the rpoA gene. Altogether, 12 and 26 non-synonymous SNPs were detected in the rpoC gene and non-hotspot rpoB region, respectively. The rpoC mutation was more common in strains with an rpoB gene S450L mutation, in concordance with a recent study.30 The rate of probable compensatory mutations found in MDR-MTBC isolates was relatively high (66.2%) compared to previous studies from other parts of the world (27–66%).29,30
A larger scale population-based research is, of course, required in the near future through employing WGS to better understand the nature and route of MTB transmission in Saudi Arabia. Although the WGS technique is powerful, lack of accessibility to other demographic and medical indicators has prevented us from epidemiologically tracing the transmission chains in individual clusters. Due to the restrictions of the study design and time frame, we could possibly have missed MDR-MTBC isolates. To conclude, for the first time our results showed that Saudi Arabia is indeed suffering from ongoing MDR-MTBC transmissions indicated by high cluster rates and identical resistance-conferring mutations. As such, effective interruption of transmission chains requires improved TB surveillance, diagnosis, and treatment. In addition, incorporating WGS into the routine TB-management plan might be a crucial step in the future in Saudi Arabia. It facilitates an accurate diagnosis, optimized case management, and a detailed estimation of ongoing transmission dynamics, which is especially important in settings with high immigration and mobility rates.
Acknowledgments
This project has been funded by the National Science, Technology, and Innovation Plan (NSTIP) of King Abdulaziz of Science and Technology (KACST, Saudi Arabia; grant no. 11-BIO-1436–20). This work was partly supported by the PathoNgenTrace (grant no. FP7-278864–2) and the German Center for Infection Research (DZIF). The sponsors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank Julia Zallet, Vanessa Mohr, and Tanja Ubben (all from Research Institute Borstel) for their help with library preparation and sequencing, Ms. Tracy Lynn AlSarhani for language editing support, and Prof. Mohammed Shoukri for support with the statistical analysis.
Author contributions
HA-G contributed to literature search, data analysis, data interpretation, and manuscript writing, as well as creation of figures and tables. TAK and MM contributed to data analysis, data interpretation, creation of tables and figures, manuscript writing, and literature search. BV contributed to study design, data collection, data analysis, data interpretation, creation of tables and figures, manuscript writing, and literature search. AH contributed to study design, data analysis, and data interpretation. SN contributed to study design, data analysis, data interpretation, manuscript writing, literature search, and creation of tables and figures. SA-H contributed to study design, data collection, data analysis, manuscript writing, and creation of tables and figures. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Global Tuberculosis Report 2017. Geneva: World Health Organization. Available from: http://apps.who.int/medicinedocs/documents/s23360en/s23360en.pdf. Accessed June 20, 2018. | ||
Walker TM, Merker M, Kohl TA, Crook DW, Niemann S, Peto TE. Whole genome sequencing for M/XDR tuberculosis surveillance and for resistance testing. Clin Microbiol Infect. 2017;23(3):161–166. | ||
Statistical Year Book: Population by Gender, Age Groups and Nationality (Saudi/Non-Saudi). Saudi Arabia: General Authority for statistics in Saudi Arabia. Available from: https://www.stats.gov.sa/en/5305. Accessed June 20, 2018. | ||
Al-Hajoj SA, Zozio T, Al-Rabiah F, et al. First insight into the population structure of Mycobacterium tuberculosis in Saudi Arabia. J Clin Microbiol. 2007;45(8):2467–2473. | ||
Varghese B, Supply P, Allix-Béguec C, et al. Admixed phylogenetic distribution of drug resistant Mycobacterium tuberculosis in Saudi Arabia. PLoS One. 2013;8(2):e55598. | ||
Bjorn-Mortensen K, Lillebaek T, Koch A, et al. Extent of transmission captured by contact tracing in a tuberculosis high endemic setting. Eur Respir J. 2017;49(3):1601851. | ||
Niemann S, Merker M, Kohl T, Supply P. Impact of Genetic Diversity on the Biology of Mycobacterium tuberculosis Complex Strains. Microbiol Spectr. 2016;4(6). | ||
Walker TM, Kohl TA, Omar SV, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15(10):1193–1202. | ||
Al-Hajoj S, Varghese B, Shoukri MM, et al. Epidemiology of antituberculosis drug resistance in Saudi Arabia: findings of the first national survey. Antimicrob Agents Chemother. 2013;57(5):2161–2166. | ||
Blom J, Jakobi T, Doppmeier D, et al. Exact and complete short-read alignment to microbial genomes using Graphics Processing Unit programming. Bioinformatics. 2011;27(10):1351–1358. | ||
Walker TM, Ip CL, Harrell RH, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13(2):137–146. | ||
Coll F, McNerney R, Guerra-Assunção JA, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812. | ||
Varghese B, Shoukri M, Memish Z, et al. Occurrence of diverse mutations in isoniazid- and rifampicin-resistant Mycobacterium tuberculosis isolates from autochthonous and immigrant populations of Saudi Arabia. Microb Drug Resist. 2014;20(6):623–631. | ||
Durmaz R, Zozio T, Gunal S, et al. Genetic diversity and major spoligotype families of drug-resistant Mycobacterium tuberculosis clinical isolates from different regions of Turkey. Infect Genet Evol. 2007;7(4):513–519. | ||
Yimer SA, Norheim G, Namouchi A, et al. Mycobacterium tuberculosis lineage 7 strains are associated with prolonged patient delay in seeking treatment for pulmonary tuberculosis in Amhara Region, Ethiopia. J Clin Microbiol. 2015;53(4):1301–1309. | ||
Torkaman MR, Nasiri MJ, Farnia P, Shahhosseiny MH, Mozafari M, Velayati AA. Estimation of Recent Transmission of Mycobacterium Tuberculosis Strains among Iranian and Afghan Immigrants: A Cluster-Based Study. J Clin Diagn Res. 2014;8(9):DC05–DC08. | ||
Ahmed MM, Mohammed SH, Nasurallah HA, Ali MM, Couvin D, Rastogi N. Snapshot of the genetic diversity of Mycobacterium tuberculosis isolates in Iraq. Int J Mycobacteriol. 2014;3(3):184–196. | ||
Samman Y, Krayem A, Haidar M, et al. Treatment outcome of tuberculosis among Saudi nationals: role of drug resistance and compliance. Clin Microbiol Infect. 2003;9(4):289–294. | ||
Al-Hajjaj MS, Al-Khatim IM. High rate of non-compliance with anti-tuberculosis treatment despite a retrieval system: a call for implementation of directly observed therapy in Saudi Arabia. Int J Tuberc Lung Dis. 2000;4(4):345–349. | ||
Ssengooba W, Meehan CJ, Lukoye D, et al. Whole genome sequencing to complement tuberculosis drug resistance surveys in Uganda. Infect Genet Evol. 2016;40:8–16. | ||
Stucki D, Brites D, Jeljeli L, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48(12):1535–1543. | ||
Al-Hajoj S, Varghese B, Al-Habobe F, Shoukri MM, Mulder A, van Soolingen D. Current trends of Mycobacterium tuberculosis molecular epidemiology in Saudi Arabia and associated demographical factors. Infect Genet Evol. 2013;16:362–368. | ||
Al-Maniri A, Singh JP, Al-Rawas O, et al. A snapshot of the biodiversity and clustering of Mycobacterium tuberculosis in Oman using spoligotyping. Int J Tuberc Lung Dis. 2010;14(8):994–1000. | ||
Drobniewski F, Balabanova Y, Nikolayevsky V, et al. Drug-resistant tuberculosis, clinical virulence, and the dominance of the Beijing strain family in Russia. JAMA. 2005;293(22):2726. | ||
Guang Xue H, Hai Ying W, Martien WB. Multidrug-Resistant Tuberculosis, People’s Republic of China, 2007–2009. Emerg Infect Dis. 2011;17(10):1831–1838. | ||
Varghese B, Hillemann A, Wijayanti DR, et al. New insight into the molecular characterization of isoniazid and rifampicin resistant Mycobacterium tuberculosis strains from Saudi Arabia. Infect Genet Evol. 2012;12(3):549–556. | ||
Kim HJ, Kwak HK, Lee J, et al. Patterns of pncA mutations in drug-resistant Mycobacterium tuberculosis isolated from patients in South Korea. Int J Tuberc Lung Dis. 2012;16(1):98–103. | ||
Whitfield MG, Soeters HM, Warren RM, et al. A global perspective on pyrazinamide resistance: systematic review and meta-analysis. PLoS One. 2015;10(7):e0133869. | ||
Comas I, Borrell S, Roetzer A, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2011;44(1):106–110. | ||
Yang C, Luo T, Shen X, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017;17(3):275–284. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.