")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Drug-likeness approach of 2-aminopyrimidines as histamine H3 receptor ligands
Authors Sadek B, Schreeb A, Schwed JS, Weizel L, Stark H
Received 14 April 2014
Accepted for publication 24 May 2014
Published 19 September 2014 Volume 2014:8 Pages 1499—1513
DOI https://doi.org/10.2147/DDDT.S66179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Bassem Sadek,1 Annemarie Schreeb,2 Johannes Stephan Schwed,2,3 Lilia Weizel,2 Holger Stark3
1Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates; 2Biocenter, Institute of Pharmaceutical Chemistry, Johann-Wolfgang Goethe University, Frankfurt, Germany; 3Institute of Pharmaceutical and Medicinal Chemistry, Heinrich Heine University, Duesseldorf, Germany
Abstract: A small series of compounds containing derivatives of 2,4-diamino- and 2,4,6-triaminopyrimidine (compounds 2–7) was synthesized and tested for binding affinity to human histamine H3 receptors (hH3Rs) stably expressed in HEK-293 cells and human H4Rs (hH4Rs) co-expressed with Gαi2 and Gβ1γ2 subunits in Sf9 cells. Working in part from the lead compound 6-(4-methylpiperazin-1-yl)-N4-(3-(piperidin-1-yl)propyl)pyrimidine-2,4-diamine (compound 1) with unsatisfactory affinity and selectivity to hH3Rs, our structure-activity relationship studies revealed that replacement of 4-methylpiperazino by N-benzylamine and substitution of an amine group at the 2-position of the 2-aminopyrimidine core structure with 3-piperidinopropoxyphenyl moiety as an hH3R pharmacophore resulted in N4-benzyl-N2-(4-(3-(piperidin-1-yl)propoxy)phenyl)pyrimidine-2,4-diamine (compound 5) with high hH3R affinity (ki =4.49±1.25 nM) and H3R receptor subtype selectivity of more than 6,500×. Moreover, initial metric analyses were conducted based on their target-oriented drug-likeness for predictively quantifying lipophilicity, ligand efficiency, lipophilicity-dependent ligand efficiency, molecular size-independent efficiency, and topological molecular polar surface. As to the development of potential H3R ligands, results showed that integration of the hH3R pharmacophore in hH4R-affine structural scaffolds resulted in compounds with high hH3R affinity (4.5–650 nM), moderate to low hH4R affinity (4,500–30,000 nM), receptor subtype selectivity (ratio hH4R/hH3R; 8–6,500), and promising calculated drug-likeness properties.
Keywords: histamine, H3 receptors, H4 receptors, drug-likeness
Introduction
Histamine effects are known to be mediated through binding to four known histamine receptor subtypes designated as histamine receptors H1 to H4 (H1R–H4R), that belong to the family of G-protein-coupled receptors. H1R and H2R antagonists have been used for many years in clinical settings in the treatment of allergic conditions and gastric ulcers, respectively, while the potential clinical applications of human H3R (hH3R) and human H4R (hH4R) ligands are currently being extensively studied. H3R, initially described in 1983, is mostly expressed in the brain.1,2 H3Rs are recognized as presynaptic autoreceptors that regulate the synthesis and release of histamine as well as heteroreceptors on non-histaminergic neurons modulating the release of many other important neurotransmitters such as acetylcholine, norepinephrine, dopamine and serotonin.3–5 Given their localization and ability to affect multiple neurotransmitter systems, it has been hypothesized that H3R antagonist/inverse agonists could be suitable for the therapeutic management of central nervous system (CNS) disorders such as sleep disorders, Alzheimer’s disease, schizophrenia, narcolepsy, epilepsy, pain, and obesity.6–9 H4Rs are the most recently recognized receptor subtype belonging to the histamine receptor family. Despite their approximate 31% sequence homology (54% in transmembrane domains) and similarity in genomic structures, hH3R and hH4R differ significantly in expression patterns. Whereas hH3R is mainly restricted to the brain, hH4R expression is found mostly in the peripheral tissues and cells of the immune system.10,11 Individual expression patterns of hH4R on various hematopoietic cells such as mast cells, basophils, eosinophils, T-cells, and dendritic cells suggests an essential role in the regulation of immune response and inflammation.8,11–16
Due to structural similarities between hH3R and hH4R, it is expected that several imidazole-containing H3R ligands such as clobenpropit, imetit, and thioperamide also have a significant affinity for hH4R.11 2,4-Diaminopyrimidines were first identified by chemists from Palau Pharma (Barcelona, Spain) and UCB Pharma (Brussels, Belgium), although without specific descriptions of pharmacological data. Pfizer (New York, NY, USA) scientists also published studies on related pyrimidines such as hH4R ligands substituted with aliphatic heterocycles, while Johnson & Johnson (New Brunswick, NJ, USA) prepared carbon-analog 2-amino-4-alkyl-pyrimidines.13 Furthermore, current medicinal chemistry efforts are predominantly focused on selective acting G protein-coupled receptor ligands, particularly histamine hH3 and hH4 selective agonists/antagonists. Previous studies resulted in the development of 6-(4-methylpiperazin-1-yl)-N4-(3-(piperidin-1-yl)propyl)pyrimidine-2,4-diamine, which had unsatisfactory affinity and selectivity for hH3R and hH4R. The blueprint structure was constructed on a 2,4,6-triaminopyrimidine core containing a central 2,4,6-triaminopyrimidine as a conjugated donor/acceptor group; a basic group in the western part of the molecule and a lipophilic moiety in the eastern part (Figure 1).13–15,17
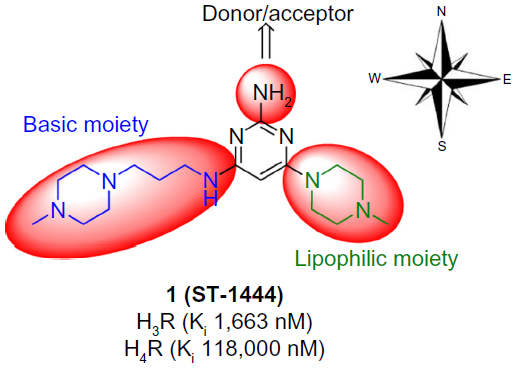 | Figure 1 Rough blueprint for hH3R ligands. |
Based on these efforts in designing ligands for hH3Rs and hH4Rs, the main objective of the current study was to link a well-established hH3R pharmacophore with an hH4R core structure, namely 2-aminopyrimidine, to determine which structural elements are necessary to create affinity to hH3Rs. As a result, a 2,4,6-triaminopyrimidine core element was chosen for further structural modifications. Affinity to hH3R stably expressed in HEK-293 cells and in hH4R transiently co-expressed with Gαi2 and Gβ1γ2 subunits in Sf9 cells was measured according to previously established methods to explore the influence of modifications to basic and lipophilic moieties in the western and eastern parts of the molecule, respectively.18,19 Several approaches are described: modifying compound 1 and related structures involved in the exchange of the lipophilic basic residue at 4-position (eastern part) by N-benzylamine moieties lacking basic character; replacement of the basic 4-methylpiperazino side chain at 6-position (western part) by unbranched basic piperidines maintaining the comparable spacer length; and substitution of the amine group at 2-position of the 2-aminopyrimidine core structure (Figure 1). Moreover, initial metric analyses were conducted to predictively quantify lipophilicity (clogP), ligand efficiency (LE), lipophilicity-dependent ligand efficiency (LELP), lipophilic efficiency (LipE), and topological molecular polar surface (TPSA), important for verification of drug-likeness of novel compounds 2–7.
Material and methods
General
Educts, reactants, and solvents were commercially obtained from Merck (Darmstadt, Germany), Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland), Alfa Aesar (Ward Hill, MA, USA), ABCR (Gainesville, FL, USA), and Acros Organics (Thermo Fisher Scientific, Waltham, MA, USA). The microwave oven used was a Biotage Initiator 2.0 (Biotage, Uppsala, Sweden). High-pressure amidation was performed using a Haberautoklav Modell IV Typ 50S (Carl Roth® GMBH, Karlsruhe, Germany). Thin layer chromatography was performed with 0.2 mm thick silica gel foil 60 F254 (Merck). For detection of precursors and final products, standard methods were performed: ultraviolet (UV) irradiation, ethanolic solution of ninhydrin reagent, and hot air. Mobile phases consisted of different mixtures of dichloromethane and methanol saturated with gaseous ammonia atmosphere. Preparative column chromatography was performed on silica gel with particle sizes of 63 μm–200 μm (Merck). Mixtures of dichloromethane and ammonia-saturated methanol were used as mobile phases. For flash chromatography, prepacked columns were used on an IntelliFlash 310™ chromatology system (Varian Medical Systems Inc., Palo Alto, CA, USA). The adsorbent agent used was 50 μm silica gel (SuperFlash™, Varian). The used mobile phases were mixtures of dichloromethane and ammonia-saturated methanol or mixtures of methanol and triethylamine (99:1). Detection was carried out using an UV detector (wavelength: λ 256 nm). Infrared spectroscopy was measured on a Bruker Alpha-T spectrometer (Bruker BioSpin MRI GmbH, Etlingen, Germany). Intensities of the bands were characterized as follows: s for strong (very intensive), m for medium (medium intensity), and w for weak (low intensity). 1H NMR and 13C NMR spectra were recorded on a Bruker AM-250 (DPX-250) spectrometer (Bruker): (1H: 250.1 MHz, 13C: 62.9 MHz); a Bruker Avance 300 (1H: 300 MHz, 13C: 75.7 MHz); and a Bruker Avance 400 (1H: 400 MHz, 13C: 100.6 MHz). Perdeuterated and dried solvents (dimethyl sulfoxide [DMSO]-d6) were used.
1H NMR and 13C NMR data were reported in the following order. 1) Chemical shift (δ) in parts per million (ppm) downfield: chemical shift signals were calibrated to non-complete perdeuterated solvent as secondary signals (signals were calibrated to internal reference tetramethylsilane). δ values were δ (1H)= 2.54 and δ (13C)= 40.45 in DMSO-d6 as solvent. 2) Signal multiplicity as follows: br = broad; s = singlet; d = doublet; dd = double doublet; pt = pseudo triplet; t = triplet; m = multiplet; approximate coupling constants (J) in hertz (Hz) and number and assignment of protons (prop = propane; pip = piperidine; Ar = aryle; ax = axial; eq = equatorial). Electrospray ionization mass spectrometry was performed on a Fisons VG Platform II mass spectrometer (Fisons Instruments Ltd, Manchester, UK) in positive polarity. Data were identified as mass number ([M+H]+) and relative intensity (%). Elemental analyses for C, H, and N were performed on a Foss Heraeus CHN-O-rapid elemental analyser (Foss Heraeus, Hanau, Germany). Melting points were determined on a Büchi® B-530 melting point apparatus (Buchi, Flawil, Switzerland).
Chemistry
Precursor syntheses of compounds P–I to P–XVII
The current study focused on the synthesis and pharmacological evaluation of piperidin-1-yl-propoxyphenyl- and 3-(4-methylpiperazin-1-yl)propoxyphenyl-substituted 2-aminopyrimidine derivatives. Consequently, the two basic linkers were introduced into the aromatic 2-aminopyrimidine skeleton while keeping the lipophilic element at 4-position with the 4-methylpiperazine or N-benzylamine substituent constant. Furthermore, type and position of the side-chain moiety at the 2-amino position and 4- and 6-positions of the pyrimidine ring were systematically modified.
P–1: 3-(Piperidin-1-yl)propan-1-ol hydrochloride
Piperidine (20 g, 236 mmol), 3-chloropropane-1-ol (14.8 g, 157 mmol), K2CO3 (32.5 g, 235 mmol), and KI (26 g, 157 mmol) were refluxed in absolute acetone (500 mL) for six days. The mixture was cooled to room temperature, inorganic components were removed by filtration, and the filtrate was concentrated by distillation. Purification of the crude orange oil was achieved by distillation under reduced pressure (15 mbar, 90°C–105°C). The oily product was crystallized as a hydrochloride from 2-propanol in isopropanol 0.5N HCl (white solid, 16.5 g, 73% yield).9,20–23
1H-NMR (DMSO-d6; 300 MHz): d=10.54 (br-s, 1 H, NH); 4.42 (s, br-s, 1 H, OH); 3.41 ( t, 3JHH =6.0 Hz, 2 H, prop-1H2); 3.37–3.33 (m, 2 H, pip-2, 6Heq); 3.02–2.95 (m, 2 H, prop-3H2); 2.82–2.78 (m, 2 H, pip-2, 6Hax); 1.88–1.64 (m; 7 H, prop-H2, pip-3, 5 H2, pip-4 Heq); 1.43–1.31 (m, 1 H, pip-4Hax) ppm. 13C-NMR (DMSO-d6; 300 MHz): d=57.98; 53.57; 51.95; 26.38; 22.27; 21.45 ppm. Electrospray ionization mass spectrometry (ESI MS): 143.8 (M + H) (100%).
P–II: 1-(3-chloropropyl)piperidine hydrochloride
3-(Piperidin-1-yl)propan-1-ol (PI, 5 g, 27.8 mmol) was suspended in 100 mL absolute toluene and an excess of thionyl chloride (10 mL, 83 mmol) was added drop-wise under inert atmosphere at 0°C. Once the exothermic reaction decayed, the mixture was stirred for 3 h at 60°C. On completion of the reaction, thionyl chloride and toluene were distilled off. The crude product was re-crystallized from ethanol (beige solid, 5.24 g, 95% yield).9,20–23
1H-NMR (DMSO-d6; 300 MHz): d=10.90 (br-s, 1 H, NH); 3.73 ( t, 3JHH =6.0 Hz, 2 H, prop-3H2); 3.40–3.35 (m, 2 H, pip-2, 6Heq); 3.10–3.03 (m, 2 H, prop-1H2); 2.89–2.77 (m, 2 H, pip-2, 6Hax); 1.87–1.65 (m; 5 H, pip-3, 5 H2, pip-4Heq); 1.41–1.31 (m, 1 H, pip-4Hax). 13C-NMR (DMSO-d6; 300 MHz): d=53.56; 51.94; 42.50; 26.13; 22.19 ppm. ESI MS: 161.6 (M + H) (100%).
P–III: 4-(3-(piperidin-1-yl)propoxy)benzonitrile
Precursor II (5 g, 25.2 mmol), 4-hydroxybenzonitrile (3.31 g, 27.8 mmol), K2CO3 (10.5 g, 76 mmol), and KI (4.2 g, 25.2 mmol) were refluxed in 100 mL absolute acetone for 48 hours. After cooling, inorganic compounds were removed by filtration and the filtrate was concentrated to dryness. The residue was dissolved in dichloromethane, filtrated, and extracted with 2M NaOH solution. Organic phases were collected, washed with saturated NaCl solution, dried with MgSO4, and concentrated to dryness (solid, 4 g, 61% yield).17
1H-NMR (DMSO-d6; 250 MHz): d=7.73 (d, 3JHH =10 Hz, 2 H, Ar-2,6H); 7.13 (d, 3JHH =10 Hz, 2 H, Ar-3,5H); 4.07 (t, 3JHH =5 Hz, 2 H, prop-1H2); 2.38–2.30 (m, 6 H, pip-2,6H2, prop-3H2); 1.88–1.83 (m, 2 H, prop-2H2); 1.47–1.35 (m, 6 H, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=164.42; 134.17; 119.05; 115.53; 102.99; 22.61; 21.46 ppm. ESI MS: 245.0 (M + H) (100%).
P–IV: (4-(3-(piperidin-1-yl)propoxy)phenyl)methanamine
Benzonitrile (P-III, 4 g, 15.5 mmol) was suspended in 50 mL ammonia-saturated methanol in an autoclave with a catalytic amount of Raney nickel and hydrated at 5bar hydrogen pressure. The catalyst was removed by filtration with Celite® 535 coarse (Sigma) and the solvent was removed under reduced pressure. The product was used for the following reactions without any further purification (solid, 4 g, 98% yield).9,20–23
1H-NMR (DMSO-d6; 300 MHz): d=7.24 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 6.88 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 4.07 (t, 3JHH =6 Hz, 2 H, prop-1H2); 3.72 (s, 2 H, CH2NH2); 2.37–2.30 (m, 6 H, pip-2, 6H2, prop-3H2); 1.86–1.74 (m, 2 H, prop-2H2); 1.51–1.43 (m, 4 H, pip-3,5H2); 1.37-1.35 (m, 2 H, pip-4H2) ppm. 13C-NMR (DMSO-d6; 300 MHz): d=160.30; 131.23; 128.07; 117.18; 69.02; 58.36; 57.24; 47.96; 29.47; 28.70; 27.26 ppm. ESI MS: 250.0 (M + H) (100%).
P–V: 6-chloro-N4-(4-(3-(piperidin-1-yl)propoxy)benzyl)pyrimidin-2,4-diamine
4,6-Dichloropyrimidine-2-amine (500 mg, 3 mmol) and precursor P-IV (757 mg, 3 mmol) were suspended in diisopropyl amine (DIPEA) (1 mL, 6.1 mmol) in a microwave vial. The reaction mixture was heated and stirred in a microwave oven at 130°C for 1 hour. Solvent was removed under reduced pressure. Purification was performed by multiple re-crystallizations in acetone (solid, 535 mg, 73% yield).
1H-NMR (DMSO-d6; 250 MHz): d=7.62 (s, 2 H, NH2); 7.23 (d, 3JHH =8 Hz, 2 H, Ar-2,6H); 6.87 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 6.43 (s, 1H, pyrimidine-H), 5.79 (s, 1 H, NH); 4.39 (s, 2H, methylene-H); 4.02–3.98 (m, 2 H, prop-1H2); 3.01–2.78 (m, 6H, prop-3H2, pip-2,6H2); 2.03–1.70 (m, 2 H, prop-2H2); 1.66–1.47 (m, 4 H, pip-3,5H2); 1.28–1.25 (m, 2 H, pip-4H2) ppm. ESI MS: 376.5 (M + H) (100%).
P–VI: 3-(piperidin-1-yl)propanitrile
Piperidine (6 mL, 58.7 mmol), 3-chloropropanenitrile (4.6 mL, 59.7 mmol), K2CO3 (24 g, 176 mmol), and KI (10 g, 59 mmol) were refluxed in 50 mL absolute acetone for 48 hours. Inorganic compounds were filtrated and the solvent was evaporated under reduced pressure. Column chromatography was performed for further purification of the product (solid phase: silica gel; mobile phase: mixture of dichloromethane and ammonia-saturated methanol 9:1) (solid, 5.2 g, 64% yield).9,20–23
1H-NMR (DMSO-d6; 250 MHz): d=2.67–2.61 (m, 2 H, prop-2H2); 2.55–2.49 (m, 2 H, prop-3H2); 2.93–2.35 (m; 4 H, pip-2,6H2); 1.52–1.38 (m; 6 H, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=56.51; 54.10; 48.45; 30.01; 25.58; 24.15 ppm. ESI MS: 138.7 (M + H) (100%).
P–VII: 3-(piperidin-1-yl)propan-1-amine
Precursor P-VI (2 g, 14.5 mmol) was suspended in 50 mL ammonia-saturated methanol in an autoclave with a catalytic amount of Raney nickel and hydrated at 5bar hydrogen pressure for 24 hours. The catalyst was removed by filtration with Celite 535 coarse and the solvent was removed under reduced pressure. Column chromatography was performed for further purification of the product (solid phase: silica gel; mobile phase: mixture of dichloromethane and ammonia-saturated methanol 95:5; 9:1) (solid, 1 g, 49% yield).9,20–23
1H-NMR (DMSO-d6; 250 MHz): d=2.55–2.49 (m, 2 H, prop-3H2); 2.25–2.19 (m, 6 H, pip-2, 6H2 prop-1H2); 1.46–1.34 (m; 8 H, pip-3, 4, 5H2, prop-2H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=56.50; 54.11; 48.46; 29.80; 25.58; 24.17 ppm. ESI MS: 142.9 (M + H) (100%).
P–VIII: 6-chloro-N4-(3-(piperidin-1-yl)propyl)pyrimidin-2,4-diamine
4,6-Dichloropyrimidin-2-amine (300 mg, 1.8 mmol) and precursor P-VII (260 mg, 1.8 mmol) were suspended in N,N-diisopropylethylamine (0.63 mL, 3.6 mmol) in a microwave vial. The reaction mixture was heated and stirred in a microwave oven at 130°C for 1.5 hours. Purification was performed by multiple recrystallizations in absolute acetone (solid, 315 mg, 37%).
1H-NMR (DMSO-d6; 250 MHz): d=7.93 (br-s, 1 H, NH); 6.38 (br-s, 2 H, NH2); 5.75 (s, 1 H, Ar-H); 3.00–2.76 (m, 8 H, prop-1, 3H2, pip-2,6H2); 1.92–1.60 (m, 8 H, prop-2H2, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=164.01; 162.87; 122.88; 54.88; 53.88; 51.95; 23.36; 22.31; 21.41 ppm. ESI MS: 270.1 (M + H) (100%).
P–IX: 1-(3-(4-nitrophenoxy)propyl)piperidine
Precursor P-II (7.4 g, 37 mmol), 4-nitrophenole (5.2 g, 37 mmol), K2CO3 (15.4 g, 111 mmol), and KI (6.2 g, 37 mmol) were refluxed in 100 mL absolute dimethylformamide for 72 hours. Inorganic compounds were removed by filtration, the solution was concentrated, and 100 mL water was added. The water phase was washed with the organic solvent ethyl acetate. The collected organic phases were washed with 50 mL 2N NaOH solution and then with 50 mL of saturated NaCl solution, dried with MgSO4, and concentrated under reduced pressure (solid, 7.6 g, 77% yield).24
1H-NMR (DMSO-d6; 400 MHz): d=8.15 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 7.09 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 4.11 (t, 3JHH =6 Hz, 2 H, prop-1H2); 2.35 (t, 3JHH =7 Hz, 2 H, prop-H2); 2.33–2.29 (m, 4H, pip-2, 6H2); 1.90–1.81 (m, 2 H, prop-2H2); 1.49–1.44 (m, 4 H, pip-3,5H2); 1.37–1.34 (m, 2 H, pip-4H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=163.96; 140.61; 126.53; 114.79; 67.04; 59.67; 54.82; 25.99; 24.08; 20.60 ppm. ESI MS: 265.51 (M + H) (100%).
P–X: 4-(3-(piperidin-1-yl)propoxy)aniline
Precursor P-IX (7.5 g, 34 mmol) was suspended in 50 mL absolute ethanol and hydrated at 5bar hydrogen pressure for 24 h in an autoclave using a catalytic amount of Palladium on carbon (10%). The used catalyst was removed by filtration with Celite 535 coarse and the solvent was removed under reduced pressure (solid, 7.5 g, 94% yield).9,20–23
1H-NMR (DMSO-d6; 250 MHz): d=6.65 (d, 3JHH =8 Hz, 2 H, Ar-2,6H); 6.53 (d, 3JHH =8 Hz, 2 H, Ar-3,5H); 3.84 (t, 3JHH =8 Hz, 2H, prop-1H2); 2.38–2.32 (m, 6 H, prop-3H2, pip-2, 6H2); 1.84–1.76 (m, 2H, prop-2H2); 1.51–1.37 (m, 6 H, pip-4H2, pip-3,5H2) ppm.13C-NMR (DMSO-d6; 250 MHz): d=150.04; 142.21; 115.34; 55.32; 54.10; 26.54; 25.59; 24.14; 18.48 ppm. ESI MS: 235.2 (M + H) (100%).
P–XI: 6-chloro-N4-(4-(3-(piperidin-1-yl)propoxy)phenyl)pyrimidin-2,4-diamin
4,6-Dichloropyrimidine-2-amine (500 mg, 3 mmol) and precursor P-X (714 mg, 3 mmol) were suspended in DIPEA (2.1 mL, 12.2 mmol) in a microwave vial. The reaction mixture was heated and stirred in a microwave oven at 160°C for 3 hours. Solvent was removed under reduced pressure. Purification was performed by multiple recrystallizations in absolute acetone and ethanol (solid, 250 mg, 22% yield).
1H-NMR (DMSO-d6; 400 MHz): d=9.09 (s, 1 H, NH); 7.58 (s, 1 H, pyrimidine-H); 7.49 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 6.85 (m, 2 H, Ar-3,5H); 4.36–4.32 (m, 2 H, prop-3H2); 3.45–3.33 (m, 6 H, prop-1H2, pip-2,6-H2); 1.49–1.17 (m, 2 H, prop-2H2); 1.07–1.01 (m, 6 H, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=162.86; 162.08; 157.88; 153.87; 133.18; 121.87; 114.65; 93.21; 65.39; 53.67; 52.89; 23.77; 22.81; 21.77 ppm. ESI MS: 361.8 (M + H) (100%).
P-XII: 3-(1-methylpiperazin-1-yl)propane-1-ol
1-methylpiperazine (12.7 g, 127 mmol), 3-chloropropan1-ol (85 g, 900 mmol), K2CO3 (18 g, 130 mmol), and KI (15 g, 90 mmol) were refluxed in absolute acetone for 6 d. Purification was performed with column chromatography (solid phase: silica gel; mobile phase: dichloromethane/ammonia-saturated methanol 9:1) and precipitated as salt of hydrochloric acid (solid, 13.7 g, 78% yield).17
1H-NMR (DMSO-d6; 250 MHz): d=4.43 (br-s, 1 H, OH); 3.40 (t; 3JHH =9 Hz; 2 H, prop-1H2); 2.49–2.48 (m, 2 H, prop-3H2); 2.49–2.17 (m, 8 H, piperazine-2,3,5,6H2); 2.12 (s; 3 H, CH3); 1.53 (qu, 2 H, prop-2H2) ppm. ESI MS: 159.22 (M + H) (100%).
P–XIII: Synthesis of 1-(3-chloropropyl)-1- methylpiperazine dihydrochloride
Thionyl chloride (5.6 mL, 77 mmol) was added dropwise to precursor P-XII (5 g, 25.7 mmol) and stirred at 60°C for 3 hours. No solvent was used. The excess of thionyl chloride was removed by distillation and the crude product was washed several times with toluene (solid, 4.4 g, 97% yield).17
1H-NMR (DMSO-d6; 250 MHz): d=3.76 (t, 3JHH =6 Hz, 2 H, prop-1H2; 3.69–3.49 (m, 10 H, piperazine-H, prop-3H2); 2.83 (s, 3 H, CH3); 2.31–2.17 (m, 2 H, prop-2H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=54.90; 48.66; 47.36; 42.15; 26.27 ppm. ESI MS: 177.33 (M + H) (100%).
P–XIV: 1-methyl-4-(3-(4-nitrophenoxy)propyl)piperazine
Precursor P-XIII (1 g, 4.7 mmol), 4-nitrophenole (0.7 g, 4.7 mmol), K2CO3 (2 g, 14 mmol), and KI (0.8 g, 4.7 mmol) were refluxed in 30 mL absolute dimethylformamide for 72 hours. Inorganic impurities were removed by filtration, the solvent was evaporated under reduced pressure, and 100 mL water was added. The water phase was extracted with dichloromethane, then collected organic phases were washed with 2N NaOH solution and saturated NaCl, dried with Na2SO4, and concentrated to dryness (solid, 600 mg, 46% yield). 24
1H-NMR (DMSO-d6; 400 MHz): d=7.95 (d, 3JHH =8 Hz, 2 H, Ar-2,6H); 6.88 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 3.91–3.88 (m, 2 H, prop-1H2); 2.29–2.27 (m, 2 H, prop-3H2); 2.18–1.09 (m, 8 H, piperazine-H); 1.90 (s, 3 H, CH3); 1.69–1.62 (m, 2 H, prop-2H2) ppm. 13C-NMR (DMSO-d6; 400 MHz): d=162.09; 134.08; 119.07; 115.46; 102.67; 66.4; 54.74; 54.14; 52.69; 45.70; 25.97 ppm. ESI MS: 280.55 (M + H) (100%).
P–XV: 4-(3-(1-methylpiperazin-1-yl)propoxy)aniline
Precursor P-XIV (7.5 g, 34 mmol) was suspended in 50 mL of absolute ethanol in an autoclave. A catalytic amount of Pd/C (10%) was added and hydration was performed under 5bar hydrogen pressure for 24 hours. The catalyst was removed by filtration with Celite 535 coarse and the solvent was evaporated under reduced pressure (solid, 490 mg, 98%).17
1H-NMR (DMSO-d6; 250 MHz): d=6.63 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 6.50 (d, 3JHH =8 Hz, 2 H, Ar-3,5H); 4.58 (s, 2 H, NH2); 3.84 (t, 3JHH =6 Hz, 2 H, prop-1H2); 2.41–2.32 (m, 10 H, piperazine-H, prop-3H2); 2.14 (s, 3 H, CH3); 1.88–1.73 (m, 2 H, prop-2H2) ppm.13C-NMR (DMSO-d6; 250 MHz): d=149.95; 142.38; 115.38; 66.35; 54.75; 54.53; 45.72; 26.44 ppm. ESI MS: 250.0 (M + H) (75%).
P–XVI: N-benzyl-2-chloropyrimidin-4-amine
2,4-Dichloropyrimidine (1 g, 6.7 mmol) and benzylamine (1.3 g, 10 mmol) were dissolved (0.7 mL, 6.7 mmol) and refluxed in isopropanol. Solvent was evaporated and the resulting residue was dissolved in dichloromethane, washed with saturated NaHCO3 and NaCl solution, dried with MgSO4, and concentrated to dryness. The crude product was purified with flash chromatography (solid phase: silica gel; mobile phase: dichloromethane/methanol (1% triethylamine, 97:3) (solid, 975 mg, 66% yield).17
1H-NMR (DMSO-d6; 250 MHz): d=8.44 (s, 1 H, NH); 8.00 (d, 3JHH =6 Hz, 1 H, pyrimidine-6H); 7.41–7.33 (m, 5 H, Ar-H); 6.58 (d, 3JHH=5 Hz, 1 H, pyrimidine-H); 4.55 (s, 2 H, methylene-H2) ppm. 13C-NMR (DMSO-d6; 300 MHz): d=134.56; 129.44; 129.12; 128.38; 127.71; 127.63; 127.35; 53.18 ppm. ESI MS: 220.19 (M + H) (100%).
P–XVII: 2-chloro-4-(4-methylpiperazin-1-yl)pyrimidine
2,4-Dichloropyrimidine (500 mg, 3.4 mmol), 1-methylpiperazine (340 mg, 3 mmol), and DIPEA (0.9 mL, 6.7 mmol) were dissolved in absolute ethanol in a microwave vial. The reaction mixture was heated and stirred at 70°C for 45 min. The solvent was removed under reduced pressure. The resulting residue was dissolved in 20 mL dichloromethane, washed with 20 mL water, dried with Na2SO4, and finally concentrated to dryness (solid, 347 mg, 18% yield).17
1H-NMR (DMSO-d6; 250 MHz): d=8.05 (d, 3JHH =6 Hz, 1 H, pyrimidine-5H); 6.82 (d, 3JHH =6 Hz, 1 H, pyrimidine-6H); 3.62–3.59 (m, 4 H, piperazine-2,6H2); 2.41–2.36 (m, 4 H, piperazine-3.5H2), 2.21 (s, 3 H, CH3) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=171.02; 167.79; 98.83; 54.77; 48.49 ppm. ESI MS: 413.16 (M + H) (100%).
Synthesis of final products (compounds 2-7)
Compound 2: 6-(4-methylpiperazin-1-yl)-N4- (3-(piperidin-1-yl)propylpyrimidin-2,4-diamine
Precursor P-VIII (200 mg, 0.7 mmol) and 1-methylpiperazine (0.17 mL, 1.5 mmol) were suspended in DIPEA (0.25 mL, 1.5 mmol) in a microwave vial. The reaction mixture was heated and stirred at 160°C for 3 hours. Purification was performed by flash chromatography (solid phase: silica gel; mobile phase: dichloromethane/ammonia-saturated methanol 97:3, 9:1). For elementary analysis and melting point determination, the product was precipitated as a salt of maleic acid (solid, 140 mg, 56% yield).
IR (KBr): =3,390.47 m; 2944.17 m; 2,697.79 m; 1,574.12 s; 1,384.70 s; 1,259.92 m; 11,94.27 m; 864.25 m; 711.22 m; 569.50 w cm−1. 1H-NMR (DMSO-d6; 400 MHz): d=6.15 (s, 1 H, NH); 5.46 (s, 2 H, NH2); 5.01 (s, 1 H, Ar-H); 3.36 (t, 4 H, piperazine-2, 6H2); 3.17–3.12 (m, 2 H, prop-1H2); 2.32–2.25 (m; 10 H, pip-2,6H2, prop-3H2, piperazine-3,5H2); 1.62–1.59 (m, 2 H, prop-2H2); 1.52–1.47 (m, 4 H, pip-3,5H2); 1.39–1.38 (m, 2 H, pip-4H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=167.11; 134.84; 103.49; 56.70; 55.12; 53.74; 52.18; 52.08; 24.66; 23.33; 21.19 ppm. ESI MS: 334.2 (M + H) (100%) base. Analytical calculation (Anal Calc) for C17H31ClN7×3C4H4O4×H2O): C, 49.68; H, 5.94; N, 12.29. Found: C, 49.94; H, 6.15; N, 13.98.
Compound 3: 6-(4-methylpiperazin-1-yl)-N4-(4-(3-(piperidin-1-yl)propoxy)phenyl)pyrimidin-2,4-diamine
Precursor P-XI (235 mg, 0.65 mmol) and 1-methylpiperazine (0.15 mL, 1.3 mmol) were suspended in DIPEA (0.22 mL, 1.3 mmol) in a microwave vial. The reaction mixture was heated and stirred at 160°C for 3 hours. The solvent was removed under reduced pressure and the product was precipitated multiple times as a hydrochloride salt in absolute ethanol (solid, 91 mg, 33% yield).
IR (KBr): n=3,283.09 m; 2,935.32 s; 2,522.62 s; 1,573.20 s; 1,505.95 s; 1,291.06 m; 1,218.53 s; 1,051.95 s; 1,002.40 m; 837.23 s; 797.83 s cm−1. 1H-NMR (DMSO-d6; 250 MHz): d=8.67 (s, 1 H, NH); 7.54 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 6.85 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 5.82 (br-s, 2 H, NH2); 5.35 (s, 1 H, pyrimidine-H); 4.44 (s, 4 H, piperazine-3,5H2); 4.04 (t, 3JHH =6 Hz, 2 H, prop-3H2); 3.23 (s, 4 H, piperazine-2,6H2); 3.04 (m, 6 H, prop-1H2, pip-2,6H2); 2.35 (s, 3 H, CH3); 2.14–1.76 (m, 6 H, pip-3,4,5H2); 1.76–1.56 (m, 2 H, prop-2H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=174.81; 172.95; 162.34; 149.29; 121.15; 114.51; 71.07; 70.48; 60.28; 52.62; 48.22; 45.41; 27.54; 26.11; 24.28 ppm. ESI MS: 426.8 (M + H) (100%). Anal Calc for C23H35N7O×HCl×H2O: C, 57.55; H, 7.98; N, 20.42. Found: C, 57.52; H, 7.82; N, 20.45.
Compound 4: 6-(4-methylpiperazin-1-yl)-N-(4-(3-(piperidin-1-yl)propoxy)benzyl)pyrimidin-2,4-diamine
Precursor P-V (750 mg, 2 mmol) and 1-methylpiperazine (0.29 mL, 2.6 mmol) were suspended in DIPEA (0.68 mL, 4 mmol) in a microwave vial. The reaction mixture was heated and stirred at 160°C for 2 hours. Purification was performed by flash chromatography (solid phase: silica gel; mobile phase: dichloromethane/methanol with 1% triethylamine; 99:1, 85:15). The crude product was dissolved in dichloromethane, washed with 2M NaOH solution, dried with MgSO4, and concentrated to dryness under reduced pressure (solid, 117 mg, 13% yield).
IR (KBr): n=3,357.41 m; 2,694.43; 1,583.63 s; 1,385.07 s; 1,257.6 m; 1,193.36 m; 973.95 s; 864.27 m; 710.27 m; 558.81 m cm−1. 1H-NMR (DMSO-d6; 400 MHz): d=7.20 (d, 3JHH =9 Hz, 2 H, Ar-2,6H); 6.85 (d, 3JHH =9 Hz, 2 H, Ar-3,5H); 5.30 (s, 1 H, NH); 5.07 (s, 1 H, pyrimidine-H); 4.32 (s, 2 H, methylene-H2); 3.96 (t, 3JHH =6 Hz, 2 H, prop-3H2); 3.35 (pt, 3JHH =5 Hz, 6 H, piperazine-2,6H2); 2.38–2.29 (m, 10 H, prop-1H2, pip-2,6H2, piperazine-2,6H2); 2.19 (s, 3 H, CH3); 1.86–1.82 (m, 2 H, prop-2H2); 1.52–1.38 (m, 6 H, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=164.28; 162.45; 162. 35; 157.33; 132.48; 128.29; 114.67; 65.86; 55.15; 54.42; 54.07; 45.48; 43.55; 26.28; 25.55; 24.10 ppm. ESI MS: 440.5 (M + H) (100%). Maldi HRMS: 440.3127 (M + H) (100%); calc. 440.3132 (M + H) (100%).
Compound 5: N4-benzyl-N2-(4-(3-(piperidin-1-yl) propoxy)phenyl)pyrimidin-2,4-diamine
The precursors P-X (340 mg, 1.5 mmol) and P-XVI (435 mg, 1.9 mmol) were suspended in trifluoroacetic acid (0.36 mL, 4.6 mmol) in a microwave vial under inert gas atmosphere. The reaction mixture was heated and stirred at 140°C for 1 hour. The solvent was removed under reduced pressure. The crude product was purified by flash chromatography (solid phase: silica gel; mobile phase: dichloromethane/methanol with 1% triethylamine; 9:1) and recrystallized multiple times in absolute ethanol. For elementary analysis and melting point determination, the product was precipitated as salt of maleic acid (solid, 92 mg, 14% yield).
IR (KBr): n=2,950.06 w; 1,508.42 s; 1,356.11 s; 1,238.79 m; 1,053.26 m; 864.63 m; 827.81 s; 731.88 s; 568.83 s cm−1. 1H-NMR (DMSO-d6; 400 MHz): d=8.80 (s, 1 H, NH); 7.87 (d, 3JHH =8 Hz, 1H, pyrimidine-6H); 7.67–7.63 (m, 2 H, Ar2-3,5H); 7.34–7.13 (m, 5 H, Ar1-H); 6.583 (d, 3JHH =9 Hz, 2 H, Ar2-2,6H2); 6.04–5.99 (m, 1 H, pyrimidine-5H); 5.84 (s, 2 H, methylene-H2); 4.03–3.99 (m; 2 H, prop-1H2); 2.46–2.36 (m, 6 H, pip-2,6H2, prop-3H2); 1.93–1.89 (m, 2 H, prop-2H2); 1.60–1.46 (m, 6 H, pip-3,4,5H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=164.44; 158.57; 158.16; 157.74; 139.14; 129.43; 129.11; 128.26; 127.18; 115.16; 114.46; 65.11; 53.44; 48.53; 41.23; 22.43; 10.89; 8.43 ppm. ESI MS: 418.5 (M + H) (100%). Anal Calc for C25H31N5O×2 C4H4O4: C, 59.36; H, 6.19; N, 10.49. Found: C, 59.00; H, 6.06; N, 10.38.
Compound 6: N4-benzyl-N2-(4-(3-(4-methylpiperazin-1-yl)propoxy)phenyl)pyrimidin-2,4-diamine
Precursors P-XV (426 mg, 1.9 mmol) and P-XVI (500 mg, 2 mmol) were suspended in trifluoroacetic acid (0.44 mL, 5.8 mmol) in a microwave vial under inert gas atmosphere. The reaction mixture was heated and stirred at 140°C for 1 hour. The solvent was removed under reduced pressure. The crude product was purified by flash chromatography (solid phase: silica gel; mobile phase: dichloromethane/methanol with 1% triethylamine; 9:1) and recrystallized multiple times in absolute ethanol. For determination of elementary analysis and melting point, the final product was precipitated as a salt of maleic acid (solid, 150 mg, 18% yield). IR (KBr): n=3056.20 s; 1356.79 s; 864.67 s; 648.96 s; 589.13 s; 452.57 m cm−1.
1H-NMR (DMSO-d6; 250 MHz): d=7.82 (d, 3JHH =7 Hz, 1 H, pyrimidine-6H); 7.44–7.25 (m, 7 H, Ar2-3,5H, Ar1-H); 6.87 (d, 3JHH =9 Hz, 2 H, Ar2-2,6H2); 6.22 (d, 3JHH =7 Hz, 1 H, pyrimidine-5H); 4.56 (d, 3JHH =6 Hz, 2 H, methylene-H2); 4.03–3.98 (m; 2 H, prop-1H2); 3.18 (s, 3 H, CH3); 3.11–3.03 (m, 6 H, pip-2,6H2, prop-3H2); 2.77–2.64 (m, 6 H, pip- 3,4,5H2); 2.10–1.96 (m, 2 H, prop-2H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=167.17; 135.76; 128.32; 127.23; 126.98; 114.48; 114.34; 79.13; 65.57; 65.54; 53.39; 52.35; 49.75; 25.52 ppm. ESI MS: 433.3 (M + H) (100%). Maldi HRMS 433.2708 (M + H) (100%), calc. 433.2710 (M+H) (100%). Anal Calc for C25H32N6O×3 C4H4O4: C, 56.92; H, 5.68; N, 10.76. Found: C, 56.56; H, 5.60; N, 10.71.
Compound 7: 4-(4-methylpiperazin-1-yl)-N-(4-(3-(4-methylpiperazin-1-yl)propoxy)phenyl)pyrimidin-2-amine
Precursors P-XV (150 mg, 0.7 mmol) and P-XVII (176 mg, 0.7 mmol) were suspended in trifluoroacetic acid (0.16 mL, 2.1 mmol) in a microwave vial under inert gas atmosphere. The reaction mixture was heated and stirred at 140°C for 1 hour. The solvent was removed under reduced pressure. The crude product was purified by flash chromatography (solid phase: silica gel; mobile phase: DCM/MeOH with 1% triethylamine; 9:1) and recrystallized multiple times in absolute ethanol. For determination of elementary analysis and melting point, the final product was precipitated as a salt of maleic acid (solid, 174 mg, 59% yield).
1H-NMR (DMSO-d6; 250 MHz): d=8.85 (br-s, 1 H, NH); 7.93 (d, 3JHH =6 Hz, 1 H, pyrimidine-5H), 7.58 (d, 3JHH =5 Hz, 2 H, Ar-3,5H); 6.83 (d, 3JHH =9 Hz, 2 H, Ar-4,6H); 6.22 (d, 3JHH =6 Hz, 1 H, pyrimidine-6H); 3.95 (t, 3JHH =6 Hz, 2 H, prop-1H2); 3.72–3.61 (m, 6 H, prop-3H2, piperazine1-3,5H2); 2.68–42.62 (m, 10 H, piperazine1-2,6H2, piperazine2-2,3,5,6H2); 2.27 (s, 3 H, CH3); 1.93–1.83 (m, 2 H, prop-2H2) ppm. 13C-NMR (DMSO-d6; 250 MHz): d=163.61; 161.97; 159.56; 156.53; 152.97; 143.35; 134.31; 120.23; 114.28; 94.64; 89.84; 65.79; 54.88; 53.98; 5340; 51.11; 48.55; 5.36; 8.52 ppm. ESI MS: 426.7 (M + H) (100%). Maldi HRMS: 426.297 (M + H) (100%); calc. 426.2976 (M + H) (100%).
Pharmacology
hH3R binding assay
Radioligand binding assays were performed on HEK-293 cell membranes as previously described with slight modifications.18,19 Shortly before beginning the experiments, cell membrane preparations were thawed, homogenized by sonification at 4°C, and kept in ice-cold binding buffer (12.5 mM MgCl2, 100 mM NaCl, 75 mM Tris/HCl; pH 7.4). Competitive binding experiments were performed as follows: membranes (20–25 μg/well in a final volume of 0.2 mL binding buffer) were incubated with [3H]Nα-methylhistamine (MeHA) (2 nM; 85 Ci/mmol) and different concentrations of test ligands. Assays were run at least in duplicates with seven concentrations of 0.01 nM–100 μM of the test compound. Incubations were performed for 90 min at 25°C and shaking at 250 rpm. Non-specific binding was determined in the presence of 10m M unlabeled HA.
In saturation binding experiments, the maximal binding concentration (Bmax) was 0.89 pmol/mg and the Kd value of [3H]Nα-MeHA was 2.98 nmol/l (pKd =8.53±0.01). Bound radioligand was separated from free radioligand by filtration through GF/B filters pre-treated with 0.3% (m/v) polyethyleneimine using an Inotech cell harvester (Inotech Bioscience, LLC, Rockville, MD, USA) Unbound radioligand was removed by three washing steps with 0.3 mL/well ice-cold 50 mM Tris-HCl buffer, pH 7.4, containing 120 mM NaCl. Filters were soaked in a vial with 9 mL PerkinElmer scintillator liquid (Betaplate Scint, PerkinElmer, Kundenunterstützung Germany) and counted using a PerkinElmer MicroBeta® Trilux scintillation counter (PerkinElmer, Germany). Competition binding data were analyzed using GraphPad Prism® v3.02 software (GraphPad Software, Inc., La Jolla, CA, USA) using non-linear least squares fit. Ki values were calculated from the IC50 values according to the Cheng–Prusoff equation.18,19,25
hH4R binding assay
Radioligand binding assays were performed on Sf9 cell membranes as previously described with slight modifications.17,18 Prior to the experiments, cell membranes were sedimented by a 10 minutes centrifugation at 4°C and 16,000 g and resuspended in binding buffer (12.5 mM MgCl2, 1 mM EDTA, 75 mM Tris/HCl, pH 7.4). Competitive binding experiments were conducted in incubating membranes with 35 μg/well prepared from Sf9 cells expressing hH4R and co-expressed with G protein Gαi2 and Gβ1γ2 subunits in a final volume of 0.2 mL containing binding buffer and [3H] Histamine (10 nM, 15.3 Ci/mmol). Assays were run in triplicate with seven concentrations of 0.1 nM–100 μM of the test compound. Incubations were performed for 60 min at 25°C and shaking at 250 rpm. Non-specific binding was determined in the presence of 100 μM unlabeled histamine. Bound radioligand was separated from free radioligand by filtration through GF/B filters pretreated with 0.3% (m/v) polyethyleneimine and washed three times with 5 mL of ice-cold binding buffer (4°C). The amount of radioactivity collected on the filter was determined by liquid scintillation counting. Competitive binding data were analyzed by GraphPad Prism 3.02 (GraphPad) software using non-linear least squares fit. Ki values were calculated from the IC50 values according to the Cheng–Prusoff equation.18,19,25
Metric analyses
The values for LE, LELP, and LipE were calculated for compounds 1–7 based on their affinities for hH3Rs and hH4Rs to further corroborate their drug-likeness and assessment as potential new leads (Table 2). These widely-used three metrics are important models in drug discovery and are easy to use for assessing whether a ligand derives its potency from optimal fit with a target protein or simply by making many contacts.26–33 Values for LE, LELP, and LipE were calculated using equations 1, 2, and 3, respectively:
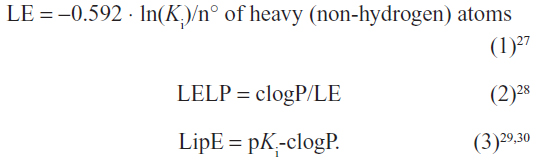
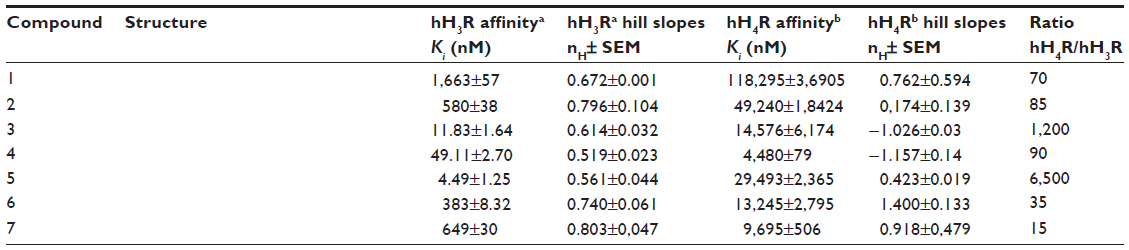 | Table 1 Histamine H3 receptor binding data of compounds 2–7 |
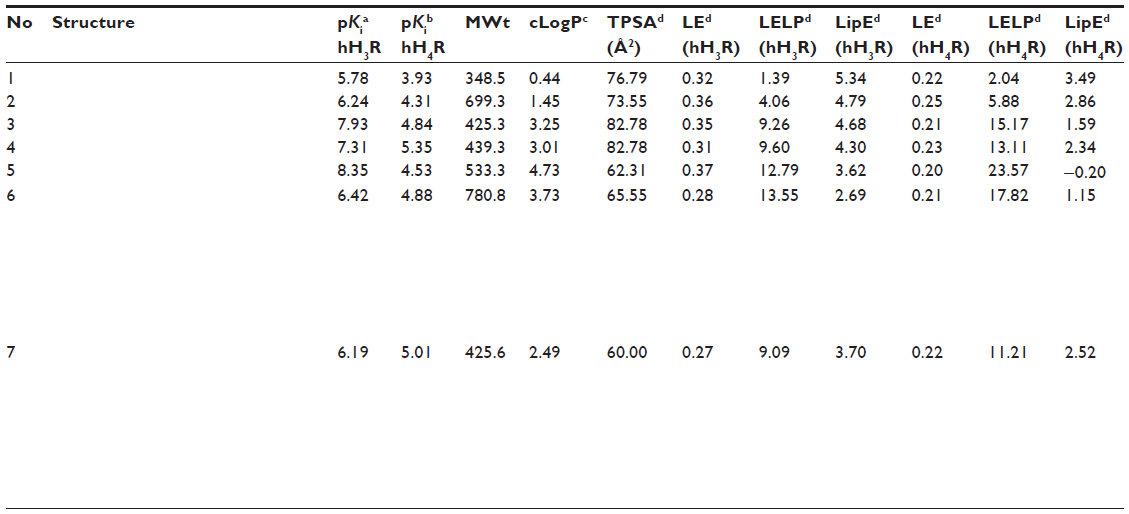 | Table 2 Molecular weight, clogP, LE, LELP, LipE, and TPSA values of compounds 2–7 |
An additional molecular descriptor, polar surface area (PSA), has been shown to correlate very well with human intestinal absorption.33 PSA is very suitable for predicting drug transport properties.32 In the current study, topological PSA (TPSA) was calculated using Molinspiration Depiction Software (Molinspiration Cheminformatics, Slovensky Grob, Slovak Republic).34 The procedure was based on the summation of the tabulated surface contributions of polar fragments, ie atoms regarding their physicochemical environment. Fragment contributions were determined by least squares fitting to the single conformer 3D PSA for 34,810 drugs from the World Drug Index. TPSA data usually provides results of virtually the same quality as classical 3D PSA; calculations, however, were 2–3 orders of magnitude faster.35,36
Results
Chemistry
Synthesis of precursors PI–XVII
In the present study, the precursors PI–IV, PVI–VII, and PX–XI containing piperidine were prepared according to previously described methods.9,20–23 The precursors PV, PVIII, and PXI were achieved through the nucleophilic reaction of 4,6-dichloropyrimidine-2-amine with precursors PIV, PVII, and PX, respectively, applying previously developed synthetic methodologies.17 The 4-methylpiperazine substituted precursors PXII–PXV and precursors PXVI and PXVII bearing benzyl or piperazin-1-yl at 4-position amine moiety were prepared in good yields. 22
Synthesis of final compounds 2–7
Compounds 2–7 were prepared in moderate to good yields using well-established conditions for microwave assisted nucleophilic substitution reactions of 4,6-dichloropyrimidin-2-amine and an appropriate basic side chain. As polar, protic solvent 2-propanol or ethanol was used and DIPEA acted as a basic auxiliar. Accordingly, precursors PVIII, PXI, and PV were intermediates for the final compounds 2, 3, and 4, respectively, while PXV was the starter precursor for final compounds 6 and 7. Finally, the nucleophilic reaction of PX with PXVI yielded compound 5. All resulting precursors PI–XVII and final compounds 2–7 described in the current study were synthesized by a series of reactions as shown in Figures 2–4.
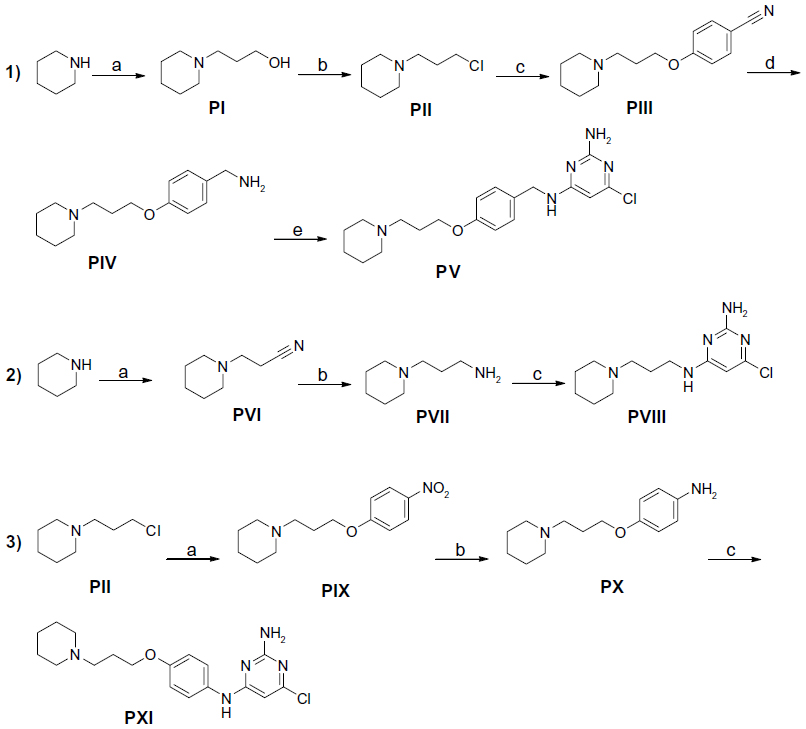 | Figure 2 Synthesis of precursors PI–PXI. |
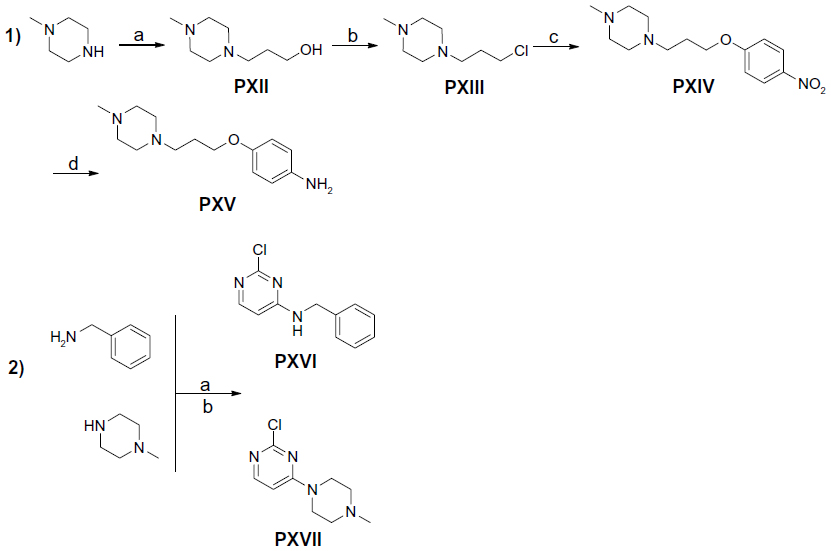 | Figure 3 Synthesis of precursors PXII–PXVII. |
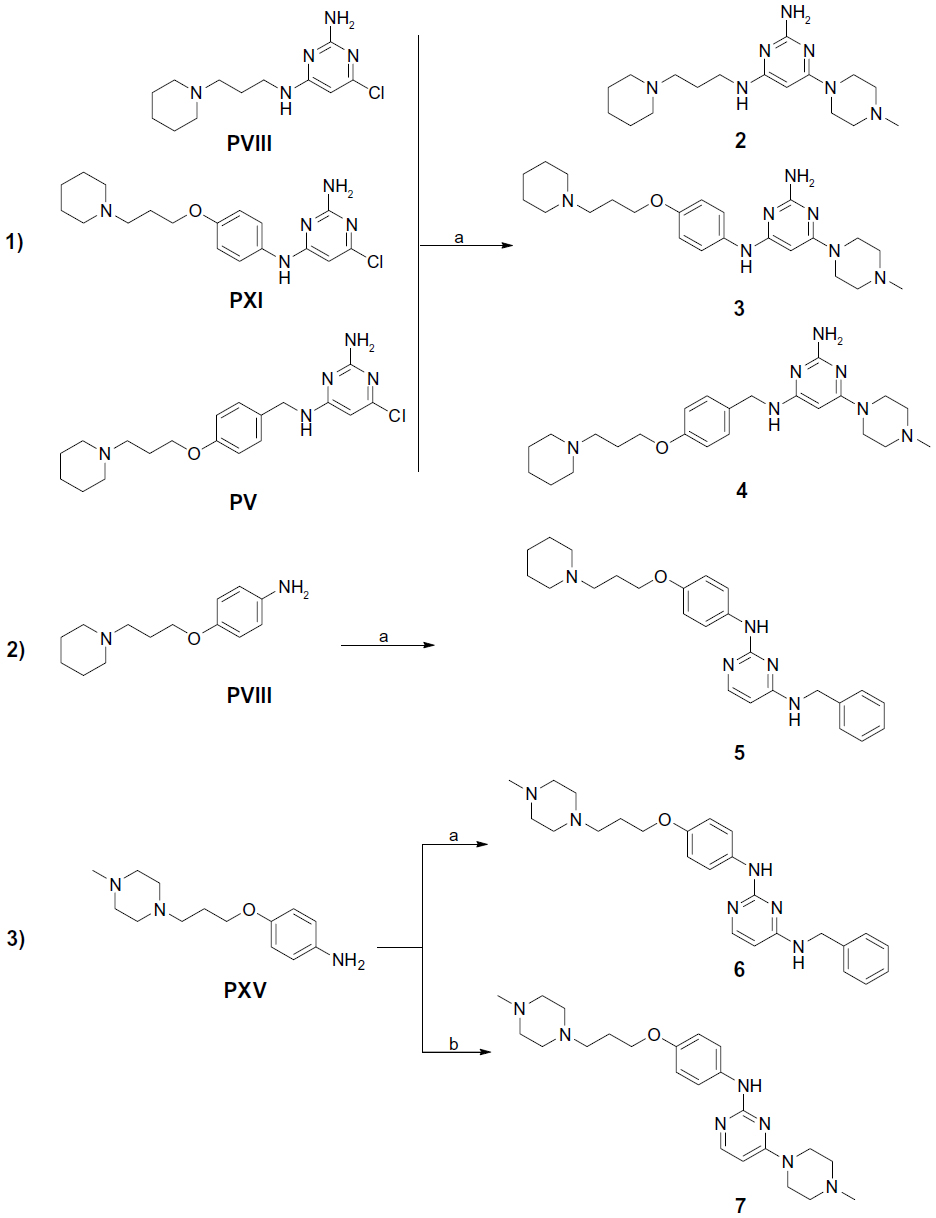 | Figure 4 Synthesis of final compounds 2–7. |
Pharmacology
H3R and H4R binding affinity
The final compounds 2–4 and 7 resulted from the combination of hH3R pharmacophore with 4-(4-methylpiperazin-1-yl)pyrimidin-2-amine. hH3R affinities of compounds 2–7 were evaluated by competition binding experiments with [3H]Nα-methylhistamine (MeHA) on membranes of HEK-293 cells stably expressing hH3R. Compounds 2–7 were further tested for their affinity to hH4R by a [3H] histamine replacement assay on membranes of Sf9 cells transiently co-expressing hH4R with Gαi2 and G1γ2 subunits.9,18–20 The H3R affinity observed for compounds 2–7 was in the nanomolar concentration range (Ki values of 4.49–649 nM), whereas affinity for hH4R was found to be evidently lower (Ki values of 4,480–49,240 nM) (Table 1).
Metric analyses
Values for LE (hH3R), LELP (hH3R), and LipE (hH3R) for compounds 1–7 were found to be in the range of 0.27–0.37, 1.39–13.55, and 2.69–5.34, respectively. Also, values for compounds 1–7 were in the range of 0.21–0.25, 2.04–17.82, and −0.20–3.49 for LE (hH4R), LELP (hH4R), and LipE (hH4R), respectively. Furthermore, the values describing the topological molecular polar surface were in the range of 60.00 Å2–82.78 Å2. Results of pharmacological screening and calculated physicochemical parameters are summarized in Tables 1 and 2.
Discussion
The synthesis of compound 2 should provide insight into whether the 2-aminopyrimidin ring can replace the aromatic center of the hH3R pharmacophore, namely piperidin-1-yl-propoxyphenyl. Indeed, compound 2 containing piperidinopropyl as the H3R pharmacophore affinity observed for hH4R (Ki (hH4R) = 49,240 nM) was decreased by approximately 2.3× compared to the lead structure 1 (Ki (hH4R) =118,295 nM), despite keeping the entire 4-(4-methylpiperazin-1-yl)pyrimidin-2-amine moiety at 4-position in the eastern part of its structure. However, affinity for hH3R was only in the three-digit nanomolar range (Ki (hH3R) = 580 nM) and was thus not satisfactory. Moreover, previous structure-activity relationships (SARs) of H3R antagonists/inverse agonists have shown that the presence of an ether function in the hH3R pharmacophore is necessary for producing high affinity for hH3R.8,9,11 The lack of an ether function, which was replaced by an amine moiety in compound 2, could explain the observed low affinity for hH3R (Ki [hH3R] =580 nM). As a result, inserting an aromatic ether linkage connecting the basic piperidinylpropyl moiety with the 4-(4-methylpiperazin-1-yl)pyrimidin-2-amine structural core resulted in compound 3 with a significant 49× increased affinity for hH3R (Ki [hH3R] =11.83 nM) and an hH3R/hH4R ratio of 1,232 towards the H3R subtype.
Conversely, the side chain elongation in the western part of the structure performed with the design and synthesis of compound 4 showed a nearly 5× weaker affinity for hH3Rs (Ki [hH3R] =49.11 nM) and a 3× higher affinity for hH4Rs (Ki [hH4R] =4,480.83 nM) compared to compound 3, indicating that such a substitution was unsuccessful and has not been explored any further. Moreover, previous studies of the SAR of H4R antagonists revealed that an amine moiety at 2-position in the 2-aminopyrimidine H4R pharmacophore was necessary to elicit affinity for hH4R.24 Thus, the substitution of the amine group at 2-position with the piperidinylpropyloxy H3R pharmacophore together with replacement of the 4-methylpiperazin-1-yl moiety in the eastern part by N-benzylamine significantly increased the affinity of the resulting compound 5 for hH3Rs (Ki [hH3R] =4.49 nM) and clearly decreased the affinity for hH4Rs (Ki [hH4R] =29,493 nM). The in-vitro observations for compound 5 at both receptor subtypes showed that proper structural derivatizations of an H4R pharmacophore, namely 2-aminopyrimidine, can result in the design and synthesis of a highly potent and selective hH3R antagonist with an hH3R/hH4R ratio of 6,500 towards hH3Rs. However, further structural modifications of compound 5, such as replacement of basic piperidine at 2-position or N-benzylamine at 4-position of the structure, resulted in compounds 6 (Ki [hH3R] = 383 nM, Ki [hH4R] =13,245) and 7 (Ki [hH3R] =649 nM, Ki [hH4R] =9,695) with a significant drop in antagonistic binding affinity towards both receptor subtypes.
Conversely, concepts of lipophilicity-related metric analyses predictively quantifying target-oriented drug-likeness have for many years been established as useful tools in the lead optimization process. Moreover, it has been shown that the lipophilicity of a respective compound is an important drug-like property, the control of which is vital for eventual success in drug development. This is not surprising since the role of LogP in altering drug potency, pharmacokinetics, and toxicity has been well established.29 However, high lipophilicity is usually linked with increased likelihood of binding to multiple targets and resultant pharmacologically based toxicology as well as poor solubility and metabolic clearance.37 It has also been suggested that compounds with a clogP <5 have a more favorable drug-likeness profile.17,26 Among the current series of compounds, clogP values were found to be <5, suggesting their applicability for oral administration. To further assist the drug discovery/development process and to guide the optimization from a lead compound to a successful drug candidate, rules for predicting drug-like physiochemical properties have been introduced.32 Lipinski’s ‘Rule of five’ and other physicochemical parameters have been shown to be useful tools in selecting oral drug candidates.29
Generally, values of LE and LELP being >0.3 and <7.5, respectively, have been validated as favorable candidates with drug-likeness.26–39 Interestingly, compounds 3, 4, and 5, the most potent hH3R antagonists among the current series, showed LE values >0.3 for hH3R whereas a significant decrease in LE values of <0.3 for hH4R was observed. These results further validate the drug-likeness of compounds 3, 4, and 5. Conversely, the correlation of LELP values (hH3R) for compounds 3, 4, and 5 of 9.26, 9.60, and 12.79, respectively, and 15.17, 13.11, and 23.57 for hH4R were not observed since both values were >7.5. However, it has been proposed that a clear understanding of probabilities in drug discovery is sometimes impossible because of a significant number of known and unknown variables.26 As a result, metric analyses have limitations except for some metrics that can be useful such as LipE and enthalpy in drug provability.38,39 It is therefore important to know when metric-based information is useful, useless, or possibly misleading so that it can be appropriately prioritized.26 Interestingly, LipE values, which are considered to be independent of molecular size of the respective compound, were found to be <5 for the three most promising compounds based on both histamine receptor subtypes. LipE results further corroborated the drug-likeness of compounds 3, 4, and 5. Moreover, it has been previously suggested that compounds with TPSA values >60 Å2 are generally regarded as poor membrane-permeable substances with predictively reduced CNS bioavailability.36,37 Among the synthesized series, the observed TPSA values for compounds 2–7 were in the range of 60 Å2–82 Å2, necessitating further structural optimization of the current class with the objective of generating newer derivatives with enhanced predictive physicochemical parameters, especially for TPSA. In this regard however, the results for compounds 2–7 showed that an increase in size and surface improved affinity and selectivity for hH3R was not successful concerning drug-likeness.
Conclusion
With respect to the development of potential histamine hH3R ligands, results observed for compounds 2–7 indicated that slight structural changes evoked extensive differences in affinities as well as histamine receptor subtype preferences. The incorporation of a piperidin-1-ylpropoxyphenyl, an established hH3R pharmacophore, in the hH4R-affine structural scaffold 2-aminopyrimidine increased hH3R affinity and subtype selectivity while hH4R affinity was decreased. Assuming reference compound 1 as starting lead, some spacer variations as well as an exchange of the lipophilic 4-methylpiperazine residue by N-benzylamine and a combination of both were performed. Compounds 3, 4, and 5 showed reduced affinities at hH4R with concentration ranges significantly lower than that of reference compound 1. These results indicate that incorporation of an H3R pharmacophore can provide greater affinity and selectivity for hH3R over hH4R. For example, the derivative with a 3-piperidinopropyloxy-substituted amine group at 2-position (compound 5) was 6,500× more selective for hH3R than hH4R, with further corroboration of these results provided by physicochemical and drug-likeness parameters.
Acknowledgments
Support to BS was provided by a UAEU Program for Advanced Research (UPAR) 2013 Grant (# 31M126), UAE University, and we greatly thank Professor JC Schwartz (Bioprojet, Saint-Grégoire, France) for providing HEK-293 cells stably expressing recombinant hH3R. We also gratefully thank Professor R Seifert (Hannover, Germany) for providing Sf9 cells and baculovirus stock solutions encoding for hH4R and G-protein Gαi2 and Gβ1γ2 subunits. Support for this work was also provided to HS by the EU COST Actions BM0806, BM1007, CM1103, and CM1207, DFG INST 208/664-1 FUGG as well as the Hesse LOEWE Schwerpunkte Fh-TMP, OSF and NEFF, Merz Pharmaceuticals, and the Else KrönerStiftung, TRIP.
Disclosure
The authors declare no conflicts of interest in this work.
References
Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302(5911):832–837. | |
Morisset S, Rouleau A, Ligneau X, et al. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408(6814):860–864. | |
Schlicker E, Fink K, Hinterthaner M, Göthert M. Inhibition of noradrenaline release in the rat brain cortex via presynaptic H3 receptors. Naunyn-Schmiedebergs Arch Pharmacol. 1989;340(6):633–638. | |
Schlicker E, Fink K, Detzner M, Göthert M. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm Gen Sect. 1993;93(1):1–10. | |
Schlicker E, Betz R, Göthert M. Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn-Schmiedeberg’s Arch Pharmacol. 1988;337:588–590. | |
Sander K, Kottke T, Stark H. Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull. 2008;31(12):2163–2181. | |
Gemkow MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today. 2009;14(9–10):509–515. | |
Tiligada E, Zampeli E, Sander K, Stark H. Histamine H3 and H4 receptors as novel drug targets. Expert Opin Investig Drugs. 2009;18(10):1519–1531. | |
Sadek B, Schwed JS, Subramanian D, et al. Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur J Med Chem. 2014;77:269–279. | |
de Esch IJ, Thurmond RL, Jongejan A, Leurs R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol Sci. 2005;26(9):462–469. | |
Więcek M, Kottke T, Ligneau X, et al. N-Alkenyl and cycloalkyl carbamates as dual acting histamine H3 and H4 receptor ligands. Bioorg Med Chem. 2011;19(9):2850–2858. | |
Smits RA, Leurs R, de Esch IJ. Major advances in the development of histamine H4 receptor ligands. Drug Discov Today. 2009;14(15–16):745–753. | |
Sander K, Kottke T, Tanrikulu Y, et al. 2,4-Diaminopyrimidines as histamine H4 receptor ligands – Scaffold optimization and pharmacological characterization. Bioorg Med Chem. 2009;17(20):7186–96. | |
Savall BM, Chavez F, Tays K, et al. Discovery and SAR of 6-Alkyl-2,4-diaminopyrimidines as histamine H4 receptor antagonists. J Med Chem. 2014;57(6):2429–2439. | |
Lazewska D, Kiec-Kononowicz K. Azines as histamine H4 receptor antagonists. Front Biosci (Schol Ed). 2012;1(4):967–987. | |
Engelhardt H, Schultes S, de Graaf C, et al. Bispyrimidines as potent histamine H4 receptor ligands: delineation of structure-activity relationships and detailed H4 receptor binding mode. J Med Chem. 2013;56(11):4264–4276. | |
Schreeb A, Walter M, Odadzic D, Schwed JS, Weizel L, Stark H. Piperazine modification in 2,4,6-triaminopyrimidine derivatives as histamine H4 receptor ligands. Pharmazie. 2013;6897:521–525. | |
Schneider EH, Schnell D, Papa D, Seifert R. High constitutive activity and a G-protein-independent high-affinity state of the human histamine H4-receptor. Biochemistry. 2009;48(6):1424–1238. | |
Kottke T, Sander K, Weizel L, Schneider EH, Seifert R, Stark H. Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur J Pharmacol. 2011;654(3):200–208. | |
Isensee K, Amon M, Galaparti A, et al. Fluorinated non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem Lett. 2009;19(8):2172–2175. | |
Meier G, Apelt J, Reichert U, et al. Influence of imidazole replacement in different structural classes of histamine H3-receptor antagonists. Eur J Pharm Sci. 2001;13(3):249–259. | |
Tomasch M, Schwed JS, Paulke A, Stark H. Bodilisant-a novel fluorescent, highly affine histamine H3 receptor ligand. ACS Med Chem Lett. 2013;4(2):269–273. | |
Amon M, Ligneau X, Camelin JC, Berrebi-Bertrand I, Schwartz JC, Stark H. Highly potent fluorescence-tagged nonimidazole histamine H3 receptor ligands. Chem Med Chem. 2007;2(5):708–716. | |
Williamson A. About the theory of the formation of ethers. Justus Liebigs Ann Chem. 1851;77:37–49. | |
Cheng YC, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. | |
Shultz MD. Improving the plausibility of success with inefficient metrics. ACS Med Chem Lett. 2014;5(1):2–5. | |
Polinsky A. In: The Practice of Medicinal Chemistry. 3rd ed. Wermuth, CG; Elsevier; Oxford, 2008; Vol 19, pp 244–254. | |
Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121. | |
Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6(11):881–890. | |
Tarcsay A, Nyíri K, Keseru GM. Impact of lipophilic efficiency on compound quality. J Med Chem. 2012;55(3):1252–1260. | |
Abad-Zapatero C. Ligand efficiency indices for effective drug discovery. Expert Opin Drug Discov. 2007;2(4):469–488. | |
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | |
Gad SC, Preclinical Development Handbook: ADME and Biopharmaceutical Properties. A John Wiley and Sons, Inc., Hoboken, New Jersey, 2008. | |
Molinspiration software or free molecular property calculation services [last accessed June 07, 2013], [2 screens], Available from: http://www.molinspiration.com/cgi-bin/properties. Accessed March 9, 2014. | |
Odović J, Marković B, Vladimirov S, Karljiković-Rajić K. In vitro modeling of angiotensin-converting enzyme inhibitor’s absorption with chromatographic retention data and selected molecular descriptors. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;15:953–954:102–107. | |
Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;15(9):430–431. | |
Meanwell NA. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24(9):1420–1456. | |
Shultz, MD. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg Med Chem Lett. 2013;23(21):5992–6000. | |
Shultz, MD. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg Med Chem Lett. 2013;23(21):5980–5991. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.