")
Back to Journals » International Journal of Nanomedicine » Volume 16
Doxorubicin Delivered via ApoE-Directed Reduction-Sensitive Polymersomes Potently Inhibit Orthotopic Human Glioblastoma Xenografts in Nude Mice
Authors Ouyang J, Jiang Y, Deng C , Zhong Z , Lan Q
Received 8 April 2021
Accepted for publication 19 May 2021
Published 15 June 2021 Volume 2021:16 Pages 4105—4115
DOI https://doi.org/10.2147/IJN.S314895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ebrahim Mostafavi
Jia Ouyang,1 Yu Jiang,2 Chao Deng,2 Zhiyuan Zhong,2 Qing Lan1
1Department of Neurosurgery, The Second Affiliated Hospital of Soochow University, Suzhou, 215004, People’s Republic of China; 2Biomedical Polymers Laboratory, College of Chemistry, Chemical Engineering and Materials Science, and State Key Laboratory of Radiation Medicine and Protection, Soochow University, Suzhou, 215123, People’s Republic of China
Correspondence: Zhiyuan Zhong; Qing Lan Tel/Fax +86-512-65880098
Email [email protected]; [email protected]
Purpose: Glioblastoma multiforme (GBM) poorly responds to chemotherapy owing to the existence of blood-brain barriers (BBB). It has been a long desire to develop BBB-permeable vehicles to facilitate drug targeting to GBM.
Method and Results: Here, we report that doxorubicin hydrochloride loaded in ApoE peptide-functionalized reduction-sensitive polymersomes (ApoE-PS-DOX) induces potent therapy of orthotopic U-87 MG model in nude mice. ApoE-PS-DOX with varying amount of ApoE (10∼ 30 mol%) all had stable DOX loading and small sizes (< 90 nm). As revealed by flow cytometry, confocal microscopy, apoptosis and MTT assays, ApoE-PS-DOX with 20 mol.% ApoE induced the best cellular uptake and inhibitory effect to U-87 MG cells, which were much better than the non-targeted PS-DOX and liposomal doxorubicin (Lipo-DOX) used in the clinic. ApoE-PS-DOX revealed a pharmacokinetic profile comparable to PS-DOX but induced considerably better growth inhibition of orthotopically xenografted U-87 MG tumors in nude mice than PS-DOX and Lipo-DOX, leading to significant survival benefits with a median survival time of 44 days, which was almost doubled relative to the phosphate-buffered saline (PBS) group. Moreover, in contrast to mice treated with Lipo-DOX and PS-DOX, ApoE-PS-DOX group exhibited little body weight loss, signifying that ApoE-PS-DOX not only has low side effects but also can effectively inhibit glioblastoma invasion.
Conclusion: This ApoE-docked multifunctional polymersomal doxorubicin induces potent and safe chemotherapy of orthotopic U-87 MG model in nude mice offering an alternative treatment modality for GBM.
Keywords: apolipoprotein E, polymersomes, doxorubicin, brain tumor, blood-brain barrier
Introduction
Glioblastoma multiforme (GBM), a leading primary brain malignancy, is hard to treat with chemotherapy owing to the existence of blood-brain barrier (BBB).1–3 Temozolomide (TMZ) is a unique drug capable of crossing BBB and currently used for treating GBM patients.4,5 However, more than half of patients with recurrent anaplastic gliomas could not benefit from TMZ treatment, and approximately 20% patients treated with TMZ suffer from the side effect such as thrombocytopenia and neutropenia,6 and the repeated administration of TMZ often induces drug resistance.7,8 Thus, design and development of novel chemotherapy become an emergent mission for GBM treatment.
BBB-permeable nanovehicles provide an intriguing platform to facilitate drug targeting to GBM. Various nanodrugs have been designed and constructed for GBM treatment.9–13 Interestingly, brain capillary endothelial cells were found to overexpress several receptors including transferrin receptor, lactoferrin receptor, and low-density lipoprotein receptor-related protein 1 (LRP1), which could be utilized to enhance the BBB crossing and brain delivery.14–17 For example, transferrin-drug conjugates, transferrin-modified liposomes and nanoparticles have been constructed for enhanced delivery of varying chemical drugs like doxorubicin (DOX) and resveratrol to treat GBM,18–22 inducing reduced tumor size and prolonged survival time in intracranial U87 glioma xenografted mice. Apolipoprotein E-installed nanoparticles have been used to assist liposomes and lipid nanoparticles to cross BBB.23,24 It should be noted, however, that chemical conjugation with large proteins to nanomedicines can be a great challenge.25 Moreover, conflicting results have been reported on transferrin-mediated brain targeting.26 Peptides such as T7, T12, iRGD, and angiopep-2 with small molecule weight, simple structure and specific target moieties have appeared to be a superior alternative for GBM targeting.27–32 Angiopep-2 demonstrated not only BBB permeability but also high affinity to GBM.33–35 Very recently, we found that apolipoprotein E derived peptide, ApoE, exhibited better transcytosis and glioblastoma accumulation than angiopep-2,36,37 which is likely a result of its multi-receptor-targeting property.
Here, we report for the first time that DOX loaded in ApoE peptide-functionalized reduction-sensitive polymersomes (ApoE-PS-DOX) induces potent therapy of orthotopic U-87 MG model in nude mice (Scheme 1). ApoE-PS-DOX was easily prepared from poly(ethylene glycol)-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) (PEG-P(TMC-DTC)) and ApoE-docked PEG-P(TMC-DTC). Our previous work showed that PS-DOX possess superior physiochemical properties to clinically used liposomal doxorubicin (Lipo-DOX), in terms of stability, tolerability, and intracellular drug release.38 The disulfide-crosslinked membrane of ApoE-PS-DOX would afford minor drug leakage during circulation and swift drug release in the cytoplasm of tumor cells that possesses 2–3 orders magnitude higher glutathione concentrations.39,40 PS-DOX has been engineered with varying ligands such as cRGD, GE11, ATN-161, and angiopep-2 for targeted treatment of different solid tumors.35,41–43 Our results showed that ApoE-PS-DOX induced significantly better growth inhibition of orthotopically xenografted U-87 MG tumors than PS-DOX and Lipo-DOX, leading to significant survival benefits with a median survival time of 44 days. Moreover, mice following the treatment with ApoE-PS-DOX exhibited little body weight loss, signifying that ApoE-PS-DOX not only has low side effects but also can effectively inhibit glioblastoma invasion.
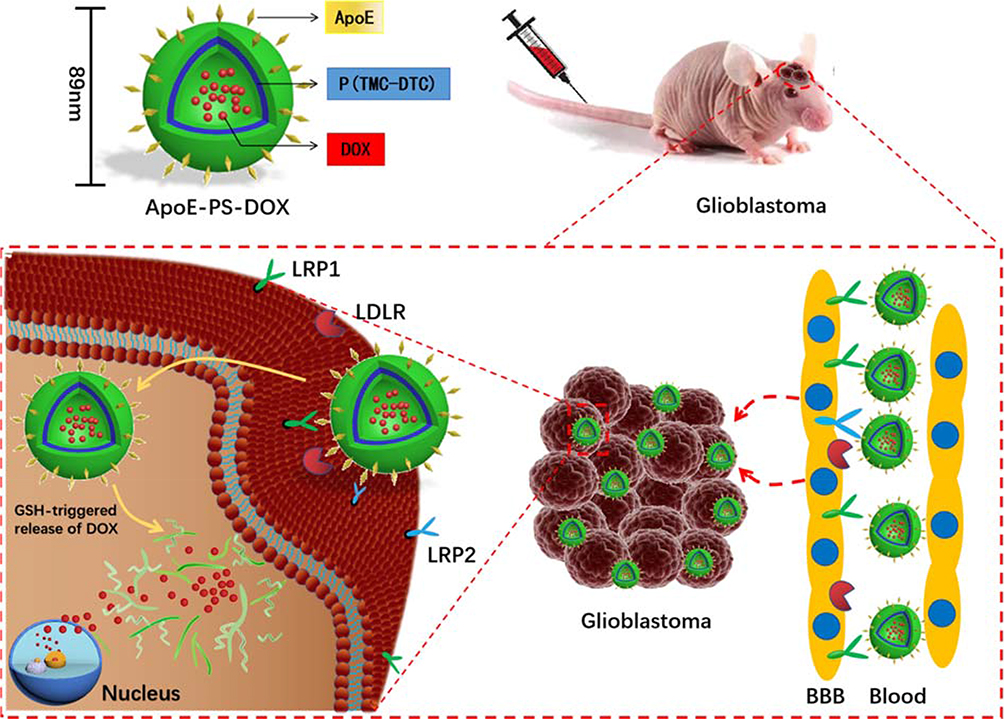 |
Scheme 1 Illustration of doxorubicin-loaded ApoE peptide-functionalized reduction-sensitive polymersomes (ApoE-PS-DOX) for the delivery of DOX to orthotopic human U-87 glioblastoma xenografts in nude mice. ApoE-PS-DOX engineered with ApoE can boost its BBB penetration and cellular uptake via LRP-1-mediated mechanism. The reduction-triggered DOX release from ApoE-PS-DOX affords significant growth suppression of orthotopically xenografted U-87 MG tumors, leading to remarkable survival benefits. |
Experimental Section
Loading and Reduction-Triggered Release of DOX·HCl
ApoE-PS was prepared through the self-assembly of PEG-P(TMC-DTC) and ApoE-PEG-P(TMC-DTC) with different molar ratios (10 mol.%, 20 mol.%, and 30 mol.%) via solvent exchange method, and the corresponding polymersomes were defined as ApoE10-PS, ApoE20-PS, and ApoE30-PS. The non-targeted PS was formed from PEG-P(TMC-DTC) only. pH-gradient method was used for DOX·HCl encapsulation. In short, PEG-P(TMC-DTC) and ApoE-PEG-P(TMC-DTC) with different molar ratios were completely dissolved in DMF to form a mixture with a polymer concentration of 10.0 mg/mL. 100 μL of the above mixture was added to 900 μL of citrate buffer (10 mM, pH 4), and the mixed solution was placed at 37 °C for 1 h followed by adjusting the pH to 7.8 using Na2HPO4 (2.0 M). After adding predetermined amount of DOX·HCl solution (5.0 mg/mL), the obtained solution was stewed at 37 °C overnight, and then exhaustingly dialyzed against phosphate buffer (PB, 10 mM, pH 7.4). The whole experiment process needs to be protected from light. In order to figure out the drug loading capacity (DLC) and drug loading efficiency (DLE), the drug-loaded vehicles were freeze-dried and dissolved in DMF to extract DOX·HCl. The amount of DOX·HCl was measured by fluorescence analysis (ex.480 nm, em. 560nm), and the DLC and DLE were calculated as reported previously.38
Cell Apoptosis Analysis
U-87 MG human GBM cells with luciferase transfection gene were purchased from Shanghai Sinochrome (China). Approximately 1.0–1.5×105 cells per well were seeded in six-well plates for overnight, and incubated with DOX·HCl, Lipo-DOX, PS-DOX, or ApoE-PS-DOX (DOX·HCl concentration: 5.0 μg/mL) for 4 h. The cells were further incubated with fresh media for 44 h for Annexin V-FITC/7-AAD assay. In short, the collected cells following complete washing were suspended in 200 μL of binding buffer, incubated with 7-AAD and Annexin V-FITC for 15 mins at room temperature, and examined using flow cytometric analysis. The experiments have been repeated three times and the data were presented as mean ± SD (n = 3).
Cell Invasion Analysis
Approximately 1.5×105 U-87 MG cells/well were plated in six-well plates overnight, treated with Lipo-DOX, PS-DOX, or ApoE-PS-DOX (DOX concentration: 0.5 μg/mL) for 4 h, and then digested. The collected cells following suspension in 100 μL of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 1.0% FBS medium were added to the upper chamber of a transwell. Prior to adding cells, the upper chamber of the transwell was covered by 30 μg of Matrigel and the lower chamber was filled with 500 μL of medium containing 10% FBS. Following 20 h incubation, the noninvasive cells in the top chamber were wiped with cotton swabs, and those invaded to the lower chamber were stained with 0.1% crystal violet and photographed under a microscope. The experiments have been repeated three times and the data were presented as mean ± SD (n = 3).
In vivo Antitumor Efficacy
The study was approved by Soochow University Laboratory Animal Center and the Animal Care and Use Committee of Soochow University (Guideline: Regulation for the Administration of Affairs Concerning Experimental Animals of Soochow University). Orthotopic glioblastoma xenograft model was built by intracranial transplantation of U-87 MG-luciferase (U-87 MG-Luc) tumor tissue. Briefly, subcutaneous tumor blocks were acquired by injection of U-87 MG-Luc cells (2×106 cells/mouse) to the flank of BALB/c nude mice and cut into small fragments. Then, the tumor fragments were administrated using a 24# trocar into the left skull (2 mm lateral to the bregma and 3 mm depth) of anesthetized BALB/c nude mice.32,37,44 The tumor growth was monitored by an IVIS Lumina II (λex/λem = 640 nm/668 nm), and the mice were separated into four groups at random (n = 8/group). PBS, ApoE20-PS-DOX, PS-DOX, or Lipo-DOX (5 mg DOX·HCl equiv./kg) was administered via tail vein on day 10, 13, 16, or 19. One day after the last treatment, one mouse from each group was sacrificed to acquire the major organs and tumors for histological analysis. The harvested tissues were fixed with a paraformaldehyde solution (4%) and implanted with paraffin. Then, the sliced tissues were stained by hematoxylin and eosin (H&E) or TUNEL and observed with a microscope (Leica QWin, Germany).
Results and Discussion
Preparation of ApoE-PS-DOX
ApoE-PS was readily self-assembled from PEG-P(TMC-DTC) and ApoE-PEG-P(TMC-DTC) with different molar ratios (10 ~ 30 mol.%) (Figure 1A). Dynamic Light Scattering (DLS) characterization illustrated that the sizes of ApoE-PS were about 80 nm, and the amount of ApoE basically did not affect the size and size distribution (Figure S1A). Taking ApoE20-PS as an example, we investigated the physiochemical properties of ApoE-docked polymersomes. Under the conditions of exhausting dilution, 10% FBS, and one-week storage in PB, ApoE20-PS displayed superb stability with a slight size change (Figure S1B), which mainly benefits from disulfide bond-crossing of DTC moieties in the polymersomal membrane.38 ApoE-PS, however, quickly swelled and exhibited broad distribution in 12 h, and presented largely increased size of over 1300 nm at 24 h in the presence of glutathione (GSH, 10 mM) (Figure S1C), signifying their rapid responsivity under intracellular reduction environment.
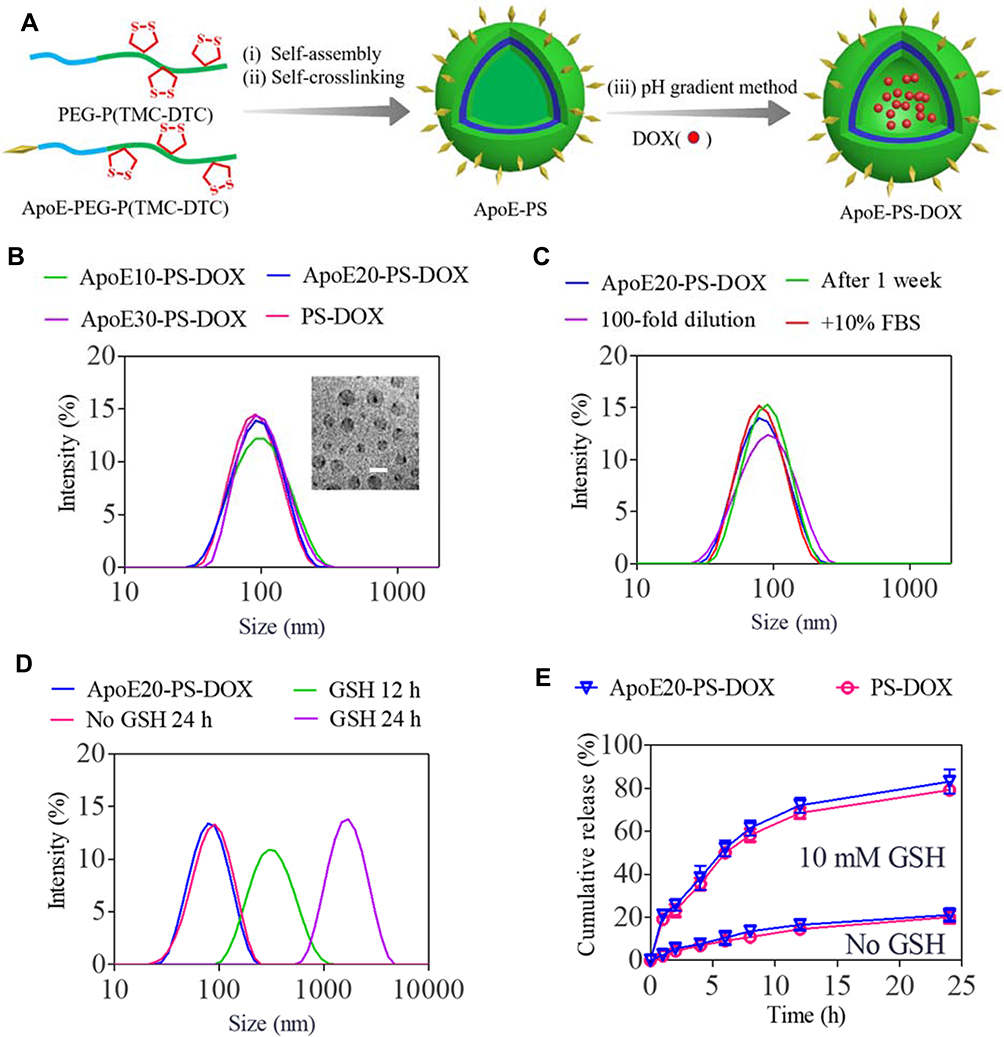 |
Figure 1 Preparation process and physicochemical properties of PS-DOX and ApoE-PS-DOX. (A) Illustration of preparation process of ApoE-PS-DOX. (B) Size distribution measured using DLS. Inset: Transmission electron microscopy (TEM) image of ApoE20-PS-DOX (bar: 100 nm). (C) Stability of ApoE20-PS-DOX under different conditions. (D) Responsiveness of ApoE20-PS-DOX under 10 mM GSH. (E) GSH-triggered drug release from ApoE20-PS-DOX in PB and PB + 10 mM GSH at 37 °C. |
DOX·HCl was actively encapsulated into ApoE20-PS through pH-gradient hydration technique that is used for clinically used Lipo-DOX at present. Both ApoE-PS and PS exhibited decent drug loading capacity with a decent DLC up to 9.3 wt.% (Table 1). In comparison with blank polymersomes, the formed ApoE20-PS-DOX displayed a slightly increased size with an average diameter of 85~89 nm (Figure 1B), comparably narrow distribution, and nearly neutral surface charge (1.5 ~ 1.9 mV) (Table 1). Meanwhile, PS-DOX without ApoE targeting ligand exhibited similar physiochemical properties including size (~80 nm), size distribution, and surface charge (0.5 mV). ApoE20-PS-DOX was robust against dilution, 10% FBS, and PB for 1 week (Figure 1C), while quickly swelled to over 1000 nm within 24 h under reductive environment (10 mM GSH) (Figure 1D). In vitro release studies displayed that ApoE-PS-DOX though possessing a minimal drug release (ca. 21%) in physiological environments within 24 h showed markedly accelerated drug release in PB solution containing 10 mM GSH, in which around 83% of drug was released although other conditions unchanged (Figure 1E). PS-DOX displayed similar drug release profiles. The remarkable features of ApoE-PS-DOX including robustly crosslinked structure, peptide conferred tumor selectivity, and redox-triggered drug release make it different from most developed DOX delivery nanosystems.45–47
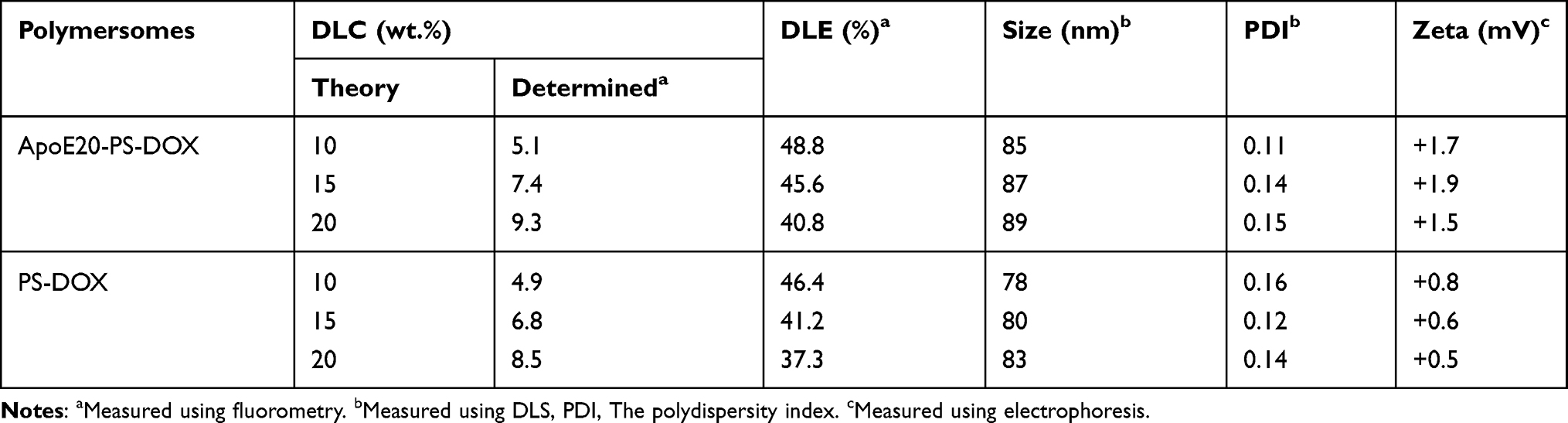 |
Table 1 Drug Loading of PS-DOX and ApoE20-PS-DOX |
In vitro Antitumor Activity and Selective Cellular Uptake of ApoE-PS-DOX
Both PS-DOX and ApoE-PS-DOX showed higher in vitro antitumor efficacy in U-87 MG cells than Lipo-DOX, mainly owing to the efficient cellular uptake and fast drug release from polymersomes under intracellular reductive environments (Figure 2A). Moreover, ApoE-PS-DOX showed stronger cytotoxic effect toward cancer cells in comparison with PS-DOX, in which ApoE20-PS-DOX displayed a lowest half-maximal inhibitory concentration (IC50) of 1.01 μg DOX⋅HCl equiv./mL. PS-DOX and Lipo-DOX revealed IC50 of 3.64 and 10.22 μg DOX⋅HCl equiv./mL, respectively. Noticeably, all empty PS and ApoE-PS (ApoE densities: 10–30 mol.%) were nontoxic toward both cancer cells (U-87 MG, Figure S2A) and healthy cells (astrocyte, Figure S2B) at concentrations of 0.1–0.5 mg/mL.
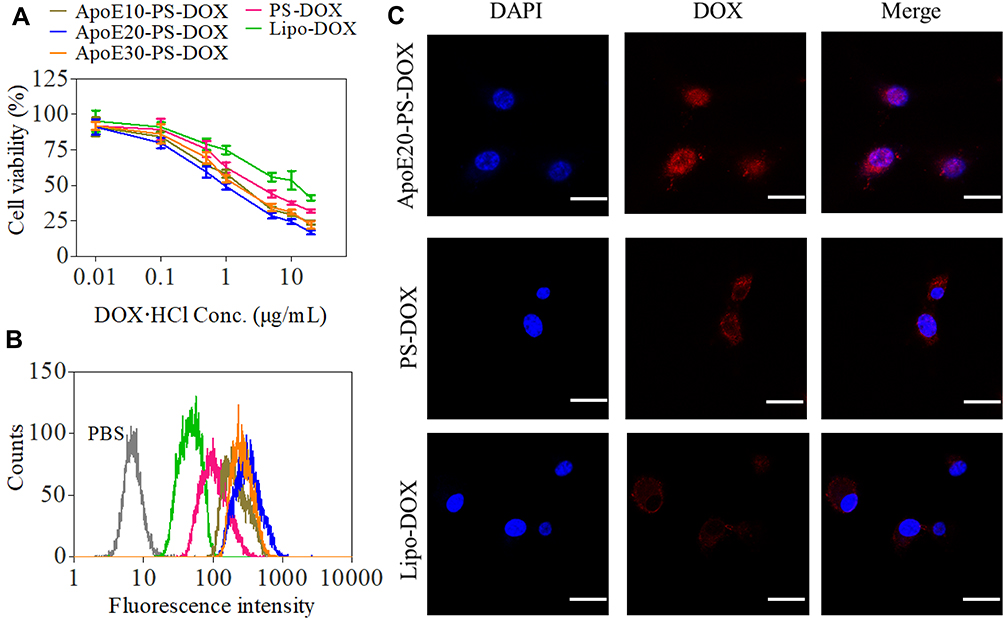 |
Figure 2 In vitro antitumor efficacy and cellular internalization of ApoE-PS-DOX in U-87 MG cells. (A) Cytotoxicity evaluated by MTT assay. Cells treated with varying formulations for 4 h were further incubated in fresh medium for 44 h (n = 4). (B) Flow cytometric analysis and (C) CLSM images of U-87 MG cells incubated with varying formulations for 4 h at a concentration of 10 μg DOX·HCl/mL. Scale bar: 30 μm. ApoE-PS-DOX with a ApoE density of 20% (ApoE20-PS-DOX) demonstrated best cellular internalization and cytotoxic capacity in U-87 MG cells. |
In comparison with Lipo-DOX and PS-DOX, ApoE-PS-DOX displayed remarkably higher cellular uptake in U-87 MG cells as measured by flow cytometry (Figure 2B), signifying that the introduction of ApoE facilitated the cellular uptake of nano-drugs via LRP and LDLR receptor-mediated mechanism. Notably, ApoE20-PS-DOX displayed highest DOX fluorescence after incubating with cells for 4 h, presenting 3.6 times higher drug accumulation in U-87 MG cells than Lipo-DOX group. ApoE20-PS-DOX was thus selected for subsequent in vitro/vivo evaluation. The cellular uptake and trafficking was further investigated using confocal laser scanning microscope (CLSM) measurement, and the results revealed that U-87 MG cells incubated with ApoE20-PS-DOX for 4 h displayed strong DOX fluorescence in both cytoplasm and nucleus (Figure 2C), signifying its efficient cellular internalization as well as fast drug release inside cancer cells. In contrast, cells incubated with Lipo-DOX or PS-DOX without targeting ligand presented faint DOX fluorescence that mostly located at perinuclear area.
Cell Apoptosis and Invasion
Cell apoptosis of U-87 MG cells following the treatment with different formulations was evaluated using Annexin V-FITC/7AAD apoptosis detection kit. Notably, both PS-DOX and ApoE10-PS-DOX caused significant apoptosis of U-87 MG cells, in which PS-DOX, ApoE10-PS-DOX, ApoE20-PS-DOX, and ApoE30-PS-DOX at a DOX·HCl concentration of 5 μg/mL displayed remarkable apoptotic rates of 49.9%, 74.6%, 85.3%, and 79.0%, respectively (Figure 3A). The most apoptotic cells generated by ApoE20-PS-DOX corresponds to its highest in vitro cytotoxic capacity towards cancer cells. Western blot assay further revealed that cells treated with ApoE20-PS-DOX had the highest expression of apoptosis-related BAX protein and the lowest expression of anti-apoptosis-associated Bcl-2 protein (Figure 3B).
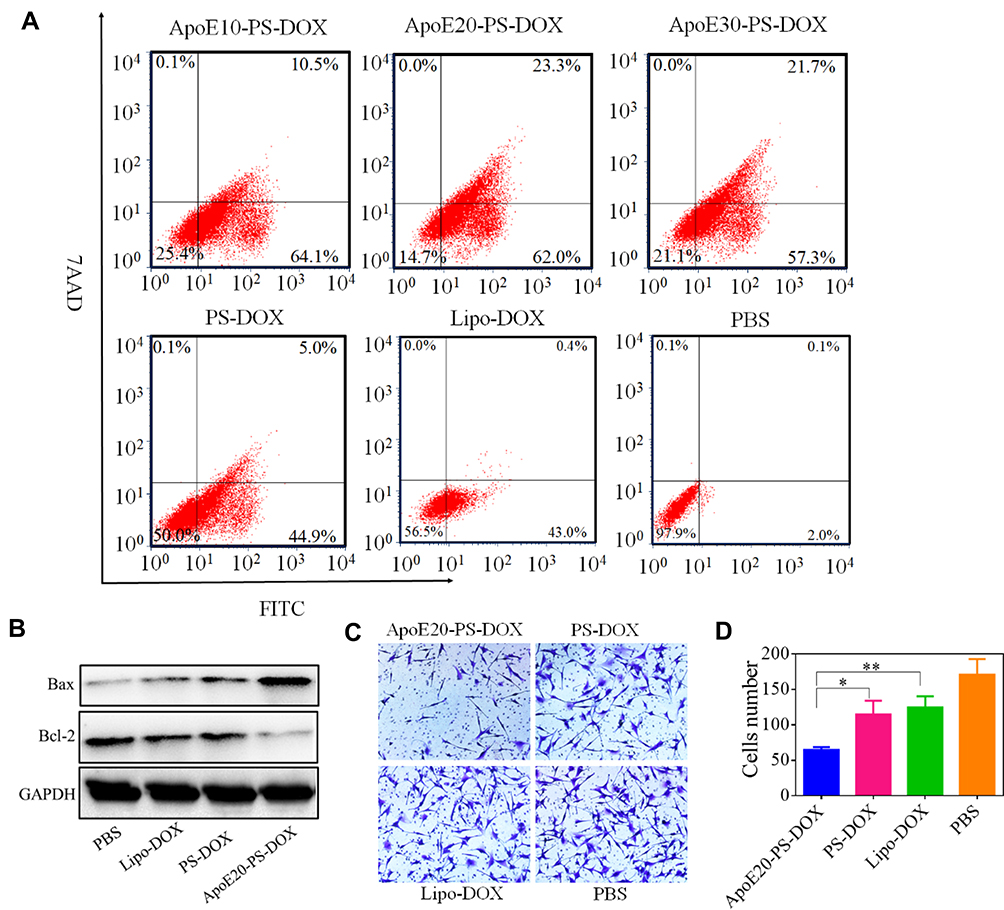 |
Figure 3 Effect of ApoE20-PS-DOX on the apoptosis and invasion glioma cells, Lipo-DOX and PS-DOX were uses as controls. (A) Cell apoptosis assay and (B) Western blot assay of the expression of Bcl-2 and BAX proteins in U-87 MG cells. (C) Effect of different drug on cell invasion. (D) Quantification of cell invasion presented in C. *p < 0.05, **p < 0.01. |
Vigorously invasive growth of GBM greatly compromises the therapeutic efficacy of surgical excision, radiotherapy and chemotherapy.48 Here, transwell assay was employed to evaluate the capacity of nanodrugs on the inhibition of cell invasion. Compare with PBS group, Lipo-DOX, PS-DOX, and ApoE20-PS-DOX showed obvious inhibition of cell invasion (Figure 3C). Interestingly, cells following the treatment with ApoE20-PS-DOX displayed around two times less invasion compared with PS-DOX and Lipo-DOX groups (Figure 3D).
Pharmacokinetics and Biodistribution
Both ApoE-PS-DOX and PS-DOX were observed to have a relatively long circulation time with a t1/2,β of ca. 4.0 h (Figure 4A), in sharp contrast with that of free DOX (ca. 0.45h49), confirming that ApoE-PS-DOX has superior in vivo stability. Comparing with Lipo-DOX, ApoE-PS-DOX achieved nearly 8-fold higher enrichment (ca. 7.2%ID/g) in the U87 glioma tumors (Figure 4B), outperforming previously reported nanodrugs.11,18,50 This high GBM accumulation of ApoE-PS-DOX is mainly attributed to the prominent BBB-crossing and cell-targeting effect of ApoE. Furthermore, ApoE-PS-DOX revealed less deposition in the liver and heart in comparison with Lipo-DOX, possibly further lessening the cardiotoxicity, a main concern of DOX in the clinics.51
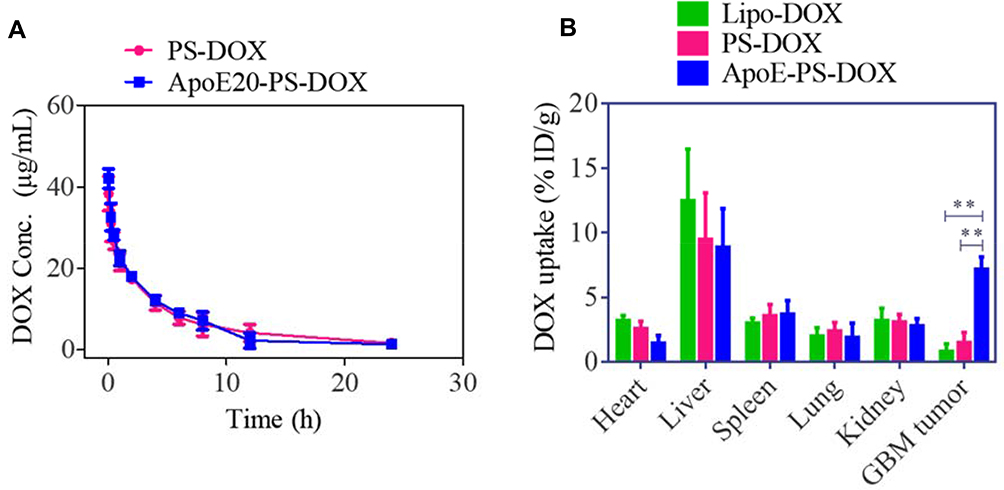 |
Figure 4 Pharmacokinetics and biodistribution studies. (A) Pharmacokinetics study of ApoE20-PS-DOX and PS-DOX in C57BL/6 mice. (B) Biodistribution study of ApoE-PS-DOX, PS-DOX, and Lipo-DOX in orthotopic xenograft mouse model at 8 h post-injection (10 mg DOX·HCl/kg). **p < 0.01. |
In vivo Therapeutic Efficacy
The anti-glioma efficacy of ApoE20-PS-DOX was assessed using orthotopic U-87 MG-luciferase (U-87 MG-Luc) tumor-bearing mouse model. The bioluminescent luciferase reporter expressed by U-87 MG cells was employed to visualize the progression of tumors. In comparison with fast tumor growth in PBS group, all DOX formulations (Lipo-DOX, PS-DOX, ApoE20-PS-DOX) could retard tumor growth as characterized by obviously lower bioluminescence intensity of intracranial tumors on days 10, 14, 18 and 22 (Figure 5A). Of note, mice treated with ApoE20-PS-DOX revealed weak tumor bioluminescence during the whole experimental period, signifying its efficient suppression on tumor progression. The bioluminescence quantification of intracranial tumors revealed that mice treated with ApoE20-PS-DOX had the lowest bioluminescence level with about 3.0-fold and 5.6-fold lower luminescence intensity than those treated with PS-DOX and PBS on day 22, respectively (Figure 5B). Moreover, ApoE-PS-DOX group exhibited little body weight loss (Figure 5C), signifying that ApoE-PS-DOX not only has low systemic toxicity but also can efficiently inhibit glioblastoma invasion. On the contrary, both Lipo-DOX and PS-DOX induced comparable body weight loss to PBS group. Remarkably, ApoE20-PS-DOX could significantly extend the survival time of orthotopic U-87 tumor-bearing mice with a median survival time (MST) of 44 days, which was over 2-fold longer than PBS group (23 days) (Figure 5D), confirming its high anti-GBM efficacy. PS-DOX and Lipo-DOX could also increase the MST to 35 and 28 days, respectively, although significantly less efficient than ApoE20-PS-DOX. In comparison, moderate survival benefits (MST = 28 ~ 38 days) were obtained from previous work on DOX and paclitaxel-loaded nanodrugs in orthotopic GBM bearing mice.12,50,52,53
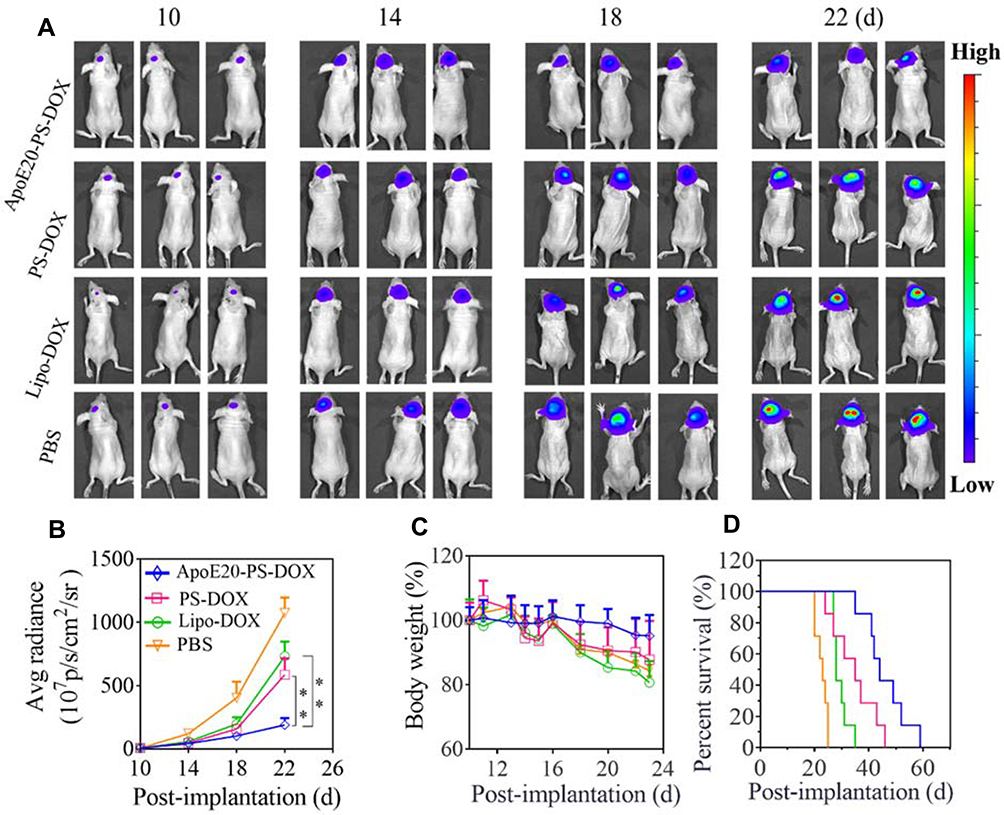 |
Figure 5 Anti-GBM efficacy of ApoE-PS-DOX, PS-DOX, and Lipo-DOX (dosage: 5.0 mg DOX·HCl equiv./kg) in orthotopic U-87 MG-Luc human glioma xenografts. ApoE-PS-DOX group demonstrated best tumor growth inhibition and survival benefits. (A) Bioluminescence images of mice following the administration on day 10, 13, 16, and 19. (B) Average radiance of U-87 MG-Luc bioluminescence in different groups (**p<0.01). (C) Body weight changes of mice within 23 d post-implantation. (D) Survival evaluation of mice. Statistical analysis: ApoE20-PS-DOX vs Lip-DOX and PBS, ** p < 0.01; Lip-DOX vs PS-DOX, ns; PS-DOX vs PBS, ** p < 0.01; Lip-DOX vs PBS, ** p<0.01 (Kaplan−Meier analysis, Log rank test). |
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays revealed that mice received the ApoE20-PS-DOX therapy induced considerable apoptosis (green) of intracranial glioblastoma cells, in sharp contrast with a few apoptotic tumor cells in both PS-DOX and Lipo-DOX groups (Figure 6A). Immunofluorescence staining was used to detect DOX distribution in intracranial tumors after treatment. Mice received the ApoE20-PS-DOX therapy displayed significant DOX fluorescence throughout the whole tumor tissue (Figure 6B), corroborating that ApoE20-PS-DOX facilitates the drug enrichment and retention in intracranial GBM tumors. On the contrary, only a small amount of DOX was perceived in the intracranial tumors of mice received the PS-DOX or Lipo-DOX therapy.
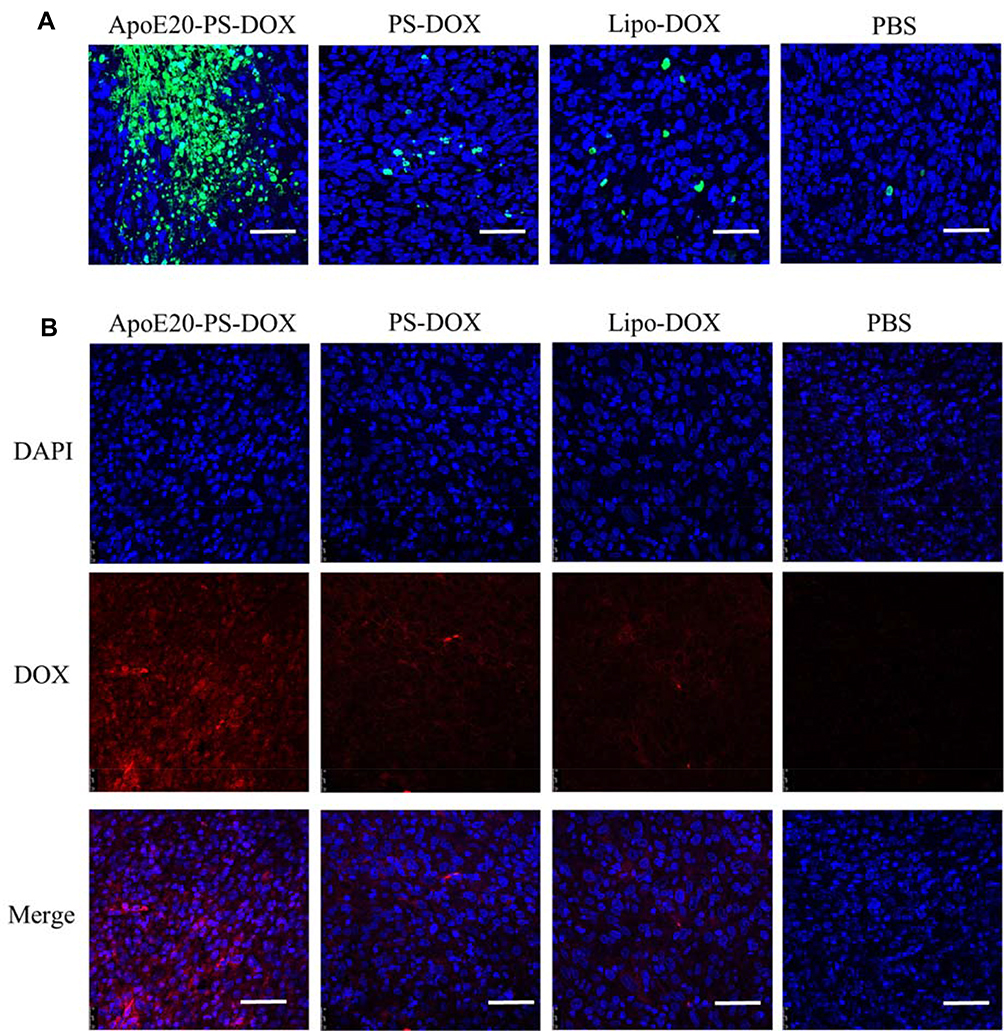 |
Figure 6 (A) Tunel analysis of brain tumor excised from mice received the therapy of ApoE20-PS-DOX, PS-DOX, Lipo-DOX and PBS. Green represents apoptotic area in tumor. Bar: 50 μm. (B) Tumor enrichment behavior of ApoE20-PS-DOX, PS-DOX and Lipo-DOX observed by CLSM. Tumor sections are obtained from GBM-bearing mice following 24 h treatment. Nuclei were stained with blue DAPI and DOX with red fluorescence. Bar: 50 μm. |
Conclusions
We have demonstrated that DOX loaded in ApoE peptide-functionalized reduction-sensitive polymersomes (ApoE-PS-DOX) induces potent and safe chemotherapy of orthotopic U-87 MG model in nude mice. ApoE-PS-DOX presents several traits including stable drug loading, small size, BBB permeability, GBM cell selectivity, and triggered drug release, giving rise to an extended circulation time and remarkably enhanced drug enrichment in the orthotopical U-87 MG tumor xenografts. Accordingly, ApoE-PS-DOX brought about significant survival benefits (median survival time doubled compared with PBS control) and reduced systemic side effects. ApoE-directed delivery of DOX appears to be an intriguing modality for the treatment of intractable GBM.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC 51973149, 51773145, and 51633005), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (19KJA220002), and the Beijing Natural Science Foundation (7204322).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149(1):36–47. doi:10.1016/j.cell.2012.03.009
2. Fine HA. New strategies in glioblastoma: exploiting the new biology. Clin Cancer Res. 2015;21(9):1984–1988.
3. Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, Phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–1532.
4. Herrlinger U, Tzaridis T, Mack F, et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): a randomised, open-label, phase 3 trial. Lancet. 2019;393(10172):678–688.
5. Perry JR, Laperriere N, O’Callaghan CJ, et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N Engl J Med. 2017;376(11):1027–1037.
6. Chamberlain MC. Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev Neurother. 2010;10(10):1537–1544.
7. Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3(3):198–210.
8. Chen X, Zhang M, Gan H, et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat Commun. 2018;9(1):2949.
9. Ganipineni LP, Danhier F, Préat V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J Control Release. 2018;281:42–57.
10. Zhao M, van Straten D, Broekman MLD, Préat V, Schiffelers RM. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics. 2020;10(3):1355–1372.
11. Kinoh H, Quader S, Shibasaki H, et al. Translational Nanomedicine Boosts Anti-PD1 Therapy to Eradicate Orthotopic PTEN-Negative Glioblastoma. ACS Nano. 2020;14(8):10127–10140.
12. Lin T, Zhao P, Jiang Y, et al. Blood-Brain-Barrier-Penetrating Albumin Nanoparticles for Biomimetic Drug Delivery via Albumin-Binding Protein Pathways for Antiglioma Therapy. ACS Nano. 2016;10(11):9999–10012.
13. Fana M, Gallien J, Srinageshwar B, Dunbar GL, Rossignol J. PAMAM Dendrimer Nanomolecules Utilized as Drug Delivery Systems for Potential Treatment of Glioblastoma: a Systematic Review. Int J Nanomedicine. 2020;15:2789–2808.
14. Moura RP, Martins C, Pinto S, Sousa F, Sarmento B. Blood-brain barrier receptors and transporters: an insight on their function and how to exploit them through nanotechnology. Expert Opin Drug Delivery. 2019;16(3):271–285.
15. Wadajkar AS, Dancy JG, Hersh DS, et al. Tumor-targeted nanotherapeutics: overcoming treatment barriers for glioblastoma. WIREs Nanomed Nanobiotechnol. 2017;9(4):e1439.
16. Xie J, Shen Z, Anraku Y, Kataoka K, Chen X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials. 2019;224:119491.
17. Sun P, Xiao Y, Di Q, et al. Transferrin Receptor-Targeted PEG-PLA Polymeric Micelles for Chemotherapy Against Glioblastoma Multiforme. Int J Nanomedicine. 2020;15:6673–6688.
18. Lakkadwala S, Dos Santos Rodrigues B, Sun C, Singh J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J Control Release. 2019;307:247–260.
19. Luo M, Lewik G, Ratcliffe JC, et al. Systematic Evaluation of Transferrin-Modified Porous Silicon Nanoparticles for Targeted Delivery of Doxorubicin to Glioblastoma. ACS Appl Mater Interfaces. 2019;11(37):33637–33649.
20. Shen Z, Liu T, Yang Z, et al. Small-sized gadolinium oxide based nanoparticles for high-efficiency theranostics of orthotopic glioblastoma. Biomaterials. 2020;235:119783.
21. Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012;161(2):429–445.
22. Jhaveri A, Deshpande P, Pattni B, Torchilin V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J Control Release. 2018;277:89–101.
23. Dal Magro R, Ornaghi F, Cambianica I, et al. ApoE-modified solid lipid nanoparticles: a feasible strategy to cross the blood-brain barrier. J Control Release. 2017;249:103–110.
24. Arora S, Layek B, Singh J. Design and Validation of Liposomal ApoE2 Gene Delivery System to Evade Blood–Brain Barrier for Effective Treatment of Alzheimer’s Disease. Mol Pharm. 2021;18(2):714–725.
25. Gu W, Meng F, Haag R, Zhong Z. Actively targeted nanomedicines for precision cancer therapy: concept, construction, challenges and clinical translation. J Control Release. 2021;329(10):676–695.
26. van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM. Comparison of five different targeting ligands to enhance accumulation of liposomes into the brain. J Control Release. 2011;150(1):30–36.
27. Oller-Salvia B, Sánchez-Navarro M, Giralt E, Teixidó M. Blood–brain barrier shuttle peptides: an emerging paradigm for brain delivery. Chem Soc Rev. 2016;45(17):4690–4707.
28. Zhao P, Wang Y, Kang X, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9(10):2674–2689.
29. Qiao C, Yang J, Shen Q, et al. Traceable Nanoparticles with Dual Targeting and ROS Response for RNAi-Based Immunochemotherapy of Intracranial Glioblastoma Treatment. Adv Mater. 2018;30(18):1705054.
30. Li B, Xiao H, Cai M, et al. Molecular Probe Crossing Blood–Brain Barrier for Bimodal Imaging–Guided Photothermal/Photodynamic Therapies of Intracranial Glioblastoma. Adv Func Mater. 2020;30(12):1909117.
31. Kim G, Kim M, Lee Y, Byun JW, Hwang DW, Lee M. Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J Control Release. 2020;317:273–281.
32. Jiang Y, Yang W, Zhang J, Meng F, Zhong Z. Protein Toxin Chaperoned by LRP-1-Targeted Virus-Mimicking Vesicles Induces High-Efficiency Glioblastoma Therapy In Vivo. Adv Mater. 2018;e1800316.
33. Kurzrock R, Gabrail N, Chandhasin C, et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol Cancer Ther. 2012;11(2):308–316.
34. Gao S, Tian H, Xing Z, et al. A non-viral suicide gene delivery system traversing the blood brain barrier for non-invasive glioma targeting treatment. J Control Release. 2016;243:357–369.
35. Shi Y, Jiang Y, Cao J, et al. Boosting RNAi therapy for orthotopic glioblastoma with nontoxic brain-targeting chimaeric polymersomes. J Control Release. 2018;292:163–171.
36. Jiang Y, Zhang J, Meng F, Zhong Z. Apolipoprotein E Peptide-Directed Chimeric Polymersomes Mediate an Ultrahigh-Efficiency Targeted Protein Therapy for Glioblastoma. ACS Nano. 2018;12(11):11070–11079.
37. Qin H, Jiang Y, Zhang J, Deng C, Zhong Z. Oncoprotein Inhibitor Rigosertib Loaded in ApoE-Targeted Smart Polymersomes Reveals High Safety and Potency against Human Glioblastoma in Mice. Mol Pharm. 2019;16(8):3711–3719.
38. Zou Y, Meng F, Deng C, Zhong Z. Robust, tumor-homing and redox-sensitive polymersomal doxorubicin: a superior alternative to Doxil and Caelyx? J Control Release. 2016;239:149–158.
39. Cheng R, Meng F, Deng C, Zhong Z. Bioresponsive polymeric nanotherapeutics for targeted cancer chemotherapy. Nano Today. 2015;10(5):656–670.
40. Fang H, Zhao X, Gu X, et al. CD44-Targeted Multifunctional Nanomedicines Based on a Single-Component Hyaluronic Acid Conjugate with All-Natural Precursors: construction and Treatment of Metastatic Breast Tumors in Vivo. Biomacromolecules. 2020;21(1):104–113.
41. Fang Y, Jiang Y, Zou Y, et al. Targeted glioma chemotherapy by cyclic RGD peptide-functionalized reversibly core-crosslinked multifunctional poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. Acta Biomater. 2017;50:396–406.
42. Fang Y, Yang W, Cheng L, Meng F, Zhang J, Zhong Z. EGFR-targeted multifunctional polymersomal doxorubicin induces selective and potent suppression of orthotopic human liver cancer in vivo. Acta Biomater. 2017;64:323–333.
43. Zhang N, Xia Y, Zou Y, et al. ATN-161 Peptide Functionalized Reversibly Cross-Linked Polymersomes Mediate Targeted Doxorubicin Delivery into Melanoma-Bearing C57BL/6 Mice. Mol Pharm. 2017;14(8):2538–2547.
44. Fan Q, Liu Y, Cui G, Zhong Z, Deng C. Brain delivery of Plk1 inhibitor via chimaeric polypeptide polymersomes for safe and superb treatment of orthotopic glioblastoma. J Control Release. 2021;329:1139–1149.
45. Lv S, Li M, Tang Z, et al. Doxorubicin-loaded amphiphilic polypeptide-based nanoparticles as an efficient drug delivery system for cancer therapy. Acta Biomaterialia. 2013;9(12):9330–9342.
46. Chan KH, Lee WH, Ni M, Loo Y, Hauser CAE. C. Terminal Residue of Ultrashort Peptides Impacts on Molecular Self-Assembly, Hydrogelation, and Interaction with Small-Molecule Drugs. Sci Rep. 2018;8(1):17127.
47. Zhao N, Woodle MC, Mixson AJ. Advances in delivery systems for doxorubicin. J Nanomed Nanotechnol. 2018;9(5):519.
48. Giese A, Bjerkvig R, Berens M, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21(8):1624–1636.
49. Zhong Y, Zhang J, Cheng R, et al. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Control Release. 2015;205:144–154.
50. Zou Y, Liu Y, Yang Z, et al. Effective and Targeted Human Orthotopic Glioblastoma Xenograft Therapy via a Multifunctional Biomimetic Nanomedicine. Adv Mater. 2018;30(51):e1803717.
51. Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–1642.
52. Ran D, Mao J, Zhan C, et al. d-Retroenantiomer of quorum-sensing peptide-modified polymeric micelles for brain tumor-targeted drug delivery. ACS Appl Mater Interfaces. 2017;9(31):25672–25682.
53. Fan K, Jia X, Zhou M, et al. Ferritin nanocarrier traverses the blood brain barrier and kills glioma. ACS Nano. 2018;12:4105–4115.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.