")
Back to Journals » OncoTargets and Therapy » Volume 13
Downregulation of USP34 Inhibits the Growth and Migration of Pancreatic Cancer Cells via Inhibiting the PRR11
Authors Lin C, Xia J, Gu Z, Meng Y, Gao D, Wei S
Received 27 August 2019
Accepted for publication 10 January 2020
Published 18 February 2020 Volume 2020:13 Pages 1471—1480
DOI https://doi.org/10.2147/OTT.S228857
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Changjie Lin,* Jing Xia,* Zhiwei Gu, Yunpeng Meng, Dekang Gao, Shaohua Wei
Department of General Surgery, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shaohua Wei
Department of General Surgery, The Second Affiliated Hospital of Soochow University, No. 1055, Sanxiang Road, Suzhou, Jiangsu 215000, People’s Republic of China
Email [email protected]
Background: Pancreatic cancer (PC) is a highly lethal malignancy worldwide. Our previous study indicated that overexpression of USP34 could promote tumor growth in PC cells. Therefore, this study aimed to further investigate the role of USP34 during the tumorigenesis of PC.
Methods: The level of USP34 in PANC-1 and MiaPaCa-2 cells transfected with USP34-shRNAs was detected by RT-qPCR. Moreover, transwell migration and Annexin V/PI analysis were conducted to detect cell migration and apoptosis, respectively.
Results: In this study, downregulation of USP34 markedly inhibited proliferation and migration, and induced apoptosis in PANC-1 cells. Moreover, silencing of USP34 obviously downregulated the levels of PRR11 and p-p38 in PANC-1 cells. An in vivo study in nude mice bearing PANC-1 cell xenografts confirmed these results.
Conclusion: Downregulation of USP34 could inhibit proliferation and migration in PANC-1 cells via inhibiting PRR11, and inactivating p38 MAPK signaling. Therefore, USP34 might be a potential therapeutic target for the treatment of PC.
Keywords: pancreatic cancer, ubiquitin-specific protease USP34, proline-rich 11, apoptosis
Introduction
Pancreatic cancer (PC) is a highly lethal malignancy and is one of the leading causes of cancer mortality worldwide.1 PC is characterized by low survival rate, insidious onset, and rapid progression.2 In addition, family history, high-fat diet, smoking, chronic pancreatitis and type 2 diabetes mellitus are the established risk factors for PC.3–5 Meanwhile, resection, chemotherapy, and radiation are the main treatment methods for patients with PC.6 However, the prognosis of PC remains poor, recurrence within 1 year occurs in approximately 50% of the patients with PC after surgery.7 To improve the health outcomes for patients with PC, it is important to search novel therapeutic targets and explore the molecular mechanisms of PC.
Ubiquitin is a 76-residue protein, which could regulate cellular processes through its post-translational attachment (ubiquitinylation) to other lysine residue on target proteins.8 Ubiquitination could regulate protein interactions via promoting protein degradation.9 Ubiquitin proteases could cleave off ubiquitin moieties and remove ubiquitin moieties from proteins.10 Ubiquitin-specific proteases (USPs) subfamily is a main class of ubiquitin proteases in human.10 In addition, ubiquitin-specific protease USP34 could encode a kind of deubiquitinating enzyme, which is located on chromosome 2p15.11 In the previous study, we found that USP34 played a pro-survival role in PC cells via activating AKT and PKC pathways.12 However, the mechanisms by which USP34 regulated the apoptosis of PC cells needs further investigation.
Proline-rich 11 (PRR11) is a cancer-associated gene, which is located on chromosome 17q22.13 Previous studies indicated that the expression of PRR11 was significantly upregulated in sever tumors compared with normal tissues, such as lung cancer, esophageal squamous cell carcinoma and pancreatic cancer.14–16 Chen et al indicated that PRR11 plays important role during the development and prognosis of cancers.17 Therefore, this study aimed to investigate whether USP34 plays the pro-survival role in PC cells via regulating the expression of PRR11.
Materials and Methods
Cell Culture
The human pancreatic cancer cell lines PANC-1 and MiaPaCa-2 were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS, Thermo Fisher Scientific), and 1% antibiotics (penicillin and streptomycin), and incubated at 37°C in a humidified incubator (5% CO2).
Lentivirus Production and Cell Transfection
Lentiviral expressing short-hairpin USP34 (USP34-shRNA1 and USP34-shRNA2) were purchased from GenePharma (Shanghai, China). The shRNA target sequences were USP34-shRNA1: forward, 5ʹ- CACCGCATATAATCCTAGACCTTTCCGAAGAAAGGTCTAGGATTATATGC-3ʹ; reverse, 5ʹ- AAAAGCATATAATCCTAGACCTTTCTTCGGAAAGGTCTAGGATTATATGC-3ʹ. USP34-shRNA2: forward, 5ʹ- CACCGGATTGAACTGTTGACGAAACCGAAGTTTCGTCAACAGTTCAATCC-3ʹ; reverse, 5ʹ- AAAAGGATTGAACTGTTGACGAAACTTCGGTTTCGTCAACAGTTCAATCC-3ʹ. The PRR11 sequence was synthesized by GenePharma, and then cloned into the pSuper (lentiviral expression vector). The USP34-shRNA1, USP34-shRNA2, and lenti-PRR11 plasmids were transfected into 293T cells respectively and incubated at 32°C for 48 h. Later on, the supernatant containing the retroviral particles were collected.
PANC-1 and MiaPaCa-2 cells (4 × 105 cells per well) were plated into cell plates (60 mm) overnight, respectively. Then, USP34-shRNA1 and USP34-shRNA2 supernatants were added directly to PANC1 cells and MiaPaCa-2 cells, respectively. Meanwhile, lenti-PRR11 supernatants were added directly to PANC1 cells. Stable PANC-1 and MiaPaCa-2 cells were then selected by puromycin (2.5 μg/mL, Sigma-Aldrich, St. Louis, MO, USA) for another 72 h.
CCK-8 Assay
Cell viability was determined by using cell counting kit-8 (CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD, USA) in accordance with the manufacturer’s instruction. A total of 5 × 103 PANC-1 or MiaPaCa-2 cells were plated into 96-well plates, respectively. Then, the cells were incubated at 37°C overnight. After that, cells were infected with USP34-shRNA1, USP34-shRNA2, or lenti-PRR1 for 72 h, respectively. Then, the culture medium was replaced by fresh DMEM medium containing 10% CCK-8 reagent and incubated for another 2 h at 37°C. The absorbance at 450 nm of each well was measured using a microplate reader (Bio-Rad, Hercules, CA, USA).
Real-Time Quantitative PCR
Total RNA from PANC-1 and MiaPaCa-2 cells were purified by using TRIzol Reagent (Thermo Fisher Scientific). The cDNA reverse transcription kit (Takara Bio Inc. Shiga, Japan) was used to synthesize the cDNA following the manufacturer’s instructions. QRT-PCR analysis was performed on Applied Biosystems Prism 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using a SYBR® Premix Ex TaqTM II (Takara) according to the manufacturer’s instructions. The gene expression was normalized to that of GAPDH using comparative cycle threshold (CT) method. The primer sequences used were as follows: USP34-F: 5ʹ-GCTGAATGTAGATTTGTTGGCC-3ʹ; USP34-R: 5ʹ- ACAGCCTGTCTTGCCTCAGG-3ʹ. PRR11-F: 5ʹ-CACCCCTGGAGACTCCAAAG-3ʹ; PRR11-R: 5ʹ-CTTTCGGCGATGGTATAAGCT-3ʹ β-actin-F: 5ʹ-GTCCACCGCAAATGCTTCTA-3ʹ and β-actin-R: 5ʹ- TGCTGTCACCTTCACCGTTC-3ʹ.
Western Blotting
Total protein was extracted from PC cells using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific). In addition, protein concentration was detected by using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins (30 μg per lane) were separated by 10% SDS‑PAGE electrophoresis and then transferred onto a polyvinylidene fluoride membrane (PVDF, Millipore, Billerica, MA, USA). After that, membranes were blocked with 5% non-fat milk in Tween 20/Tris-buffered saline (TBST) at room temperature for 1 h and then incubated with the primary antibodies overnight at 4°C: anti-USP34 (1:1000, Abcam), anti-PRR11 (1:1000, Abcam), anti-p-ERK (1:1000, Abcam), anti-ERK (1:1000, Abcam), anti-p-p38 (1:1000, Abcam), anti-p38 (1:1000, Abcam) and anti-β-actin (1:1000, Abcam). Later on, the membranes were incubated with anti-rabbit horseradish peroxidase-conjugated IgG secondary antibody (1:5000, Abcam) at room temperature for 1 h. Chemiluminescence Substrate Reagent Kit (Thermo Fisher Scientific) was used to develop signal on a ChemiDoc XRS + system (Bio-Rad, Berkeley, CA, USA). β-Actin was used as an endogenous control.
Flow Cytometric Analysis of Cell Apoptosis
PANC-1 cells were washed twice with pre-cold PBS (Thermo Fisher Scientific). Then, the cells were resuspended in 100 μL binding buffer (Thermo Fisher Scientific). After that, the cells were stained with 5 μL of propidium iodide and 5 μL of Annexin V-FITC for 15 min in darkness at room temperature. Subsequently, apoptotic cells were assessed using a flow cytometer (FACScan™, BD Biosciences, Franklin Lakes, NJ, USA).
Transwell Migration Assay
PANC-1 cells (5 × 104 cells) were suspended in 200 μL serum-free DMEM medium and seeded into the upper chamber of 24-well transwell insert (Corning New York, NY, USA). Meanwhile, DMEM medium with 10% FBS (600 μL) was added into the lower compartment to induce cell migration. After 24 h of incubation, cells attached to the upper surface of the chamber were removed using cotton swabs. Then, the cells that migrated through the Transwell membrane were stained with 0.2% crystal violet. After washing with PBS, cells were observed using a laser confocal microscope (Olympus).
Animal Study
Four- to six-week-old nude mice (BALB/c, 15–20 g) were purchased from the Shanghai SLAC Animal Center (Shanghai, China) and animals were allowed free access to normal food and water following the guidelines of the Institutional Animal Care and Use Committee. Animals were randomly divided into three groups (n = 3 each): control, USP34-shRNA1, and USP34-shRNA1 + Lenti-PRR11 group. To establish PC xenograft model, USP34-shRNA1-infected PANC-1 cells, or USP34-shRNA1 and Lenti-PRR11-co-infected PANC-1 cells (5 × 106 cells per mouse) were resuspended in 50 μL PBS and injected into the left flank of nude mice, respectively. Tumor volume in each group was monitored every week with a caliper and analyzed using the formula: TV = (length × width2)/2. All nude mice were sacrificed in 4 weeks, and the entire tumors were dissected out and weighed. All animal experiments were performed according to the rules approved by the Institutional Ethical Committee of the Second Affiliated Hospital of Soochow University. All animal experiments in this study were approved Institutional Ethical Committee of the Second Affiliated Hospital of Soochow University.
Immunohistochemistry (IHC) Assay
PC tissues were embedded in paraffin and then cut into 5 μm slices. Later on, the sections were incubated with the anti-Ki67 (Abcam) primary antibodies overnight at 4°C. Then, the slides were incubated with peroxidase combined secondary antibody (Abcam) for 20 min. Then, DAB solution was added for signal reactions according to the manufacturer’s specification. Ki67 staining was imaged under a fluorescence microscope (Olympus CX23 Tokyo, Japan).
Statistical Analysis
All data were repeated in triplicate. Data are presented as mean ± standard error (S. D.). All statistical analyses were performed using GraphPad Prism software (version 7.0, La Jolla, CA, USA). One-way analysis of variance (ANOVA) and Tukey’s tests were carried out for multiple group comparisons. A P value <0.05 was considered to be statistically significant.
Results
Downregulation of USP34 Inhibited the Proliferation of PC Cells
To investigate the role of USP34 in PC, human PC cell lines PANC-1 and MiaPaCa-2 were infected with USP34-shRNA1 and USP34-shRNA2, respectively. As shown in Figure 1A and C, the level of USP34 was decreased in the most following infection with USP34-shRNA1 in two PC cells, compared with the NC group. Therefore, USP34-shRNA1 was utilized in the following experiments. Next, CCK-8 assay was performed to determine the viability of PC cells after infection with USP34-shRNA1. As indicated in Figure 1B and D, downregulation of USP34 significantly inhibited the proliferation of PANC-1 and MiaPaCa-2 cells and PANC-1 cells were more sensitive to USP34-shRNA1 transfection. Thus, PANC-1 cells were utilized in the following studies. These results suggested that downregulation of USP34 could inhibit the proliferation of PC cells.
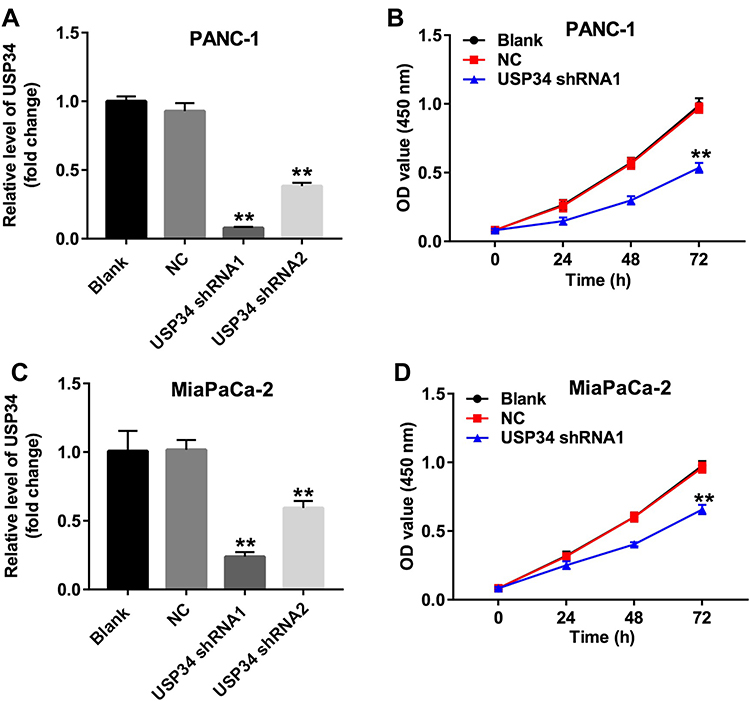 |
Figure 1 Downregulation of USP34 inhibited the proliferation of PC cells. (A) PANC-1 cells were transfected with USP34-shRNA1 or USP34-shRNA2 for 72 h. The level of USP34 in PANC-1 cells was detected using RT-qPCR. (B) PANC-1 cells were transfected with NC or USP34-shRNA1 for 24, 48 and 72 h. CCK-8 assay was used to determine cell viability. (C) MiaPaCa-2 cells were transfected with USP34-shRNA1 or USP34-shRNA2 for 72 h. The level of USP34 in MiaPaCa-2 cells was detected using RT-qPCR. (D) MiaPaCa-2 cells were transfected with NC or USP34-shRNA1 for 24, 48 and 72 h. CCK-8 assay was used to determine cell viability. **P<0.01 vs NC group. |
Downregulation of USP34 Inhibited the Proliferation of PC Cells via Inhibiting PRR11 and Inactivating P38 Signaling
PRR11 was is a newly identified oncogene, which could promote the proliferation of PC cells.18 Therefore, we aimed to explore whether downregulation of USP34 could inhibit the proliferation of PANC-1 cells via regulating the expression of PRR11. As shown in Figure 2A, the level of PRR11 was obviously upregulated in PANC-1 cells following infection with lenti-PRR11. However, overexpression of PRR11 had no effect on cell viability and apoptosis (Supplementary Figure 1A and 1B). In addition, Western blot results indicated that upregulation of PRR11 had no effect on the expression of USP34 in PANC-1 cells (Figure 2B and C). In contrast, downregulation of USP34 significantly decreased the levels of PRR11 and p-p38 in PANC-1 cells, which were markedly reversed after infection with lenti-PRR11 (Figure 2B, D and E). Meanwhile, the level of p-ERK in PANC-1 cells was not altered following infection with USP34-shRNA1 or lenti-PRR11 (Figure 2B and F). These data indicated that downregulation of USP34 could inhibit the proliferation of PC cells via inhibiting the PRR11, and inactivating p38 signaling.
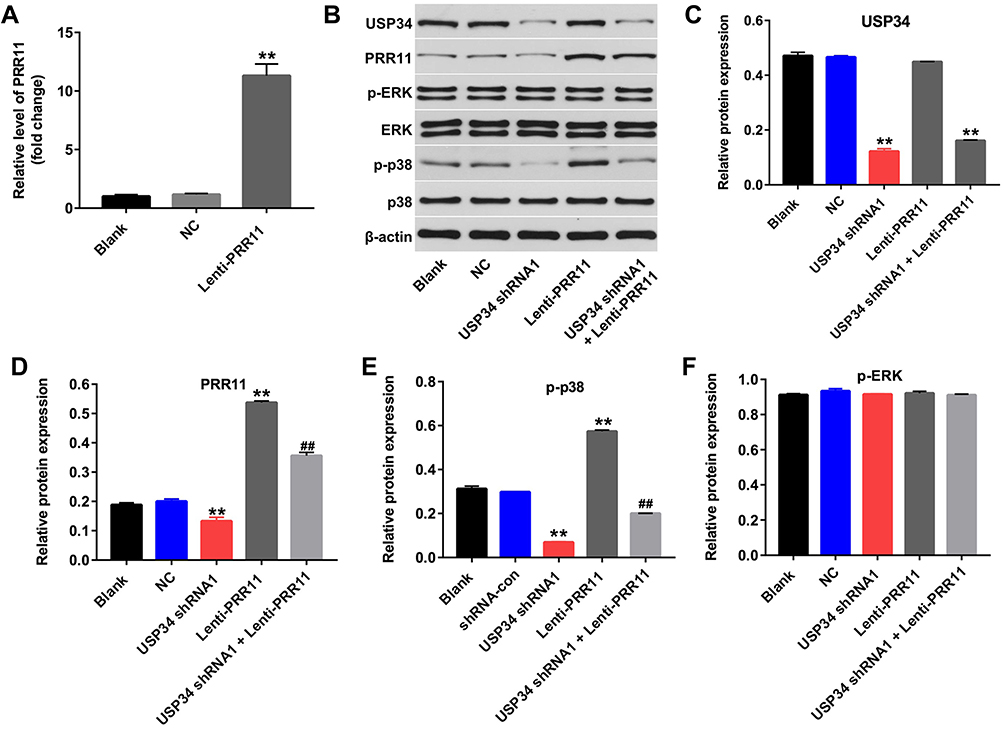 |
Figure 2 Downregulation of USP34 inhibited the proliferation of PC cells via inhibiting PRR11, and inactivating p38 signaling. (A) PANC-1 cells were transfected with NC or lenti-PRR11 for 72 h. The level of PRR11 in PANC-1 cells was detected using RT-qPCR. (B) PANC-1 cells were transfected with USP34-shRNA1 or lenti-PRR11 for 72 h. Meanwhile, PANC-1 cells were co-transfected with USP34-shRNA1 and lenti-PRR11 for 72 h. Expression levels of USP34, PRR11, p-ERK, ERK, p-p38 and p38 in PANC-1 cells were detected with Western blotting. (C, D) The relative expressions of USP34 and PRR11 in PANC-1 cells were quantified via normalization to β-actin. (E, F) The relative expressions of p-ERK, p-p38 in PANC-1 cells were quantified via normalization to ERK and p38, respectively. **P<0.01 vs NC group, ##P<0.01 vs USP34-shRNA1 group. |
Downregulation of USP34 Inhibited the Migration and Induced the Apoptosis in PANC-1 Cells
To further explore the function of USP34 in PC, flow cytometric analysis was used. As indicated in Figure 3A and B, downregulation of USP34 significantly induced apoptosis in PANC-1 cells, which was significantly reversed after infection with lenti-PRR11. In addition, silencing of USP34 obviously inhibited the migration ability of PANC-1 cells. However, the inhibitory effect of USP34-shRNA1 on the migration of PANC-1 cells was markedly reversed following infection with lenti-PRR11 (Figure 3C and D). These results indicated that downregulation of USP34 could inhibit the migration and induce the apoptosis in PANC-1 cells.
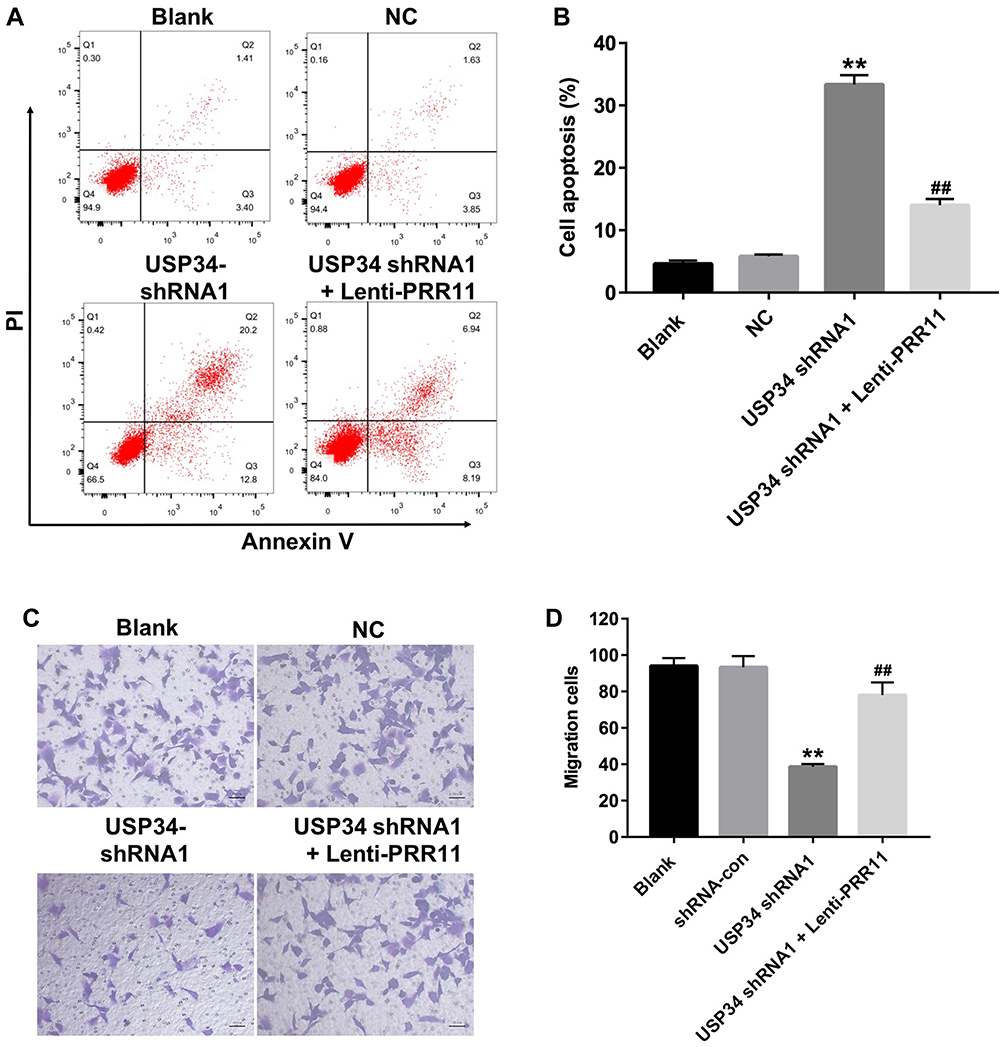 |
Figure 3 Downregulation of USP34 inhibited migration and induced apoptosis in PANC-1 cells. PANC-1 cells were transfected with USP34-shRNA1 or co-transfected with USP34-shRNA1 and lenti-PRR11 for 72 h. (A, B) Apoptotic cells were detected with Annexin V and PI double staining. (C, D) Transwell invasion assay was used to detect the cell invasion. **P<0.01 vs NC group, ##P<0.01 vs USP34-shRNA1 group. |
Downregulation of USP34 Inhibited the Tumor Growth of PANC-1 Xenograft in vivo
PANC-1 xenograft tumor model was established to further investigate the role of USP34 on tumor growth of PC in vivo. As indicated in Figure 4A–C, downregulation of USP34 significantly suppressed the tumor growth of PANC-1 xenograft. However, when the mice treated with USP34-shRNA1 plus lenti-PRR11, the unfavorable role of USP34-shRNA1 on tumor volume and tumor weight was abolished by PRR11 overexpression. In addition, the results of immunohistochemistry assay indicated that downregulation of USP34 significantly decreased the number of Ki67 positive PANC-1 cells in tumor tissues, which was markedly reversed after infection with lenti-PRR11 (Figure 4D and E). These data indicated that downregulation of USP34 could inhibit the tumor growth of pancreatic tumors in vivo.
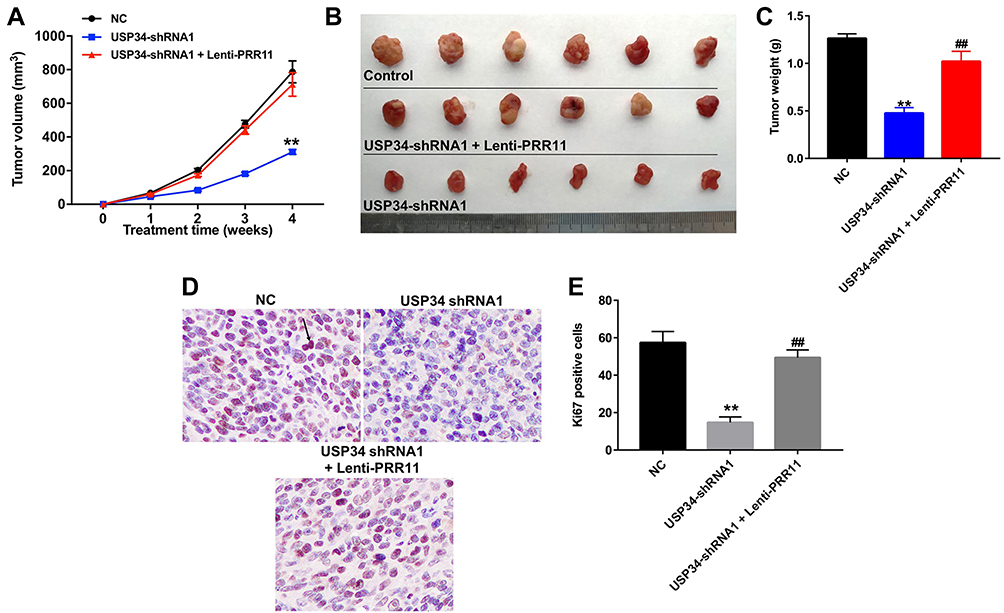 |
Figure 4 Downregulation of USP34 inhibited the tumor growth of PANC-1 xenograft in vivo. PANC-1 cells were subcutaneously implanted with USP34-shRNA1 infected PANC-1 cells, or USP34-shRNA1 and lenti-PRR11 co-infected PANC-1 cells to establish a xenograft model of PC. (A) Tumor volumes of animals were monitored and measured with calipers weekly. (B) Representative photographs of xenograft tumor were captured after 4 weeks. (C) Tumor weights in each animal were calculated. (D and E) Representative images of Ki67 expression of xenograft tumor were photographed. (800× magnification). Arrow: Ki67-positive cell. **P<0.01 vs NC group, ##P<0.01 vs USP34-shRNA1 group. |
Downregulation of USP34 Inhibited the Tumor Growth of PANC-1 Xenograft in vivo via Inhibiting PRR11, and Inactivating P38 Signaling
Next, Western blotting assay was used to detect the expression of USP34 in vivo. As revealed in Figure 5A and B, downregulation of USP34 significantly decreased the level of USP34 in tumor tissues. In addition, downregulation of USP34 significantly downregulated the levels of PRR11, p-p38, p-Akt, p-mTOR and p-PKC in tumor tissues, which were obviously reversed in the presence of lenti-PRR11 (Figure 5A, C and D, Supplementary Figure 2A, 2B, 2C and 2D). Meanwhile, the levels of p-JNK in PANC-1 cells were not altered by USP34-shRNA1 or USP34-shRNA1 plus lenti-PRR11 (Supplementary Figure 2A and 2E). These data illustrated that downregulation of USP34 could inhibit the tumor growth of PANC-1 xenograft in vivo via inhibiting the PRR11, and inactivating p38, Akt/mTOR and PKC signaling pathways.
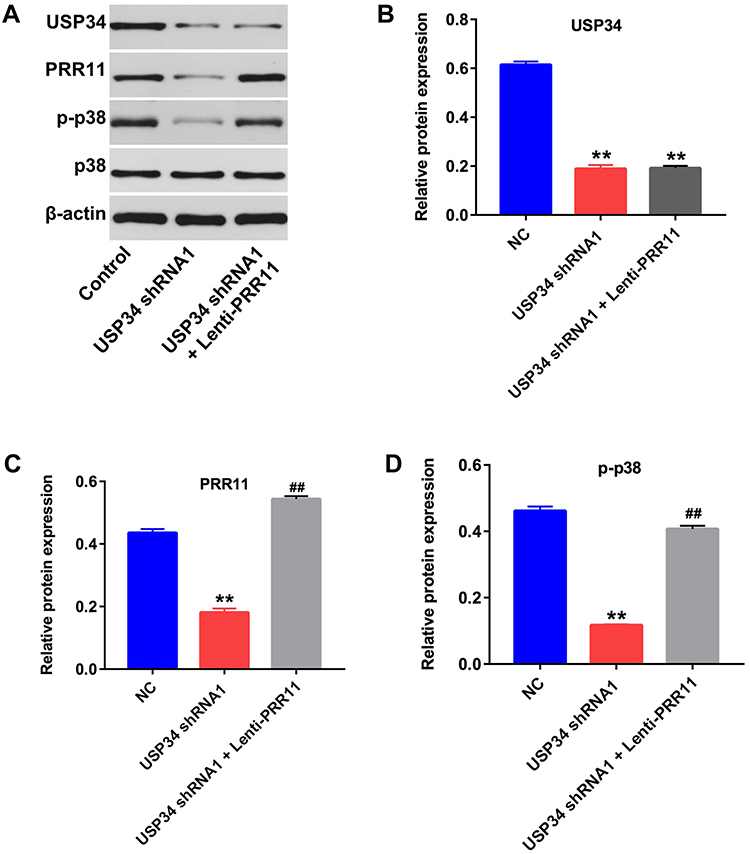 |
Figure 5 Downregulation of USP34 inhibited the tumor growth of PANC-1 xenograft in vivo via inhibiting PRR11 and inactivating p38 signaling. (A) Expression levels of USP34, PRR11, p-p38 and p38 in tumor tissues were detected with Western blotting. (B, C) The relative expressions of USP34 and PRR11 in tumor tissues were quantified via normalization to β-actin. (D) The relative expressions of p-p38 in tumor tissues were quantified via normalization to p38. **P<0.01 vs NC group, ##P<0.01 vs USP34-shRNA1 group. |
Discussion
Our previous study indicated that USP34 could promote human PC cell survival via activating AKT and PKC pathways.12 In this study, we found that downregulation of USP34 could inhibit the tumor growth in PANC-1 cells via inhibiting the PRR11 and inactivating p38 signaling in vitro and in vivo. To sum up, we demonstrated that USP34 plays an oncogenic role in PC.
Ubiquitin–proteasome system is responsible for the degradation of the majority of proteins in eukaryotic cells.9,19 The ubiquitin-specific proteases are also called deubiquitinating enzymes, which could remove ubiquitin conjugates from target proteins.20 The deubiquitinating enzymes may play an oncogenic or a tumor-suppressor functions in different types of cancers.21 Previous study indicated that the balance between ubiquitination and deubiquitination is important for cellular survival.22 In our previous study, we found that the expression of USP34 was markedly upregulated in PC tissues, compared with para-cancerous tissue samples.12 Moreover, downregulation of USP34 could inhibit proliferation and migration, and induce apoptosis in PANC-1 cells. Meanwhile, downregulation of USP34 could inhibit the tumor growth of PANC-1 xenograft in vivo. These data indicated that downregulation of USP34 could inhibit the tumorigenesis of PC cells in vitro and in vivo.
However, the mechanisms by which USP34 regulated the tumorigenesis of PC cells have not yet been elucidated. It has been shown that the sequence of PRR11 protein included the ubiquitination site and phosphorylation sites.23 Evidence has been shown that PRR11 was served as an oncogene gene, which plays an important role in tumorigenesis.24 Hu et al found that MAPK signaling could maintain PRR11 protein stability via suppression of ubiquitin–proteasome activity.25 In this study, we found that downregulation of USP34 could inhibit the level of PRR11 in PANC-1 cells, while overexpression of PRR11 had no effects on the expression of USP34 in PANC-1 cells. We speculated that downregulation of USP34 could suppress the level of PRR11 via releasing the ubiquitin–proteasome activity in PANC-1 cells. These data indicated that downregulation of USP34 induced apoptosis of PANC-1 cells via inhibiting PRR11 expression.
MAPK/p38, a member of the MAPK superfamily, which exhibited an important role in cell apoptosis.26 Evidences have been shown that p38 MAPK could function as a tumor suppressor or a oncogene in human cells.27,28 Cao et al indicated resveratrol could inhibit the migration of PC cells via suppression of p38 MAPK signaling.29 In contrast, Li et al indicated that miR-144-3p induced apoptosis in PC cells by suppressing PRR11, and upregulating the expressions of p-JNK and p-p38.18 In this study, downregulation of USP34 significantly suppressed the level of p-p38 in PANC-1 cells, indicating that downregulation of USP34 induced the apoptosis of PC cells via inactivating p38 signaling. Meanwhile, overexpression of PRR11 markedly increased the level of p-p38 in PANC-1 cells, indicating that PRR11 could activate p38 MAPK in PANC-1 cells. Taken together, we indicated that downregulation of USP34 induced apoptosis of PANC-1 cells via inhibiting PRR11, and inactivating p38 signaling.
Evidence has been shown that ERK plays a major role in the proliferation of cancer cells.30 Our data indicated that knockdown of USP34 or overexpression of PRR11 had no effects on the expression of p-ERK in PANC-1 cells. Moreover, downregulation of USP34 could inhibit the proliferation of PC cells. These data indicated that USP34 may not exert its oncogenic effect in PC cells by regulating ERK signaling. In addition, we found that downregulation of USP34 could decrease the levels of p-Akt, p-mTOR and p-PKC in tumor tissues, which was obviously reversed following infection with lenti-PRR11. Therefore, our data suggested that downregulation of USP34 could inhibit the growth of PC cells in vivo via inhibiting the PRR11, and inactivating p38, Akt/mTOR and PKC signaling pathways.
Zhang et al indicated that downregulation of PRR11 could induce autophagy and inhibit the proliferation of non-small-cell lung cancer cells through inactivating Akt/mTOR signaling pathway.31 Therefore, it will be important to investigate whether downregulation of USP34 inhibits the growth of PC cells via inhibiting the PRR11, and then inducing autophagy.
Conclusion
The current study showed that downregulation of USP34 could inhibit the proliferation and migration in PANC-1 cells via inhibiting PRR11, and inactivatingp38 MAPK signaling. Therefore, USP34 might be a potential therapeutic target for the treatment of PC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bai X, Lu D, Lin Y, Lv Y, He L. A seven-miRNA expression-based prognostic signature and its corresponding potential competing endogenous RNA network in early pancreatic cancer. Exp Ther Med. 2019;18(3):1601–1608. doi:10.3892/etm.2019.7728
2. Xia L, Song M, Sun M, Chen W, Yang C. miR-486 promotes Capan-2 pancreatic cancer cell proliferation by targeting phosphatase and tensin homolog deleted on chromosome 10 (PTEN). Front Genet. 2019;10:541. doi:10.3389/fgene.2019.00541
3. Matsubayashi H, Takaori K, Morizane C, Kiyozumi Y. Familial pancreatic cancer and surveillance of high-risk individuals. Gut Liver. 2019;13:498–505. doi:10.5009/gnl18449
4. Wang D, Bi Y, Hu L, et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun Signal. 2019;17(1):19. doi:10.1186/s12964-019-0333-7
5. Wang C, Li J. Pathogenic microorganisms and pancreatic cancer. Gastrointest Tumors. 2015;2(1):41–47. doi:10.1159/000380896
6. Mohammed S, Van Buren G
7. Beger HG, Thorab FC, Liu Z, Harada N, Rau BM. Pathogenesis and treatment of neoplastic diseases of the papilla of Vater: Kausch-Whipple procedure with lymph node dissection in cancer of the papilla of Vater. J Hepatobiliary Pancreat Surg. 2004;11(4):232–238. doi:10.1007/s00534-004-0895-6
8. Su S, Zhang Y, Liu P. Roles of ubiquitination and SUMOylation in DNA damage response. Curr Issues Mol Biol. 2019;35:59–84. doi:10.21775/cimb.035.059
9. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi:10.1152/physrev.00027.2001
10. Engel E, Viargues P, Mortier M, et al. Identifying USPs regulating immune signals in Drosophila: USP2 deubiquitinates Imd and promotes its degradation by interacting with the proteasome. Cell Commun Signal. 2014;12:41. doi:10.1186/1478-811X-12-2
11. Zhao S, Tian Y, Zhang W, et al. An association study between USP34 and polycystic ovary syndrome. J Ovarian Res. 2015;8:30. doi:10.1186/s13048-015-0158-y
12. Gu Z, Lin C, Hu J, et al. USP34 regulated human pancreatic cancer cell survival via AKT and PKC pathways. Biol Pharm Bull. 2019;42(4):573–579. doi:10.1248/bpb.b18-00646
13. Wang C, Yu L, Ren X, et al. The oncogenic potential of PRR11 gene in tongue squamous cell carcinoma cells. J Cancer. 2019;10(11):2541–2551. doi:10.7150/jca.29265
14. Zhou L, Deng ZZ, Li HY, et al. Overexpression of PRR11 promotes tumorigenic capability and is associated with progression in esophageal squamous cell carcinoma. Onco Targets Ther. 2019;12:2677–2693. doi:10.2147/OTT.S180255
15. Huang Y, Ni R, Wang J, Liu Y. Knockdown of lncRNA DLX6-AS1 inhibits cell proliferation, migration and invasion while promotes apoptosis by downregulating PRR11 expression and upregulating miR-144 in non-small cell lung cancer. Biomed Pharmacother. 2019;109:1851–1859. doi:10.1016/j.biopha.2018.09.151
16. Tan S, Jiang Z, Hou A, et al. Expression of PRR11 protein and its correlation with pancreatic cancer and effect on survival. Oncol Lett. 2017;13(6):4117–4122. doi:10.3892/ol.2017.5974
17. Chen Y, Cha Z, Fang W, et al. The prognostic potential and oncogenic effects of PRR11 expression in hilar cholangiocarcinoma. Oncotarget. 2015;6(24):20419–20433. doi:10.18632/oncotarget.3983
18. Li J, Sun P, Yue Z, et al. miR-144-3p induces cell cycle arrest and apoptosis in pancreatic cancer cells by targeting proline-rich protein 11 expression via the mitogen-activated protein kinase signaling pathway. DNA Cell Biol. 2017;36(8):619–626. doi:10.1089/dna.2017.3656
19. Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315(5809):201–205. doi:10.1126/science.1127085
20. Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. doi:10.1016/j.cell.2005.11.007
21. Luise C, Capra M, Donzelli M, et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One. 2011;6(1):e15891. doi:10.1371/journal.pone.0015891
22. McClurg UL, Robson CN. Deubiquitinating enzymes as oncotargets. Oncotarget. 2015;6(12):9657–9668. doi:10.18632/oncotarget.v6i12
23. Larance M, Ahmad Y, Kirkwood KJ, Ly T, Lamond AI. Global subcellular characterization of protein degradation using quantitative proteomics. Mol Cell Proteomics. 2013;12(3):638–650. doi:10.1074/mcp.M112.024547
24. Proline-rich protein 11 regulates epithelial-to-mesenchymal transition to promote breast cancer cell invasion [Retraction]. Int J Clin Exp Pathol. 2016;9(1):407.
25. Hu H, Song Z, Yao Q, et al. Proline-rich protein 11 regulates self-renewal and tumorigenicity of gastric cancer stem cells. Cell Physiol Biochem. 2018;47(4):1721–1728. doi:10.1159/000491005
26. Lu X, Li C, Wang YK, Jiang K, Gai XD. Sorbitol induces apoptosis of human colorectal cancer cells via p38 MAPK signal transduction. Oncol Lett. 2014;7(6):1992–1996. doi:10.3892/ol.2014.1994
27. Kong X, Li M, Shao K, et al. Progesterone induces cell apoptosis via the CACNA2D3/Ca2+/p38 MAPK pathway in endometrial cancer. Oncol Rep. 2019. doi:10.3892/or
28. Chen L, Gong X, Huang M. YY1-activated long noncoding RNA SNHG5 promotes glioblastoma cell proliferation through p38/MAPK signaling pathway. Cancer Biother Radiopharm. 2019;34(9):589–596. doi:10.1089/cbr.2019.2779
29. Cao L, Chen X, Xiao X, Ma Q, Li W. Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and migration of pancreatic cancer cells via suppression of the ERK and p38 MAPK signaling pathways. Int J Oncol. 2016;49(2):735–743. doi:10.3892/ijo.2016.3559
30. Salaroglio IC, Mungo E, Gazzano E, Kopecka J, Riganti C. ERK is a pivotal player of chemo-immune-resistance in cancer. Int J Mol Sci. 2019;20(10):2505. doi:10.3390/ijms20102505
31. Zhang L, Lei Y, Zhang Y, et al. Silencing of PRR11 suppresses cell proliferation and induces autophagy in NSCLC cells. Genes Dis. 2018;5(2):158–166. doi:10.1016/j.gendis.2017.12.003
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.