")
Back to Journals » OncoTargets and Therapy » Volume 12
Downregulation of NSD3 (WHSC1L1) inhibits cell proliferation and migration via ERK1/2 deactivation and decreasing CAPG expression in colorectal cancer cells
Authors Yi L, Yi L, Liu Q, Li C
Received 22 October 2018
Accepted for publication 14 March 2019
Published 21 May 2019 Volume 2019:12 Pages 3933—3943
DOI https://doi.org/10.2147/OTT.S191732
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Lanjuan Yi,1 Lanjie Yi,2 Qing Liu,3 Chen Li4
1Department of gastroenterology, Yantai Shan Hospital, Yantai, Shandong 264001, People’s Republic of China; 2Research Office of Clinical literature, Nanjing University of Chinese Medicine, Nanjing, Jiangsu 210023, People’s Republic of China; 3Department of Nosocomial Infection Control, Xuzhou Hospital of Traditional Chinese Medicine Affiliated to Nanjing University of Chinese Medicine, Xuzhou, Jiangsu 310015, People’s Republic of China; 4Department of Gastroenterology, Xuzhou Chinese Medicine Hospital Affiliated to Nanjing University of Chinese Medicine, Xuzhou, Jiangsu 310015, People’s Republic of China
Purpose: NSD3 (WHSC1L1) is a protein lysine methyltransferase that is recurrently amplified (8p11.23) in several cancer types, and its upregulation is involved in tumor cell proliferation, metastasis, and epithelial-mesenchymal transition (EMT). We aimed to evaluate its potential function as an oncogenic force in colorectal cancer (CRC), and to elucidate relevant mechanisms of its oncogenic activity.
Materials and methods: NSD3 levels were analyzed in human CRC and adjacent normal tissues or cells by Western blot analysis and RT-qPCR. Expression levels of the proteins were detected by Western blot analysis and RT-qPCR.
Results: NSD3 was significantly upregulated in both CRC tissues and cell lines. Knockdown of NSD3 expression resulted in significant decreases in CRC cell proliferation, migration, and EMT process marker proteins vimentin, simultaneously reducing E-cadherin and N-cadherin expression. The opposite results were observed when NSD3 was overexpressed. Additionally, overexpressing of NSD3 dramatically activated the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway and enhanced actin-capping protein (CAPG) expression. Furthermore, the proliferation and migration abilities evidently facilitated by pcDNA3.1(+) expression vector containing full-length CDS of NSD3 (pcDNA3.1(+)-NSD3, or NSD3) were partially decreased after incubation with ERK1/2 signaling pathway inhibitor (PD98059) and/or specific siRNA against CAPG (siCAPG) in SW480 and HT-29 CRC cells.
Conclusion: NSD3 overexpression stimulated CRC cell proliferation and migration through targeting the ERK1/2 signaling pathway and downstream CAPG. Thus, NSD3 could serve as a promising target for anticancer drug development for patients with CRC.
Keywords: colorectal cancer, histone methyltransferase NSD3, extracellular signal-regulated kinase 1/2, actin-capping protein (CAPG)
Plain Language Summary
The purpose of the experiment is to evaluate potential function of NSD3 (WHSC1L1), aprotein lysine methyltransferase, as an oncogenic force in colorectal cancer (CRC), and to elucidate relevant mechanisms of its oncogenic activity. Our invitro studies revealed that the expression of NSD3 was obviously higher in CRC tissues than in normal tissues. Knockdown of NSD3 expression resulted in significant decreases in CRC cell proliferation, migration, and epithelial-mesenchymal transition (EMT) process marker proteins vimentin, simultaneously reducing E-cadherin and N-cadherin expression. The opposite results were observed when NSD3 was overexpressed. NSD3 overexpression in CRC cells facilitated cell proliferation and migration, and promoted cell EMT progress via increasing CAPG expression and ERK1/2 signaling pathway activation. Additionally, we also demonstrated that ERK1/2 signaling pathway could facilitate CAPG-induced proliferation and migration in CRC cells. These results imply NSD3 may become a reliable CRC biomarker for diagnoses and atarget for precise therapy.
Introduction
Colorectal cancer (CRC) is a type of gastrointestinal neoplasm with the highest rates both in incidence and mortality.1 More than 49 million patients worldwide die of CRC each year.2,3 In China, the morbidity of CRC has been increasing since 2000.4 As most CRC cases are diagnosed at metastatic stages, the prognosis of CRC patients is poor.5 Therefore, it is urgent to develop early diagnose and effective intervene preventing tumors from metastasize in CRC.
It has been widely identified that aberrant expression of histone methylation and its regulator histone methyltransferase are closely related to various cancers.6 NSD3, also known as Wolf - Hirschhorn syndrome candidate 1-like 1 (WHSC1L1), is a nuclear protein mapped at chromosome 8p11.23 and functions as a chromatin regulator by modulating the expression of genes through demethylation of lysine 36 on histone H3 (H3K36me2).7 NSD3 plays a critical role in the expansion of various tumors.8 It can interact with NUP98,9 NUT,10 E2F2,11 BRD4, or CHD8,12 resulting in occurrence of tumors or proliferation and invasion of cancer cells in leukemogenesis, NUT midline carcinoma (NMC), and breast cancer. Collectively, NSD3 plays an important role in human carcinogenesis, and it may serve as a potential druggable target for selective CRC therapy in the future. However, the functional mechanisms of NSD3 in CRC have not been clarified yet.
Current findings show that epithelial-mesenchymal transition (EMT) is an important mechanism for the metastasis of malignant tumors.13 By EMT epithelial-derived malignant cells acquire the phenotype of mesenchymal cell and the ability of invasion and metastasis.14 Herein, we investigated the effects of NSD3 on CRC cell proliferation, metastasis and EMT progress, and then explored the molecular mechanism of NSD3 in CRC. Specifically, we examined its effect on the regulation of extracellular signal regulated kinases 1 and 2 (ERK1/2) signaling pathway and actin-capping protein (CAPG), which are strongly associated with tumor cell proliferation, apoptosis, metastasis, and EMT.15–18
Materials and methods
Human CRC tissues and cell lines
In total, CRC tissues (n =24) and adjacent normal tissues (n =26) were collected after surgical resection at the Xuzhou Hospital of Traditional Chinese Medicine Affiliated to Nanjing University of Chinese Medicine (Xuzhou, Jiangsu, China).All samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C until total RNA was extracted. The researches were approved by the Independent Ethics Committee (IEC) of Xuzhou Hospital of Traditional Chinese Medicine Affiliated to Nanjing University of Chinese Medicine, and all patients provided written informed consent. The study methodologies conformed to the standards set by the Declaration of Helsinki.
The human normal colorectal mucosa cell line (FHC) and seven human CRC cell lines, including Lovo, SW480, SW620, HT-29, HCT-116, Caco-2, and SW48, were all purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Hyclone Laboratories, Inc., South Logan, UT, USA) and 100 μg/mL streptomycin sulfate (Gibco, Rockville, MD, USA) in plastic tissue culture plates. All cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
RNA extraction and quantitative real-time reverse transcription PCR (RT-qPCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized from 2 μg of RNA using the PrimeScriptTM RT Reagent Kit (Takara Biotechnology, Dalian, China) according to the manufacturer’s instructions. The sequences for sense (S) and antisense (AS) primers are as follows: human-NSD3-S, 5′-AAG AGC CAC CGC CTG TTA AA-3′, human-NSD3-AS, 5′- GCT GTC ACA AAT GGA GGT GC-3′; human-E-cadherin-S, 5′-CGG GAA TGC AGT TGA GGA TC-3′, human-E-cadherin-AS, 5′-AGG ATG GTG TAA GCG ATG GC-3′; human-N-cadherin-S, 5′-AAG AGG CCG AGA CTT GTG AA-3′, human-N-cadherin-AS, 5′-CAC TGG GGA TAA GGG AAG GT-3′; human-vimentin-S, 5′-GAG AAC TTT GCC GTT GAA GC-3′, human-vimentin-AS, 5′-GCT TCC TGT AGG TGG CAA TC-3′; human-CAPG-S, 5′-GGG GAC TCC TAC CTA GTG CTG-3′, human-CAPG-AS, 5′-CAC CAC CTT CCT GGT ACT TGA-3′. RT-qPCR was conducted using SYBR Premix Ex Taq™ (Takara, Dalian, China) at 95 °C for 30 sec, followed by 40 cycles of 95 °C for 5 sec and 60 °C for 34 sec in the ABI StepOnePlus Real-time PCR system. The relative fold changes in mRNA expression were calculated using the 2−ΔΔCT method.
Protein extraction and western blot analysis
Cells were lysed in radioimmunoprecipitation assay lysis buffer with protease inhibitors (Sigma, St. Louis, MO, USA) on ice for 30 min. The cell lysates were centrifuged at 12,000× g for 30 min at 4 °C. Total proteins from each lysate were separated by SDS-PAGE and transferred onto PVDF membranes and then blocked with 5% nonfat milk for 1 hr. The membranes were then probed with the indicated primary antibodies at 4 °C with gentle shaking overnight and incubated with horseradish peroxidase- (HRP-) conjugated secondary antibodies. Then, the proteins were visualized by chemiluminescence, and signals were quantified by Image J software. Antibodies used in this study are as follows:NSD3 (Cell Signaling Technology (CST), Boston, MA, USA; cat. no. 92056); N-Cadherin (CST, Boston, MA, USA; cat. no. 13116); E-Cadherin (CST, Boston, MA, USA; cat. no. 3159); vimentin (CST, Boston, MA, USA; cat. no. 13116); (CST, Boston, MA, USA; cat. no. 5741); p-ERK1/2 (CST, Boston, MA, USA; cat. no. 4370); ERK1/2 (CST, Boston, MA, USA; cat. no. 4695); CAPG (Abcam, Cambridge, UK; cat. no. ab155688). The dilution factor for antibodies was 1:1000.
Small interfering RNA transfection
NSD3 and CAPG gene sequences were searched in GenBank. Small interfering RNA (siRNA) oligonucleotides targeting NSD3 (siNSD3, sense: 5′- GUA CUG AAA UUC GGA GAG CA-3′ and antisense: 5′-UGU CUC CGA AUU UCA GUA C-3′) and CAPG (siCAPG, sense: 5′- CCC AGA GUG GCU CUC CAU UC-3′ and antisense: 5′-GAA GAC GCC CUG GUU UCU UUG-3′), and an unrelated control siRNA (NC) as a negative control were designed and synthesized by GenDiscovery Biotechnology (Taipei, Taiwan). One day before transfection, SW480 and HT-29 cells were seeded in a 24-well plate at l×106/well, and siRNA were transfected with Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) when the cell abundance was up to 70%~80%. The final concentration of siRNA transfection was 200 nm/L. After transfection for 48 h, the knockdown efficiency was evaluated by Western blot analysis.
Construction and transfection of overexpression vectors NSD3
A pair of primers for the known NSD3 coding sequences was designed as mentioned earlier. The PCR conditions consisted of 5 min at 95 °C 1 cycle, 30 sec at 95 °C, 30 sec at 60 °C, 60 sec at 72 °C, and 7 min at 72 °C 40 cycles. The target genes and pcDNA3.1(+) were connected. pcDNA3.1(+)-NSD3 (NSD3) was transfected into CRC cells.
Cell viability assay
Cell proliferation reagent kit I (MTT) (Roche, Basel, Switzerland) was used to assess cell viability. Transfected cells were plated in each well of a 96-well plate and assessed every 24 h (0 h, 24 h, 48 h, 72 h, 96 h) according to the manufacturer’s instructions. Detected optical density at 450 nm using the ultraviolet spectrophotometer. Cell proliferation was detected using a BrdU proliferation kit (Biovision, Milpitas, CA, USA) in accordance with the manufacturer’s instructions, and positive BrdU-labeled cells were thus detected using a fluorescence microscope (Olympus, Tokyo, Japan) under 100× magnification.
Cell migration assay
To assess the migratory ability of CRC cells, they were plated onto 6-well plates and cultured at 37°C to 80% confluence. The cell layers were scratched linearly using a cell scraper. After 24 h, the number of cells that migrated over the scratched line was calculated under a light microscope (magnifcation, ×100; Nikon Corporation). The mean of random 5 felds was calculated. In addition, the expression levels of EMT marker proteins E-cadherin, N-cadherin, and vimentin were examined by RT-qPCR and Western blot analysis. The cells were allowed to migrate for 72 h at 37 °C.
Statistical analysis
The data are expressed as means ± SD and each experiment was performed in triplicate in this study. After the homogeneity test for variance, comparisons between groups were performed by one-way analysis of variance (ANOVA) using SPSS 13.0 software, and then post hoc test was determined by LSD test. A significant difference was indicated when the p-value <0.05.
Result
NSD3 is upregulated in CRC tissues and cell lines
To assess potential roles of NSD3, we determined the levels of NSD3 in tumors from 24 patients with CRC and 26 samples of adjacent normal colorectal epithelial tissues. As shown in Figure 1A, increased NSD3 mRNA expression was detected in all CRC tissues as compared with adjacent normal tissues. This significantly elevated expression was confirmed at the protein level using three pairs of CRC samples (Figure 1B). Furthermore, to evaluate the baseline expression level of NSD3 in human CRC cell lines, mRNA and protein expression analysis of NSD3 were performed in a panel of 7 CRC cell lines (Lovo, SW480, SW620, HT-29, HCT-116, caco-2, and SW48) by RT-qPCR and Western blot analysis, respectively. Our results evidenced that the expression of NSD3 in seven human CRC cells was significantly higher than that in a normal colorectal mucosa cell line (FHC) at both mRNA and protein levels (Figure 1C and D).
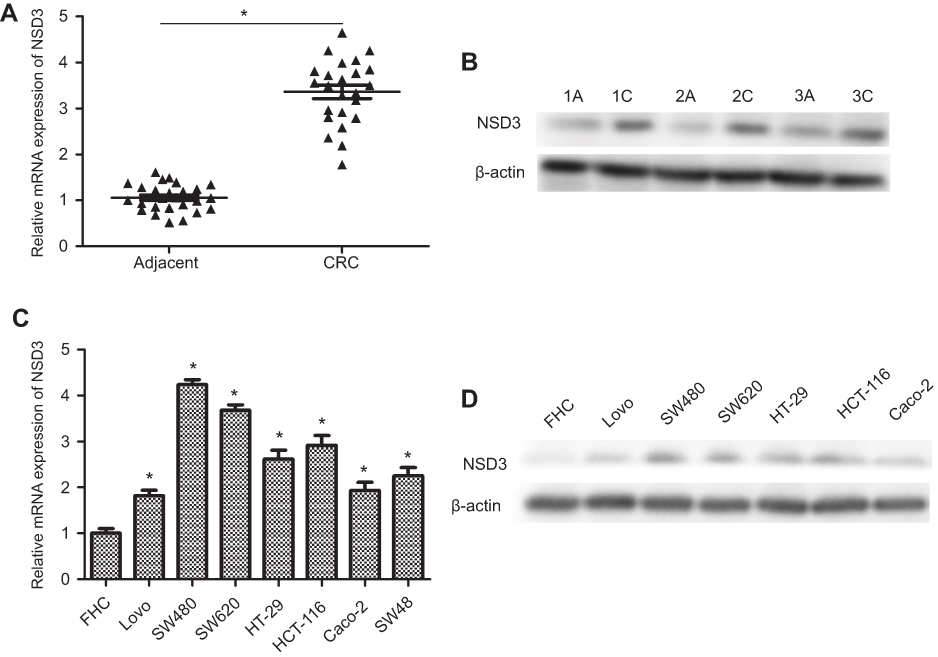 | Figure 1 Expression of NSD3 in CRC patients and human CRC cells. (A) The expression of NSD3 in human CRC tissues (n =24) and adjacent normal tissues (n =26) was compared by RT-qPCR (paired t test) .(B) Random three pairs of CRC samples were used to validate NSD3 expression by Western blot analysis. (C, D) NSD3 and its mRNA expression in seven CRC cell lines (Lovo, SW480, SW620, HT-29, HCT-116, caco-2, and SW48) were detected by RT-qPCR and Western blot analysis. FHC is human normal colonic epithelial cells. The bands were presented as the mean ± SEM. β-actin as a loading control. *P<0.05 vs adjacent normal tissues or FHC.Abbreviations: CRC, colorectal cancer ; RT-qPCR, real-time reverse transcription PCR. |
Knockdown of NSD3 inhibits cell proliferation and migration
To explore the potential role of NSD3 in progression of CRC, we chose to silence NSD3 expression in SW480 and HT-29 cell lines, which had salient and moderate NSD3 expression individually (Figure 1C and D). Western blot analysis revealed that the level of NSD3 was reduced by specific siRNA against NSD3 (siNSD3) compared with a control siRNA (NC) both in SW480 and HT-29 cells (Figure 2A). To examine the important of NSD3 in CRC cell viability and migration, we performed MTT assay 、BrdU assay and scratch wound healing, respectively. As a result, silencing of NSD3 in SW480 and HT-29 cells decreased the ability of cell viability and migration (Figure 2B–D). Likewise, scratch wound healing assay showed that NSD3 knockdown also weakened SW480 and HT-29 cell migration (Figure 2E). Next, the expressions of EMT marker proteins E-cadherin (epithelial), N-cadherin (neural) and vimentin (mesenchymal) were detected using RT-qPCR and Western blot analysis. The results demonstrated that the silencing of NSD3 increased vimentin expression, simultaneously reduced E-cadherin and N-cadherin expression at both protein and mRNA levels (Figure 2F–I). The data above support that NSD3 knockdown decreases the cell proliferation, migration and diminishes EMT in CRC.
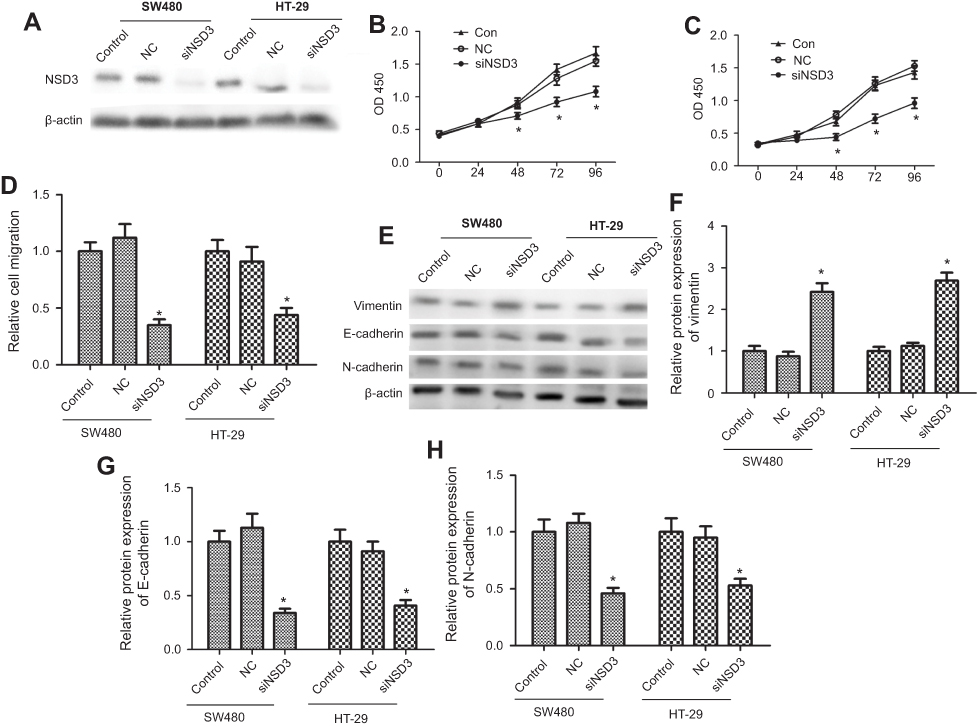 | Figure 2 NSD3 knockdown inhibited CRC cells proliferation and metastasis in vitro. (A) Suppressive capacity of specific siRNA against NSD3 (siNSD3, 50 nM) transfected in SW480 and HT29 cells (5.0×106/cm2) after 48 h. (B, C) MTT assay results respectively showed the trend of SW480 and HT29 cells (5.0×104/cm2) viability within 96 h after silencing NSD3 (siNSD3, 50 nM). (D) Proliferation of SW480 and HT29 cells were evaluated by BrdU incorporation after silencing NSD3. Brdu, DNA fluorescent dye; PI, nuclear fluorescent dye. (E) The migration ability of SW480 and HT29 cells were evaluated by scratch wound healing assay revealing. Wild-type cells and cells transfected with unrelated control siRNA (NC) were used as controls. (F–I) Western blot and RT-qPCR analysis of the E-cadherin, N-cadherin, and vimentin expression in wild-type cells (control), unrelated control cells (NC), and in cells with stable knockdown of NSD3 (siNSD3) after 72 h. Reverse transfection procedure was used to deliver 50 nM siRNA to 5.0×106 cells in a 6-well plate. β-actin as a loading control. The bands were presented as the mean ± SEM. *P<0.05 vs control or NC.Abbreviations: CRC, colorectal cancer; NC, normal control. |
Overexpression of NSD3 facilitates cell proliferation and migration
To confirm that NSD3 affects the proliferation and migration of CRC cells, a pcDNA3.1(+)-NSDS3 (NSD3) was established. Western blot analysis was employed to confirm the expression levels of NSD3 both in SW480 and HT-29 cells. The results showed that NSD3 expression was significantly increased in the NSD3 group compared with the expression in the control vector (pcDNA) and blank groups (Figure 3A). MTT 、BrdU and scratch wound healing assays indicated that NSD3 overexpression in SW480 and HT-29 cells increased the ability of cell viability and migration (Figure 3B–D). Meanwhile, NSD3 overexpression also enhanced SW480 and HT-29 cell migration (Figure 3E). The expression level of EMT marker proteins E-cadherin and N-cadherin were drastically increased while the expression of vimentin was decreased after overexpression NSD3 at both protein and mRNA levels (Figure 3F–I). The data show that NSD3 overexpression increases the cell proliferation, migration, and EMT progress in CRC.
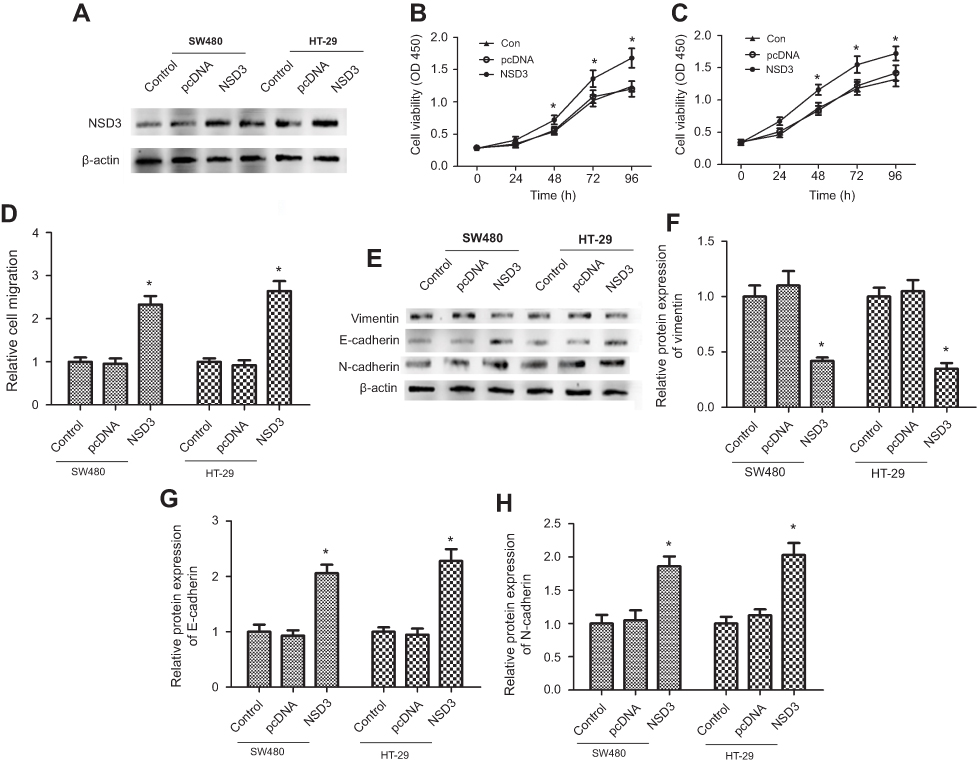 | Figure 3 Overexpression of NSD3 facilitated CRC cell proliferation and metastasis in vitro. (A) Western blot analysis showed the expression level of NSD3 was significantly upregulated both in SW480 and HT-29 cells (5.0×106/cm2) transfected with pcDNA3.1(+)-NSD3 (NSD3) after 48 h. (B, C) MTT assay results respectively showed the trend of SW480 and HT-29 cells (5.0×104/cm2) viability within 96 h after overexpressing NSD3 (NSD3). (D) Proliferation of SW480 and HT29 cells were evaluated by BrdU incorporation after overexpressing NSD3 (NSD3). Brdu, DNA fluorescent dye; PI, nuclear fluorescent dye.(E) The migration ability of SW480 and HT29 cells were evaluated by scratch wound healing assay revealing. Wild-type cells and cells transfected with an empty pcDNA3.1(+) expression vector (pcDNA) were used as controls. (F–I) Western blot and RT-qPCR analysis of the E-cadherin, N-cadherin, and vimentin expression in wild-type cells (control), cells transfected with an empty pcDNA3.1(+) (pcDNA), and in cells with stable overexpression of NSD3 (NSD3) after 72 h. β-actin as a loading control. The bands were presented as the mean ± SEM. *P<0.05 vs control or pcDNA.Abbreviations: CRC, colorectal cancer; RT-qPCR, reverse transcription PCR |
ERK1/2 and CAPG are positively regulated by NSD3 overexpression
We next explored the possible mechanisms of NSD3 eliciting these biological effects in CRC. It has been shown that ERK1/2 signaling pathway plays an important role in the process of tumor cell growth and metastasis.19 Notably, ERK1/2 facilitates CRC cell proliferation, migration,20,21 and the elevated levels of ERK1/2 phosphorylation can induce cell cycle arrest.22 Hence, the phosphorylation level of ERK1/2 in CRC cells was examined, an increase in phosphorylated ERK1/2 (p-ERK) was observed in both SW480 and HT-29 cell lines treated with pcDNA3.1(+)-NSD3 (NSD3) compared with an empty pcDNA3.1(+) expression vector (pcDNA) (Figure 4A).
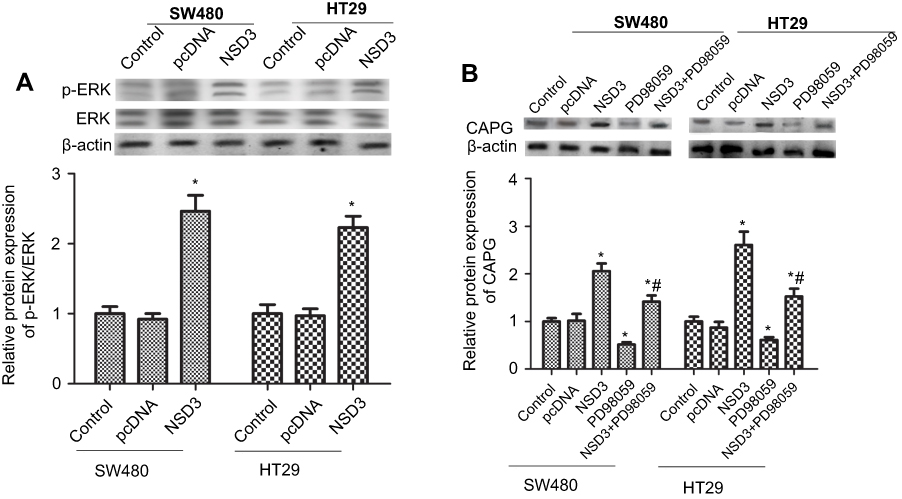 | Figure 4 NSD3 overexpression stimulated ERK1/2 and CAPG axis. RT-qPCR and Western blot analysis of the p-ERK1/2. (A) and CAPG (B) expression in wild-type cells (control), cells transfected with an empty pcDNA3.1(+) expression vector (pcDNA), and in cells with stable overexpression of NSD3 (NSD3). ERK1/2 signaling pathway inhibitor (PD98059) was used to weaken p-ERK. Cells were analyzed for mRNA levels 48 h after transfection. β-actin as a loading control. The bands were presented as the mean ± SEM. *P<0.05 vs bands without an asterisk, *#P<0.05 vs bands with an asterisk but without a hashtag. |
On the other hand, previous studies have shown that CAPG is closely related to cancer cell dissemination and migration.23 Therefore, we simultaneously examined the effects of NSD3 on activation of CAPG. As expected, overexpression of NSD3 enhanced the expression level of CAPG, while the ERK1/2 signaling pathway inhibitor (PD98059) was capable of silencing CAPG (Figure 4B). Furthermore, PD98059 treatment significantly attenuated the activatory effect of pcDNA3.1(+)-NSD3 (NSD3) on CAPG expression (Figure 4B). These results indicate that CAPG is positively regulated by NSD3 overexpression through activating ERK1/2 in CRC cells.
ERK1/2 signaling pathway and CAPG were involved in the NSD3-mediated CRC proliferation and migration
To further studied whether NSD3 could enhance vitality and metastasis of CRC cells through its potential downstream targets ERK1/2 and CAPG, we detected the proliferation and migration ability of CRC cells after overexpressing NSD3, in the presence of specific siRNA against CAPG (siCAPG) or/and PD98059. The proliferation and migration of CRC cells could be dramatically increased through transfected with pcDNA3.1(+)-NSD3 (NSD3). Deactivation of the ERK1/2 signaling pathway or knocking down of CAPG individually could dramatically decrease the proliferation and migration of CRC cells. However, when pcDNA3.1(+)-NSD3 (NSD3) was cooperated with siCAPG or/and PD98059, the ability of CRC cell proliferation and metastasis was partially decreased compared with only pcDNA3.1(+)-NSD3 (NSD3) transfected but still higher than wild-type cells both in SW480 and HT-29 cells (Figure 5A–D). These findings suggest that overexpress of NSD3 could enhance the proliferation and migration of CRC cells through regulating CAPG and ERK1/2 signaling pathway.
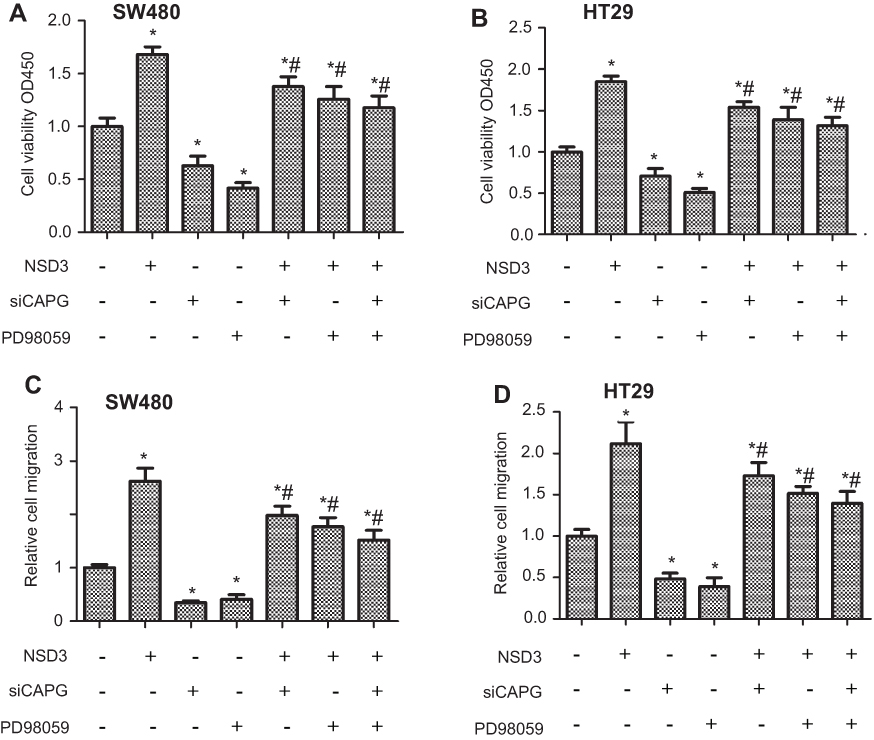 | Figure 5 NSD3 overexpression facilitated CRC cell proliferation and migration by CAPG in an ERK1/2 signaling pathway manner. The SW480 and HT-29 proliferation (A, B) and migration (C, D) ability were performed in cells transfected with specific siRNA against CAPG (siCAPG), PD98059-added cells, and/or in cells with stable overexpression of NSD3 (NSD3). Reverse transfection procedure was used to deliver 50 nM siRNA to 5.0×104 (A, B)/5.0×106 (C, D) cells in a 6-well plate. Cells were analyzed for mRNA levels 48 h after transfection. *P<0.05 vs bands without an asterisk, *#P<0.05 vs bands with an asterisk but without a hashtag. |
Discussion
Histone methyltransferases (HMTases), the key components in the regulation of histone methylation, are commonly disrupted in multiple malignancies.24 The NSD protein lysine methyltransferase family (PKMT) is composed of three members, NSD1 (KMT3B), NSD2 (WHSC1/MMSET) and NSD3 (WHSC1L1), which is primarily known to regulate gene expression through methylation of lysine 36 on histone H3 (H3K36).25 As a member of the NSD PKMTs, NSD3 directly contributes to the production and development of multiple human cancers through a variety of mechanisms, including DNA repair, direct regulation of histone methylation, either the protein-protein interactions by the specific domains. In NUT midline carcinoma (NMC), the NSD3-NUT fusion oncogene encoded a protein that was important for the blockade of differentiation.10Additionally, NSD3 was observed as a translocation partner of NUP98 in acute myeloid leukemia (AML).9 NSD3 led to tumorigenesis by cooperating with chromatin remodeling enzyme CHD8 and bromodomain-containing protein 4 (BRD4), which was a potential therapeutic target in AML.12 Taken together, it appears that NSD3 could become a valuable therapeutic target for a wide variety of cancers using selective NSD3 inhibitors.
Notably, NSD3 has been found to abnormally express in various types of malignant tumors, such as osteosarcoma,26 breast,27 head and neck,28 lung,29 and bladder cancers.30 Actually, the amplification of NSD3 is closely related to various behaviors such as tumor cell cycle stagnation, proliferation, and metastasis.9–12 NSD3 could obviously suppress cell proliferation and invasion through affecting the expression of cell cycle regulator E2F2 and Arl2 in breast cancer.11 However, after knocking down NSD3, cell proliferation was effectively reduced and expression of cell cycle enhancers, cyclin G1 (CCNG1) and NEK7 was decreased.30 Nonetheless, the biological roles of NSD3 in CRC are not clear and remain to be clarified. Our studies revealed that the expression level of NSD3 in CRC was obviously higher than that in normal tissues and cells. Likewise, we demonstrated that the depletion of NSD3 weakened CRC cell proliferation and migration. As the metastasis of various cancer cells is related to the EMT process,13 we firstly studied the effect of NSD3 on EMT processes in CRC cells. EMT marker proteins N-cadherin and E-cadherin were downregulated, and vimentin was upregulated due to NSD3 knockdown, whereas after NSD3 was overexpressed, the opposite results were observed. That was NSD3 was participated in the CRC cell EMT process. Thus, NSD3 is likely to be a promising target for CRC therapy.
Emerging evidence has showed that NSD3 inhibited cancer cell differentiation and induced proliferation and invasion through various signaling pathways.31 In breast cancer, the enhanced expression of NSD3 markedly promoted the regulatory factors of WNT signaling pathway, iroquois homeobox 3 (IRX3), and TBL1X, accompanied by the decreased expression of a negative factor SFRP1.32 Furthermore, NSD3 mono-methylated lysine 721 in the tyrosine kinase domain of epidermal growth factor receptor EGFR, and this methylation led to enhanced activation of its downstream ERK1/2 signaling pathway cascade.28 ERK1/2 have been found to be associated with the development of cancer and progression to distant metastasis, including CRC.33,34 Notably, ERK1/2 pathway was involved in the regulation of EMT process.35,36 However, the function of NSD3 in regulating the ERK1/2 signaling pathway has not been studied yet in CRC. Our studies showed NSD3 overexpression enhanced the phosphorylation level of ERK1/2 in CRC cells. And the inhibited ERK1/2 significantly reduced CRC cell proliferation and migration, implying that NSD3 may regulate CRC cell activities through the ERK1/2 signaling pathway.
Many studies supported CAPG, an important actin-capping protein, is associated with the clinicopathological features of tumor cells such as motility and proliferation.37,38 CAPG expression in ovarian carcinoma tissues was significantly higher than that in normal tissues, and a single nucleotide polymorphism (SNP) rs6886 inside the CAPG gene affected CAPG phosphorylation and further influenced cell invasion and migration.39 CAPG inhibition strongly reduced cell metastasis, and nanobody-CAPG may become a drug target in breast cancer.37 Our studies proved that NSD3 overexpression could increase the expression of CAPG and siCAPG could obviously hinder cell migration in CRC. On the other hand, various oncology studies have found that CAPG induces different effects on cell proliferation in different tumors. Tonack et al40 suggested that the overexpression of CAPG promoted pancreatic cancer cell migration but did not affect cell cycle and proliferation. The same results were confirmed in gastric41 and liver cancers.42 However, the proliferation capability of prostate cancer cells was significantly lower than that of the control cells after inhibition of CAPG gene.43 In the present study, knockdown of CAPG could dramatically suppress CRC cell proliferation, which is the first discovery that CAPG promotes the cell proliferation in CRC. Furthermore, we detected that deactivation of the ERK1/2 significantly decreased the expression of CAPG. And this data initially showed that CAPG expression was regulated by the ERK1/2 signaling pathway.
Conclusion
The expression of NSD3 was obviously higher in CRC tissues than in normal tissues. NSD3 overexpression in CRC cells facilitated cell proliferation and migration, and promoted cell EMT progress via increasing CAPG expression and ERK1/2 activation. In addition, we also demonstrated that ERK1/2 signaling pathway could facilitate CAPG-induced proliferation and migration. These results imply that NSD3 may become a reliable CRC biomarker for diagnoses and a target for precise therapy.
Acknowledgments
The present study was supported by the Project of Administration of Traditional Chinese Medicine of Jiang Su Province of China (grant no. YB2015085), Project of Xuzhou Science and Technology Bureau of Jiangsu Province of China (grant no. KC15SH035), Special Foundation of Xuzhou medical youth reserve talents Project (grant no. 2014), and Special Foundation of outstanding TCM clinical talents of Jiangsu province (grant no. 2018).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chan AT, Giovannucci EL. Primary prevention of colorectal cancer. Gastroenterology. 2003;66(1):20.
2. Siegel RL, Miller KD
3. Weitz J, Koch M, Debus J, Höhler T, Galle PR, Büchler MW. Colorectal cancer. Lancet. 2005;365(9454):153–165. doi:10.1016/S0140-6736(05)17706-X
4. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132.
5. Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet. 2010;375(9719):1030–1047.
6. Chen F, Kan H, Castranova V. Chapter 10 - Methylation of Lysine 9 of Histone H3: Role of Heterochromatin Modulation and Tumorigenesis. Elsevier Inc; Handbook of Epigenetics, 2011;15(3):149–157.
7. Wang C, Wang Q, Xu X, et al. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J Exp Med. 2017;214:12.
8. Han X, Piao L, Zhuang Q, Yuan X, Liu Z, He X. The role of histone lysine methyltransferase NSD3 in cancer. Onco Ther. 2018;11:3847–3852.
9. Rosati R, La RS, Veronese A, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood. 2002;99(10):3857–3860.
10. French CA, Rahman S, Walsh EM, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 2014;4(8):928. doi:10.1158/2159-8290.CD-13-0646
11. Zhou Z, Thomsen RS, Nielsen AL, Nielsen AL. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochem Biophys Res Commun. 2010;398(3):565–570. doi:10.1016/j.bbrc.2010.06.119
12. Shen C, Ipsaro JJ, Shi J, et al. NSD3-short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol Cell. 2015;60(6):847–859. doi:10.1016/j.molcel.2015.10.033
13. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi:10.1038/nrm3758
14. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675. doi:10.1016/j.tcb.2015.07.012
15. Ding C, Luo J, Li L, et al. Gab2 facilitates epithelial-to-mesenchymal transition via the MEK/ERK/MMP signaling in colorectal cancer. J Exp Clin Cancer Res Cr. 2016;35(1):5. doi:10.1186/s13046-015-0280-0
16. Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. doi:10.1016/S1470-2045(05)70168-6
17. Nomura H, Uzawa K, Ishigami T, et al. Clinical significance of gelsolin-like actin-capping protein expression in oral carcinogenesis: an immunohistochemical study of premalignant and malignant lesions of the oral cavity. BMC Cancer. 2008;8(1):39. doi:10.1186/1471-2407-8-172
18. Yun DP, Wang YQ, Meng DL, et al. Actin-capping protein CapG is associated with prognosis, proliferation and metastasis in human glioma. Oncol Rep. 2018;39(3):1011–1022. doi:10.3892/or.2018.6225
19. Ding C, Tang W, Fan X, et al. Overexpression of PEAK1 contributes to epithelial–mesenchymal transition and tumor metastasis in lung cancer through modulating ERK1/2 and JAK2 signaling. Cell Death Dis. 2018;9:8. doi:10.1038/s41419-018-0817-1
20. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344):re8. doi:10.1126/scisignal.2005189
21. Ning S, Liang P, Xia B, et al. Lyn is involved in CD24-induced ERK1/2 activation in colorectal cancer. Mol Cancer. 2012;11(1):1. doi:10.1186/1476-4598-11-1
22. Jeon HK, Choi SU, Jung NP. Association of the ERK1/2 and p38 kinase pathways with nitric oxide-induced apoptosis and cell cycle arrest in colon cancer cells. Cell Biol Toxicol. 2005;21(2):115–125. doi:10.1007/s10565-005-0148-8
23. Impe B, Cool S, Impens F, et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast cancer research. 2013;15(6): R116.
24. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–113. doi:10.1126/science.1060118
25. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126. doi:10.1038/nrm3274
26. Liu Z, Piao L, Zhuang M, et al. Silencing of histone methyltransferase NSD3 reduces cell viability in osteosarcoma with induction of apoptosis. Oncol Rep. 2017;38(5):2796–2802. doi:10.3892/or.2017.5936
27. Turner-Ivey B, Smith EL, Rutkovsky AC, Spruill LS, Mills JN, Ethier SP. Development of mammary hyperplasia, dysplasia, and invasive ductal carcinoma in transgenic mice expressing the 8p11 amplicon oncogene NSD3. Breast Cancer Res Treat. 2017;164(2):349–358. doi:10.1007/s10549-017-4258-9
28. Saloura V, Vougiouklakis T, Zewde M, et al. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci Rep. 2017;7:40664. doi:10.1038/srep40664
29. Tonon G, Wong KK, Maulik G, et al. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102(27):9625–9630. doi:10.1073/pnas.0504126102
30. Kang D, Cho HS, Toyokawa G, et al. The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer. 2012;52(2):126–139. doi:10.1002/gcc.22012
31. Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics. 2015;7(5):863. doi:10.2217/epi.15.32
32. Zeng-Quan Y, Gang L, Aliccia BF, Giroux CN, Ethier SP. Transforming properties of 8p11–12 amplified genes in human breast cancer. Cancer Res. 2010;70(21):8487–8497. doi:10.1158/0008-5472.CAN-10-1013
33. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: A new perspective. Cancer. 2015;120(22):3446–3456.
34. Ning S, Liang P, Xia B, et al. Lyn is involved in CD24-induced ERK1/2 activation in colorectal cancer. Mol Cancer. 2012;11(1):1–13.
35. Elsum IA, Martin C, Humbert PO. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J Cell Sci. 2013;126(17):3990–3999.
36. Ichikawa K, Kubota Y, Nakamura T, et al. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial-mesenchymal transition by regulating the co-repressor CtBP. Mol Cell. 2015;58(1):35–46.
37. Impe KV, Bethuyne J, Cool S, et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Res Bcr. 2013;15(6):R116.
38. Renz M, Betz B, Niederacher D, Bender HG, Langowski J. Invasive breast cancer cells exhibit increased mobility of the actin‐binding protein CapG. Int J Cancer. 2008;122(7):1476–1482.
39. Niederacher D, Fleisch MC. Macrophage capping protein CapG is a putative oncogene involved in migration and invasiveness in ovarian carcinoma. Biomed Res Int. 2014;2014(5):379847.
40. Tonack S, Patel S, Jalali M, et al. Tetracycline-inducible protein expression in pancreatic cancer cells: effects of CapG overexpression. World J Gastroenterol. 2011;17(15):1947.
41. Ichikawa H, Kanda T, Kosugi S, et al. Laser microdissection and two-dimensional difference gel electrophoresis reveal the role of a novel macrophage-capping protein in lymph node metastasis in gastric cancer. J Proteome Res. 2013;12(8):3780–3791.
42. Kimura K, Ojima H, Kubota D, et al. Proteomic identification of the macrophage-capping protein as a protein contributing to the malignant features of hepatocellular carcinoma. J Proteomics. 2013;78(1):362–373.
43. Li BK, Guo K, Li CY, et al. Influence of suppression of CapG gene expression by siRNA on the growth and metastasis of human prostate cancer cells. Genet Mol Res Gmr. 2015;14(4):15769.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.