")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Downregulation of MALAT1 alleviates saturated fatty acid-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis
Authors Jia P, Wu N, Jia D, Sun Y
Received 28 January 2019
Accepted for publication 4 March 2019
Published 7 May 2019 Volume 2019:12 Pages 655—665
DOI https://doi.org/10.2147/DMSO.S203151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Pengyu Jia,1 Nan Wu,2 Dalin Jia,1 Yingxian Sun1
1Department of Cardiology, the First Affiliated Hospital of China Medical University, Shenyang, Liaoning, People‘s Republic of China; 2The Central Laboratory of the First Affiliated Hospital of China Medical University, Shenyang, Liaoning, People‘s Republic of China
Purpose: The increased level of saturated fatty acids (SFAs) is found in patients with diabetes, obesity, and other metabolic disorders. SFAs can induce lipotoxic damage to cardiomyocytes, but the mechanism is unclear. The long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) acts as a key regulator in palmitic acid (PA)-induced hepatic steatosis, but its role in PA-induced myocardial lipotoxic injury is still unknown. The aim of this study was to explore the role and underlying mechanism of MALAT1 in PA-induced myocardial lipotoxic injury.
Methods: MALAT1 expression in PA-treated human cardiomyocytes (AC16 cells) was detected by RT-qPCR. The effect of MALAT1 on PA-induced myocardial injury was measured by Cell Counting Kit-8, lactate dehydrogenase (LDH), and creatine kinase-MB (CK-MB) assays. Apoptosis was detected by flow cytometry. The activities of cytokines and nuclear factor (NF)-κB were detected by enzyme-linked immunosorbent assay. The interaction between MALAT1 and miR-26a was evaluated by a luciferase reporter assay and RT-qPCR. The regulatory effects of MALAT1 on high mobility group box 1 (HMGB1) expression were evaluated by RT-qPCR and western blotting.
Results: MALAT1 was significantly upregulated in cardiomyocytes after PA treatment. Knockdown of MALAT1 increased the viability of PA-treated cardiomyocytes, decreased apoptosis, and reduced the levels of LDH, CK-MB, TNF-α, and IL-1β. Moreover, we found that MALAT1 specifically binds to miR-26a and observed a reciprocal negative regulatory relationship between these factors. We further found that the downregulation of MALAT1 represses HMGB1 expression, thereby inhibiting the activation of the Toll-like receptor 4 (TLR4)/NF-κB-mediated inflammatory response. These repressive effects were rescued by an miR-26a inhibitor.
Conclusion: We demonstrate that MALAT1 is induced by SFAs and its downregulation alleviates SFA-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Our findings provide new insight into the mechanism underlying myocardial lipotoxic injury.
Keywords: metastasis-associated lung adenocarcinoma transcript 1, saturated fatty acids, microRNA, high mobility group box-1 protein, inflammation
Introduction
The excessive accumulation of lipids or lipid intermediates in non-adipose tissues, such as the liver and kidney, leads to cellular dysfunction and death.1 This pathophysiological process is termed lipotoxicity.1 It usually develops in patients with diabetes, obesity, and other metabolic disorders.2 The heart can also be affected by lipotoxicity, predominantly manifesting as myocardial fibrosis and even heart failure.3 The main cause may be the deposit of excess saturated fatty acids (SFAs) in cardiomyocytes.3 We and others have provided direct evidence that palmitic acid (PA), a major SFA, leads to myocardial lipotoxic injury in vitro and in vivo.4–6 Although several mechanisms, including inflammation, endoplasmic reticulum stress, alterations in autophagy, and oxidative stress, are responsible for SFA-induced myocardial lipotoxic injury,7 the exact mechanism is still not well understood.
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1, also named NEAT2) is a highly evolutionarily conserved long noncoding RNA (lncRNA) and was initially identified in early-stage non-small cell lung cancer.8 MALAT1 is involved in multiple pathophysiological processes, including tumorigenesis and metastasis,9,10 ischemic stroke,11 myocardial ischemia reperfusion injury,12 and pulmonary fibrosis.13 Notably, a recent study has found that MALAT1 acts as a key regulator in PA-induced hepatic steatosis by promoting lipid accumulation in hepatocytes.14 Furthermore, MALAT1 plays a role in the regulation of inflammatory responses.15–17 However, the role of MALAT1 in SFA-induced myocardial lipotoxic injury is unknown.
Recently, lncRNAs have been reported to bind to microRNAs, as a competitive endogenous RNA (ceRNA), to further regulate target mRNA expression at the post-transcriptional level.18 Using a bioinformatics approach, we found that MALAT1 transcript sequences contained an miR-26a binding region. Moreover, miR-26a has been demonstrated to bing to high mobility group box 1 (HMGB1) and inhibited HMGB1 expression, resulting in the inhibition of the Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB signaling pathway-mediated inflammatory response.19 Therefore, it is hypothesized that MALAT1 regulates HMGB1 expression through binding to miR-26a in a ceRNA mechanism.
In this study, we evaluated the role of MALAT1 in SFA-induced myocardial lipotoxic injury and its underlying mechanism. Our results indicated that MALAT1 is significantly induced in cardiomyocytes after treatment with PA and the knockdown of MALAT1 alleviates PA-induced myocardial inflammatory injury. Mechanistically, we found that MALAT1 acts as a competing ceRNA to regulate HMGB1 expression by binding to miR-26a, thereby inhibiting the activation of the TLR4/NF-κB signaling pathway-mediated inflammatory response.
Materials and methods
Cell culture and PA treatment
Human adult ventricular cardiomyocytes (AC16 cell line) purchased from the American Type Culture Collection (Manassas, VA) were grown in Dulbecco’s modified Eagle medium (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (TBD, Tianjin, China), 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere at 37 °C with 5% CO2. AC16 cells were treated with 300 µM PA (Sigma-Aldrich, St. Louis, MO) to induce myocardial lipotoxic injury as described in a previous article.20
Cell transfection
MALAT1-specific small interfering RNA (siRNA), scrambled siRNA (negative control, NC), miR-26a mimics, NC mimics, miR-26a inhibitor, NC inhibitor, and TNF-α-specific siRNA (si-TNF-α) were designed and synthesized by GenePharma Co., Ltd. (Shanghai, China). Cells were transfected with the siRNAs and microRNAs (miRNAs) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were harvested and processed for further analysis after 24 h or 48 h of transfection.
qRT-PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen). To detect MALAT1 and mRNA expression, the PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa, Kusatsu, Japan) and SYBR Premix Ex Taq II (TaKaRa) were used for reverse transcription and quantitative PCR according to the manufacturer’s instructions. GAPDH was used as an internal control. To detect miR-26a expression, the Mir-X™ miRNA First Strand Synthesis Kit (TaKaRa) and Mir-X™ miRNA qRT-PCR SYBR® Kit (TaKaRa) were used for reverse transcription and quantitative PCR according to the manufacturer’s instructions. U6 was used as an internal control. All oligonucleotide primers were designed by Sangon Biotech Co., Ltd. (Shanghai, China) (Table 1). Relative expression was analyzed using the 2‑ΔΔCt method.
 | Table 1 The information of primer sequences |
Cell Counting Kit-8 assay
Cell viability was determined using the Cell Counting Kit-8 (CCK-8) assay (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Absorbance was detected at 450 nm using the Ultramicro Microporous Plate Spectrophotometer (BioTek, Winooski, VT).
Lactate dehydrogenase and creatine kinase-MB assays
Lactate dehydrogenase (LDH) and creatine kinase-MB (CK-MB) released from cardiomyocytes into the culture medium were determined using the LDH Assay Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and CK-MB Assay Kit (Nanjing Jiancheng Bioengineering Institute), respectively, according to the manufacturer’s protocols. Absorbance was detected at 450 nm using the Ultramicro Microporous Plate Spectrophotometer (BioTek).
Apoptosis assay
Cell apoptosis was determined using the Annexin V-Fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Kit (KeyGEN BioTECH, Jiangsu, China) according to the manufacturer’s instructions for flow cytometry (BD Biosciences, Franklin Lakes, NJ).
Enzyme-linked immunosorbent assay of cytokine and NF-κB activity
The concentrations of tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) in the culture medium were detected using a commercial human TNF-α enzyme-linked immunosorbent assay (ELISA) kit (MultiSciences, Hangzhou, China) and human IL-1β ELISA kit (MultiSciences), respectively, according to the manufacturer’s instructions. The activity of NF-κB in cell lysates was detected using the NF-κB p65 Transcription Factor Assay Kit (Abcam, Cambridge, UK) according to the recommended experimental protocol.
Luciferase activity assay
Luciferase reporter plasmids (pmirGLO-MALAT1 and pmirGLO-MALAT1-mut) were obtained from Shanghai GenePharma Co., Ltd. The cells were plated in 24-well plates and transfected with pmirGLO-MALAT1 or pmirGLO-MALAT1-mut together with miR-26a mimics or NC mimics. At 48 h post-transfection, luciferase activity was analyzed using the Dual-Glo Luciferase Assay System (Promega, Madison, WI) according to the manufacturer’s instructions.
Western blotting analysis
Proteins were extracted from cell lysates using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China) and the protein concentration was measured using the Enhanced BCA Protein Assay Kit (Beyotime) according to the manufacturer’s instructions. Heat-denatured proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes, followed by blocking with 1% bovine serum albumin solution for 1 h. The membranes were then incubated with primary antibodies, including anti-HMGB1 (1:1000; Abcam), anti-TLR4 (1:1000; Abcam), anti-NF-κB p65 (1:1000; Abcam), anti-cleaved caspase-8 (1:1000; Abcam), anti-cleaved caspase-3 (1:1000; Abcam), anti-GAPDH (1:1000; Zhongshan Jinqiao Biotechnology, Beijing, China), and anti-Lamin A (1:1000; Abcam) at 4 °C overnight, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin G (1:4000; EarthOx, Millbrae, CA) at room temperature for 30 min. Protein bands were detected using an enhanced chemi-luminescence kit for western blotting (Beyotime) according to the manufacturer’s instructions. GAPDH or Lamin A was used as an internal control. Relative densitometry was calculated using Image J2x analysis software (National Institutes of Health, Bethesda, MD).
Statistical analysis
Data are expressed as the mean ± standard error. Statistical analysis was performed using SPSS version 17.0 software (SPSS Inc., Chicago, IL). Differences between groups were first evaluated using one-way analysis of variance, and if the differences were significant, multiple comparison testing was further performed using Fisher’s least significant difference test. P-values less than 0.05 were considered statistically significant.
Results
MALAT1 was induced in myocardial cells by PA
Expression of MALAT1 in cardiomyocytes increased by 2.57-, 4.70-, 7.41-, and 2.87-fold after treatment with 300 µM PA for 6, 12, 24, and 48 h, respectively (Figure 1). These results demonstrated that MALAT1 is induced by PA in AC16 cells.
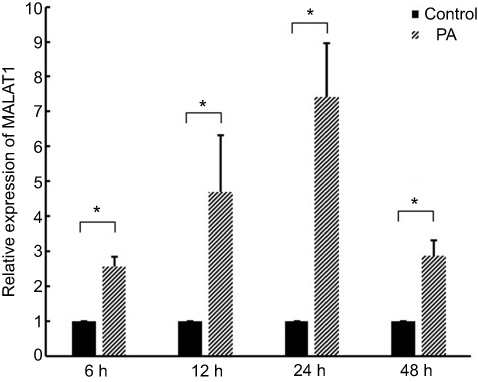 | Figure 1 Expression of MALAT1 in cardiomyocytes after treatment with palmitic acid (PA). *P<0.05. Data are presented as means ± standard error from three independent experiments. |
Knockdown of MALAT1 expression alleviated PA-induced myocardial injury in vitro
Expression of MALAT1 in AC16 cells decreased by 63% after transfection with MALAT1-specific siRNA (Figure 2A), indicating the effective repression of the endogenous expression of MALAT1 in cardiomyocytes. Cell viability increased remarkably (Figure 2B), the activity of LDH (Figure 2C) and CK-MB (Figure 2D) decreased significantly, and the ratio of apoptosis (Figure 2E and F) decreased significantly in response to the downregulation of MALAT1. These results demonstrated that the downregulation of MALAT1 protects against PA-induced myocardial injury in vitro.
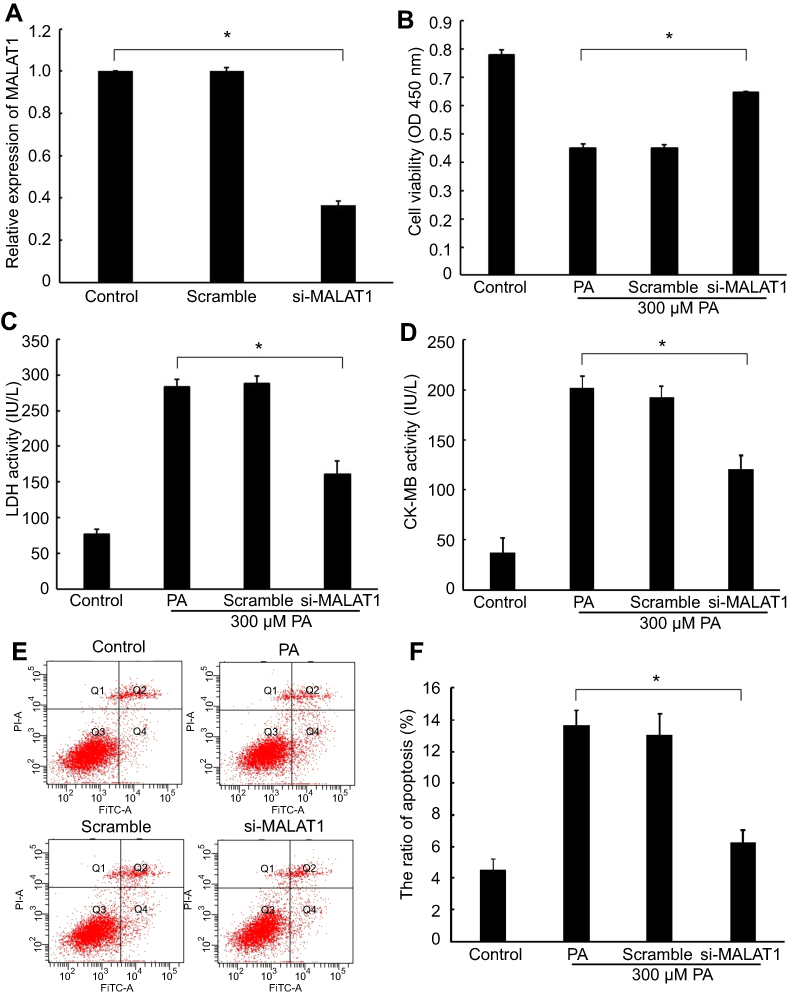 | Figure 2 Effect of the knockdown of MALAT1 on palmitic acid (PA)-induced myocardial injury. (A) The expression of MALAT1 in AC16 cells was detected by RT-qPCR after transfection with MALAT1-specific siRNA (si-MALAT1) and a negative control (scramble). (B) Cell viability was determined by a Cell Counting Kit-8 assay. (C) Lactate dehydrogenase (LDH) activity in the culture medium was detected by spectrophotometry. (D) Creatine kinase-MB (CK-MB) activity in the culture medium was detected by spectrophotometry. (E) Apoptosis was measured by flow cytometry. (F) Statistical analysis of the ratio of apoptotic cells. *P<0.05. Data are presented as the mean ± standard error from three independent experiments.Abbreviations: FiTC, Annexin V-Fluorescein Isothiocyanate; IL-1β, interleukin β; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; HMGB1, high mobility group box 1; TNF-α, tumour necrosis factor α. |
Interaction between MALAT1 and miR-26a
LncBase Predicted v.2 tool21 (
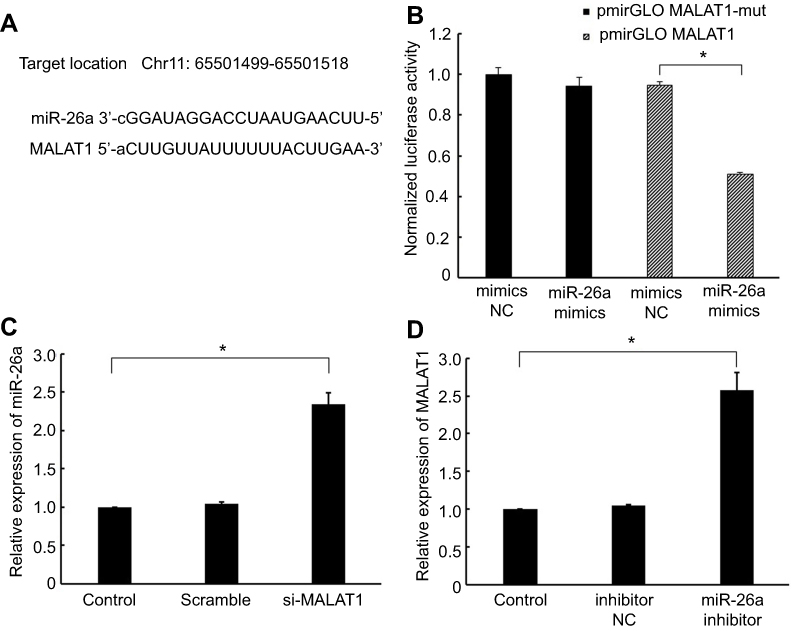 | Figure 3 Interaction between MALAT1 and miR-26a. (A) miRNA binding sites in the MALAT1 sequence. (B) Luciferase reporter gene assay validated the interaction between MALAT1 and miR-26a in AC16 cells. pmirGLO-MALAT1, luciferase vector of MALAT1; pmirGLO-MALAT1-mut, MALAT1 mutant vector. (C) Effect of MALAT1 on the level of miR-26a. (D) Effect of miR-26a on the level of MALAT1. *P<0.05. Data are presented as the mean ± standard error from three independent experiments.Abbreviations: NC, negative control; MALAT1, metastasis-asociated lung adenocarcinoma transcript 1. |
Knockdown of MALAT1 inhibited HMGB1/TLR4/NF-κB signaling via miR-26a
HMGB1 is a known target of miR-26a;19 accordingly, we explored whether MALAT1, acting as a ceRNA, regulates HMGB1 expression via miR-26a. The mRNA and protein expression levels of HMGB1 were repressed by transfection with MALAT1 siRNA alone. However, co-transfection with MALAT1 siRNA and an miR-26a inhibitor did not inhibit HMGB1 at the mRNA and protein expression levels (Figure 4A and B). Furthermore, we detected the effect of MALAT1 on the TLR4/NF-κB signaling pathway. Transfection with MALAT1 siRNA alone decreased the protein expression of TLR4, reduced the protein expression of NF-κB in the nucleus, inhibited the activity of NF-κB, and increased the protein expression of NF-κB in the cytoplasm. However, these effects were abolished by the co-transfection of MALAT1 siRNA and an miR-26a inhibitor (Figure 4C–F). Taken together, these results demonstrated that the knockdown of MALAT1 inhibits the HMGB1/TLR4/NF-κB signaling pathway via miR-26a.
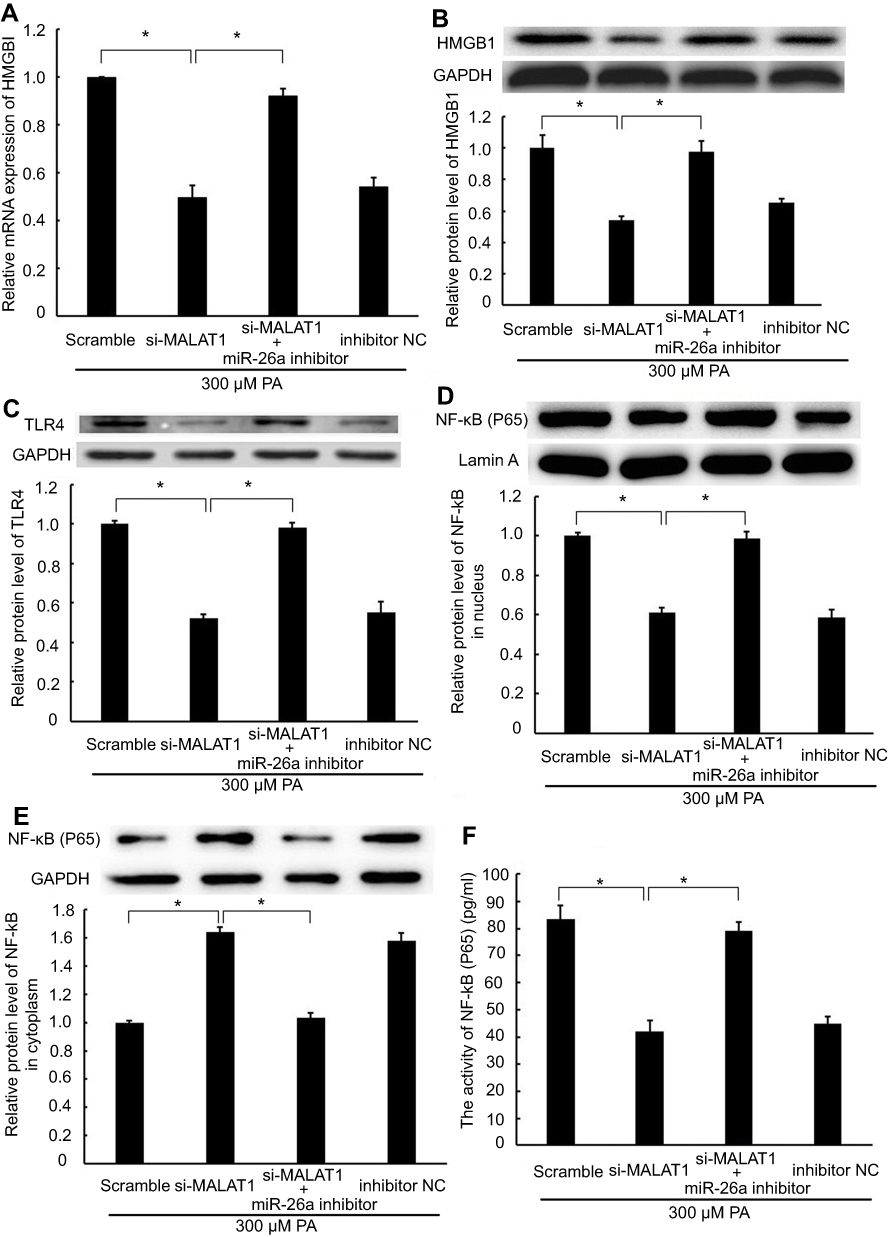 | Figure 4 The regulatory effect of MALAT1 on the HMGB1/TLR4/NF-κB signaling pathway was mediated by miR-26a. MALAT1 negative control (scramble) and MALAT1-specific siRNA (si-MALAT1) alone or in combination with an miR-26a inhibitor or miR-26 negative control (inhibitor NC) was transfected into AC16 cells, followed by palmitic acid treatment. (A) The mRNA expression of HMGB1 was detected by RT-qPCR. (B) The protein expression of HMGB1 was detected by western blotting. (C) The protein expression of TLR4 was detected by western blotting. (D) The protein expression of NF-κB (P65) in the nucleus was detected by western blotting. (E) The protein expression of NF-κB (P65) in the cytoplasm was detected by western blotting. (F) The activity of NF-κB was detected by ELISA. *P<0.05. Data are presented as the mean ± standard error from three independent experiments.Abbreviations: NC, negative control; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; HMGB1, high mobility group box 1; TLR4, Toll-like receptor 4. |
Effect of MALAT1 on PA-induced myocardial inflammation was mediated by miR-26a
To evaluate whether MALAT1 regulates PA-induced inflammation, we detected the mRNA expression levels and concentrations of the pro-inflammatory cytokines TNF-α and IL-1β in AC16 cells and culture medium, respectively. The mRNA expression and concentrations of TNF-α and IL-1β were notably reduced by the downregulation of MALAT1. The repression of TNF-α and IL-1β by the downregulation of MALAT1 was rescued by an miR-26a inhibitor (Figure 5A–D). Taken together, these results demonstrated that the downregulation of MALAT1 inhibits PA-induced inflammation, and miR-26a mediates the suppressive effect of MALAT1 on PA-induced myocardial inflammation.
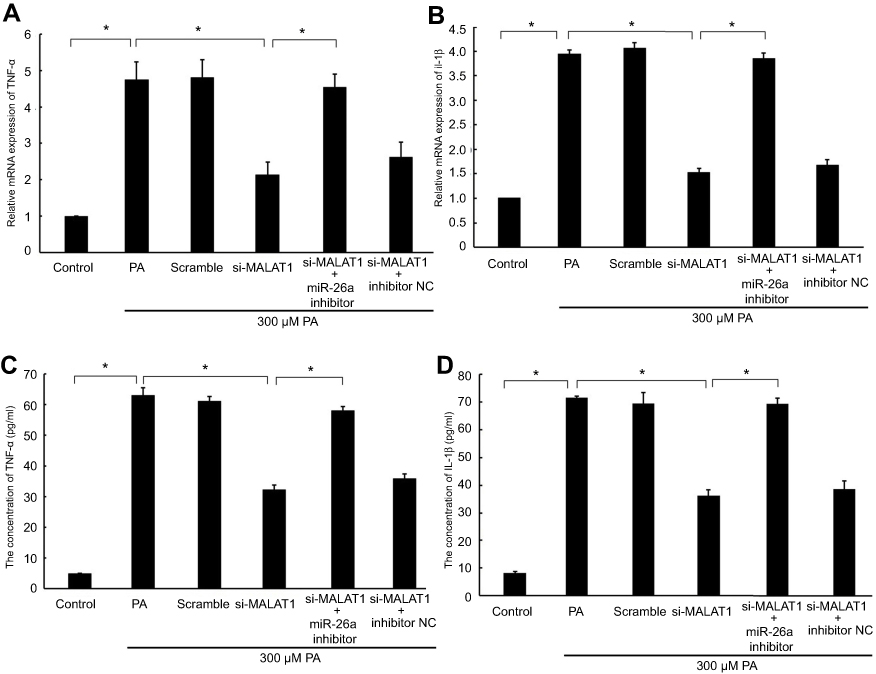 | Figure 5 The effect of MALAT1 on PA-induced myocardial inflammation was mediated by miR-26a. MALAT1 negative control (scramble) and MALAT1-specific siRNA (si-MALAT1) alone or in combination with an miR-26a inhibitor or miR-26 negative control (inhibitor NC) and TNF-α-specific siRNA (si-TNF-ɑ) were transfected into AC16 cells, followed by palmitic acid (PA) treatment. (A) The mRNA expression of TNF-α was detected by RT-qPCR. (B) The mRNA expression of IL-1β was detected by RT-qPCR. (C) The concentration of TNF-α in the culture medium was detected by ELISA. (D) The concentration of IL-1β in the culture medium was detected by ELISA. *P<0.05. Data are presented as the mean ± standard error from three independent experiments.Abbreviations: IL-1β, interleukin β; MALAT1, metastasis-associated lung adenocarcinoma transcript 1;TNF-α, tumour necrosis factor α; NC, negative control. |
MALAT1/miR-26a manipulation regulated PA-induced cell death via the TNF-α-induced apoptosis pathway
Given that the changes in TNF-α and IL-1β induced by PA are regulated by MALAT1/miR-26a manipulation and TNF-α can induce apoptosis by the death-inducing signaling complex/FADD pathway, we inferred that MALAT1/miR-26a manipulation may influence PA-induced cell death by regulating the TNF-α-induced apoptosis pathway. As expected, we found that the activation of caspase-8, an essential effector of the TNF-α-induced apoptosis pathway, was downregulated by si-MALAT1, similar to the knockdown of TNF-α (si-TNF-α). The repression of caspase-8 by si-MALAT1 was rescued by an miR-26a inhibitor (Figure 6A). Moreover, the activation of caspase-3 was also regulated by MALAT1/miR-26a manipulation (Figure 6B). These results suggest that the activation of caspase-8/caspase-3 by PA is regulated by MALAT1/miR-26a manipulation. To further determine the effect of MALAT1/miR-26a manipulation on TNF-α-induced apoptosis, we used TNF-α to induce cardiomyocyte apoptosis (Figure 6C). The downregulation of MALAT1 by si-MALAT1 inhibited TNF-α induced apoptosis in cardiomyocytes and the inhibition of apoptosis by si-MALAT-1 was rescued by an miR-26a inhibitor (Figure 6D). This result suggests that MALAT1/miR-26a manipulation has a direct regulatory effect on TNF-α-induced apoptosis. Lastly, we found that the downregulation of MALAT1 by si-MALAT1 increases cell viability and decreases the activity of LDH, consistent with the effects of TNF-α knockdown (si-TNF-α) (Figure 6E and F).
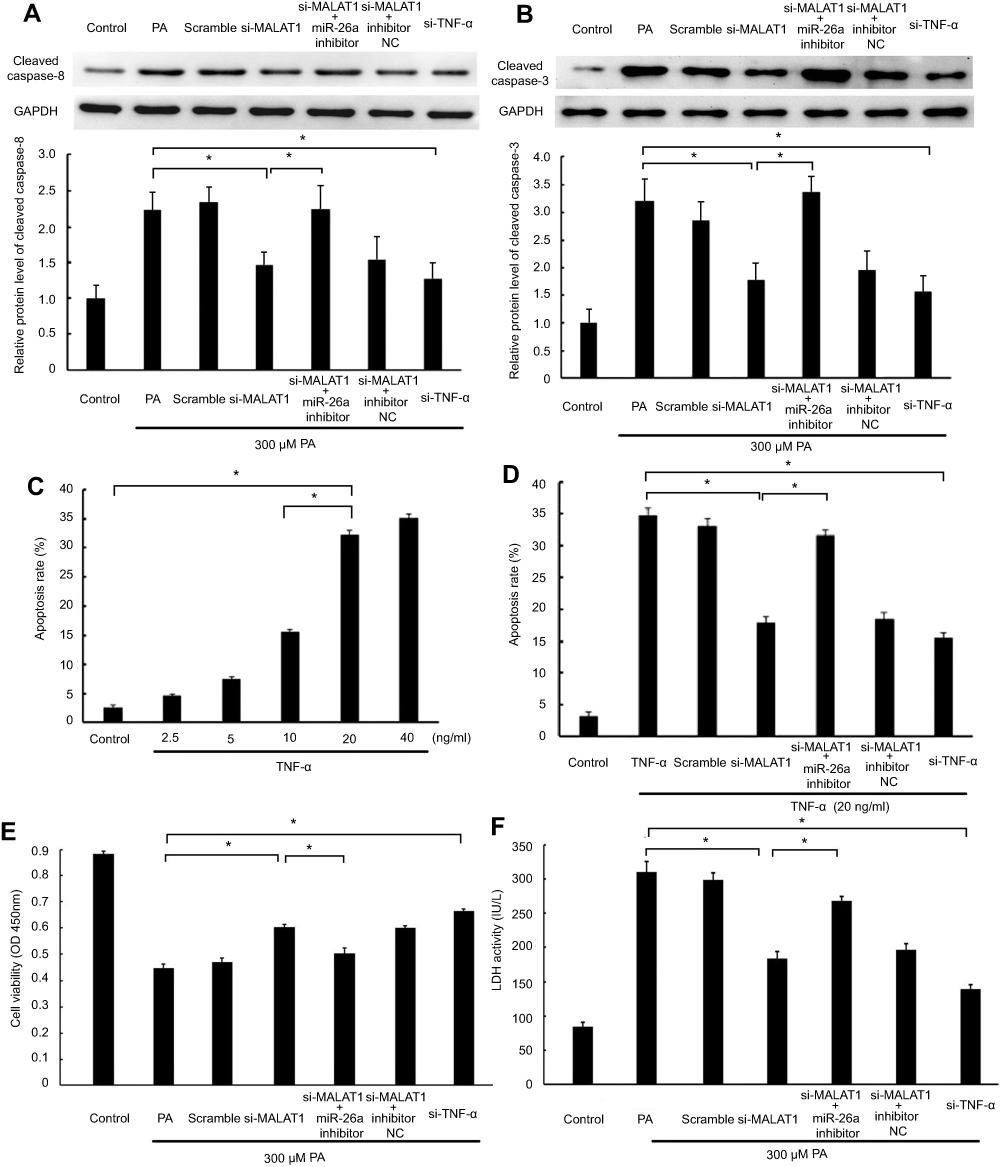 | Figure 6 MALAT1/miR-26a manipulation regulated PA-induced cell death via a TNF-α-induced apoptosis pathway. MALAT1 negative control (scramble) and MALAT1-specific siRNA (si-MALAT1) alone or in combination with an miR-26a inhibitor or miR-26 negative control (inhibitor NC) and TNF-α-specific siRNA (si-TNF-ɑ) were transfected in AC16 cells, followed by palmitic acid (PA)/TNF-α treatment. (A) The protein expression of cleaved caspase-8 was detected by western blotting. (B) The protein expression of cleaved caspase-3 was detected by western blotting. (C) AC16 cells were treated with different concentrations of TNF-α for 12 h to induce apoptosis. (D) MALAT1/miR-26a manipulation regulated TNF-α-induced apoptosis. (E) Cell viability was determined by CCK8 assay. (F) Lactate dehydrogenase (LDH) activity in the culture medium was detected by spectrophotometry. *P<0.05. Data are presented as the mean ± standard error from three independent experiments.Abbreviations: si-TNF-α, TNF-α-specific siRNA; NC, negative control, MALAT1, metastasis-associated lung adenocarcinoma transcript 1; CCK8, cell counting kit-8. |
Taken together, these results suggest that MALAT1/miR-26a manipulation may control PA-induced cell death, via the TNF-α-induced apoptosis pathway, at least in part.
Discussion
In our study, we reported the expression and role of MALAT1 on SFA-induced myocardial lipotoxic injury for the first time. MALAT1 is highly conserved among mammals and is ubiquitously expressed in all tissues.22 It is usually induced under various stress conditions, such as ischemia or hypoxia,11,12,23 dust exposure,13 and hyperglycemia.16 Thus, we inferred that MALAT1 may be induced in myocardial cells by SFAs. As expected, we found that MALAT1 was upregulated in PA-treated human adult ventricular cardiomyocytes (AC16 cells). We further found that the downregulation of MALAT1 alleviates the extent of PA-induced myocardial injury in vitro. This result is similar to those of Wang et al, who found that the knockdown of MALAT1 expression dramatically suppresses PA-induced lipid accumulation in HepG2 cells.14 Taken together, MALAT1 is a new putative therapeutic target for SFA-induced myocardial lipotoxic injury.
There is growing evidence that myocardial lipotoxic injury induced by SFAs, especially PA, is involved in the activation of inflammatory and innate immune responses.5,6 PA can promote the development of myocardial injury by direct interactions with myeloid differentiation protein 2, a TLR4 accessory protein, thereby resulting in the activation of TLR4/NF-κB signaling for the regulation of pro-inflammatory molecules.5 Several studies have demonstrated that MALAT1 is a modulator of hyperglycemia or the lipopolysaccharide-induced inflammatory response and the overexpression of MALAT1 upregulates the expression of pro-inflammatory molecules, including TNF-α, IL-1β, and IL-6.16,17 In the present study, we demonstrated that the downregulation of MALAT1 inhibits PA-induced inflammatory responses in myocardial cells by repressing the levels of TNF-α and IL-1β. Thus, our results provide additional evidence that MALAT1 is a modulator of the inflammatory process and regulates the expression of pro-inflammatory molecules.
Like other lncRNAs, MALAT1 could regulate gene expression by various mechanisms. For instance, MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation.24 Moreover, MALAT1 could also bind to several miRNAs, such as miR-144,9 miR-125,25 miR-142,26 and miR-129,26 to regulate target gene expression by a ceRNA regulatory mechanism. In the present study, a luciferase reporter assay and RT-qPCR confirmed that MALAT1 specifically binds to miR-26a and there exists a reciprocal negative regulatory relationship between miR-26a and MALAT1. These findings lay a foundation for further analyzes of a potential ceRNA regulatory network.
HMGB1 is a known target gene of miR-26a and miR-26a inhibits HMGB1 expression, resulting in the inhibition of the TLR4/NF-κB-mediated inflammatory response.19 Accordingly, we hypothesized that MALAT1 regulates HMGB1 expression by acting as a ceRNA for miR-26a. As expected, we found that the downregulation of MALAT1 inhibited the expression of HMGB1, leading to the repression of the TLR4/NF-κB signaling pathway. These repressive effects of the downregulation of MALAT1 were mediated by miR-26a. Similarly, Liu et al reported that MALAT1 regulates the expression of HMGB1 via miR-142-3p and miR-129-5p in osteosarcoma cells.26 We consider that MALAT1 regulates HMGB1 expression by miR-26a binding, thereby inhibiting the activation of the TLR4/NF-κB signaling pathway. Furthermore, we found that the phenotypes of myocardial inflammatory injury by the downregulation of MALAT1 were rescued by an miR-26a inhibitor, which confirmed that miR-26a is required for the suppressive effect of MALAT1 on PA-induced myocardial inflammatory injury. Taken together, these results demonstrated that MALAT1 regulates PA-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis.
However, this study had two limitations. First, the role of MALAT1 in PA-induced myocardial injury was only confirmed in a single myocardial cell line. Thus, these results should be verified in more myocardial cell lines and animal models. Second, we did not illuminate how PA induces MALAT1 expression in cardiomyocytes. Previous studies have suggested that various transcription factors, such as specificity protein 1 and hypoxia-inducible factor-2a,27,28 could recognize the promoter region of MALAT1 to induce expression. Thus, further studies should evaluate whether PA promotes the interaction between transcription factors and the promoter region of MALAT1.
Conclusion
We demonstrated that MALAT1 is induced in cardiomyocytes by SFAs. The knockdown of MALAT1 alleviates SFA-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Our findings provide new insight into the mechanism underlying myocardial lipotoxic injury.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81800232 and 81670320) and Natural Science Foundation of Liaoning Province (No. 201602826).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14(3):281–287. doi:10.1097/01.mol.0000073508.41685.7f
2. Zlobine I, Gopal K, Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta. 2016;1861(10):1555–1568. doi:10.1016/j.bbalip.2016.02.011
3. Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801(3):311–319. doi:10.1016/j.bbalip.2009.09.023
4. Zou L, Li X, Wu N, Jia P, Liu C, Jia D. Palmitate induces myocardial lipotoxic injury via the endoplasmic reticulum stress-mediated apoptosis pathway. Mol Med Rep. 2017;16(5):6934–6939. doi:10.3892/mmr.2017.7404
5. Wang Y, Qian Y, Fang Q, et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat Commun. 2017;8:13997. doi:10.1038/ncomms13997
6. Zeng C, Zhong P, Zhao Y, et al. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-κB both in vitro and in vivo. J Mol Cell Cardiol. 2015;79:1–12. doi:10.1016/j.yjmcc.2014.10.002
7. Sletten AC, Peterson LR, Schaffer JE. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med. 2018;284(5):478–491. doi:10.1111/joim.12728
8. Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–8041. doi:10.1038/sj.onc.1206928
9. Wang Y, Zhang Y, Yang T, et al. Long non-coding RNA MALAT1 for promoting metastasis and proliferation by acting as a ceRNA of miR-144-3p in osteosarcoma cells. Oncotarget. 2017;8(35):59417–59434. doi:10.18632/oncotarget.19727
10. Zhang ZC, Tang C, Dong Y, Zhang J, Yuan T, Li XL. Targeting LncRNA-MALAT1 suppresses the progression of osteosarcoma by altering the expression and localization of β-catenin. J Cancer. 2018;9(1):71–80. doi:10.7150/jca.22113
11. Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ. Long noncoding RNA Malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci. 2017;37(7):1797–1806. doi:10.1523/JNEUROSCI.3389-16.2017
12. Zhao ZH, Hao W, Meng QT, Du XB, Lei SQ, Xia ZY. Long non-coding RNA MALAT1 functions as a mediator in cardioprotective effects of fentanyl in myocardial ischemia-reperfusion injury. Cell Biol Int. 2017;41(1):62–70. doi:10.1002/cbin.10701
13. Yan W, Wu Q, Yao W, et al. MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K p85 and is sponged by lncRNA MALAT1. Sci Rep. 2017;7(1):11313. doi:10.1038/s41598-017-11904-8
14. Yan C, Chen J, Chen N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability. Sci Rep. 2016;6:22640. doi:10.1038/srep22640
15. Yang H, Liang N, Wang M, et al. Long noncoding RNA MALAT-1 is a novel inflammatory regulator in human systemic lupus erythematosus. Oncotarget. 2017;8(44):77400–77406. doi:10.18632/oncotarget.20490
16. Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015;19(6):1418–1425. doi:10.1111/jcmm.12576
17. Zhao G, Su Z, Song D, Mao Y, Mao X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-κB. FEBS Lett. 2016;590(17):2884–2895. doi:10.1002/1873-3468.12315
18. Sen R, Ghosal S, Das S, Balti S, Chakrabarti J. Competing endogenous RNA: the key to posttranscriptional regulation. ScientificWorldJournal. 2014;2014:896206. doi:10.1155/2014/896206
19. Yao L, Lv X, MicroRNA WX. 26a inhibits HMGB1 expression and attenuates cardiac ischemia-reperfusion injury. J Pharmacol Sci. 2016;131(1):6–12. doi:10.1016/j.jphs.2015.07.023
20. Oh CC, Nguy MQ, Schwenke DC, Migrino RQ, Thornburg K, Reaven P. p38α mitogen-activated kinase mediates cardiomyocyte apoptosis induced by palmitate. Biochem Biophys Res Commun. 2014;450(1):628–633. doi:10.1016/j.bbrc.2014.06.023
21. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(1):231–238. doi:10.1093/nar/gkv1270
22. Wu Y, Huang C, Meng X, Li J. Long noncoding RNA MALAT1: insights into its biogenesis and implications in human disease. Curr Pharm Des. 2015;21(34):5017–5028.
23. Michalik KM, You X, Manavski Y, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389–1397. doi:10.1161/CIRCRESAHA.114.303265
24. Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. doi:10.1016/j.molcel.2010.08.011
25. Chen H, Wang X, Yan X, Cheng X, He X, Zheng W. LncRNA MALAT1 regulates sepsis-induced cardiac inflammation and dysfunction via interaction with miR-125b and p38 MAPK/NF-κB. Int Immunopharmacol. 2017;55:69–76. doi:10.1016/j.intimp.2017.11.038
26. Liu K, Huang J, Ni J, et al. MALAT1 promotes osteosarcoma development by regulation of HMGB1 via miR-142-3p and miR-129-5p. Cell Cycle. 2017;16(6):578–587. doi:10.1080/15384101.2017.1288324
27. Li S, Wang Q, Qiang Q, et al. Sp1-mediated transcriptional regulation of MALAT1 plays a critical role in tumor. J Cancer Res Clin Oncol. 2015;141(11):1909–1920. doi:10.1007/s00432-015-1951-0
28. Yuan P, Cao W, Zang Q, Li G, Guo X, Fan J. The HIF-2α-MALAT1-miR-216b axis regulates multi-drug resistance of hepatocellular carcinoma cells via modulating autophagy. Biochem Biophys Res Commun. 2016;478(3):1067–1073. doi:10.1016/j.bbrc.2016.08.065
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.