")
Back to Journals » OncoTargets and Therapy » Volume 11
Downregulation of lncRNA ANRIL suppresses growth and metastasis in human osteosarcoma cells
Authors Guan H, Mei Y, Mi Y, Li C, Sun X, Zhao X, Liu J, Cao W, Li Y, Wang Y
Received 6 April 2018
Accepted for publication 25 May 2018
Published 14 August 2018 Volume 2018:11 Pages 4893—4899
DOI https://doi.org/10.2147/OTT.S170293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Hongya Guan,1 Yingwu Mei,2 Yang Mi,2 Cheng Li,3 Xiaoya Sun,2 Xuefeng Zhao,2 Jia Liu,1 Wei Cao,1 Yuebai Li,2 Yisheng Wang3
1Translational Medical Center, Zhengzhou Central Hospital Affiliated to Zhengzhou University, Zhengzhou 450007, People’s Republic of China; 2Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Zhengzhou University, Zhengzhou 450001, People’s Republic of China; 3Department of Orthopedic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, People’s Republic of China
Background: This study was designed to research the potential function of lncRNA ANRIL in osteosarcoma (OS).
Materials and methods: Quantitative real-time PCR, cell counting kit-8, wound healing assay, Transwell assay, flow cytometric analysis, caspase activity analysis, and Western blot were carried out.
Results: ANRIL was remarkably upregulated in human OS tissues and cells, and knockdown of ANRIL significantly suppressed MG63 cell proliferation, migration, and invasion and promoted apoptosis. Moreover, our mechanistic research findings verified that ANRIL-influenced growth and apoptosis may be partly through regulation of caspase-3 and Bcl-2. Migration and invasion were influenced via ANRIL-mediated regulation of MTA1, TIMP-2, and E-cadherin.
Conclusion: Our finding demonstrates that ANRIL plays vital roles in OS growth and metastasis.
Keywords: osteosarcoma, ANRIL, proliferation, invasion, apoptosis, long noncoding RNA
Introduction
Osteosarcoma (OS) is a common primary malignant bone tumor that occurs frequently in young children and adolescents. This condition has a global incidence of ~1–3 per million annually and is characterized by high levels of invasiveness and early systemic metastasis.1,2 The prognosis of OS has significantly improved due to improvements in surgical methods and the application of new chemotherapy drugs, but ~40% of patients still experience tumor metastasis. Therefore, it is urgently necessary to identify diagnostic and prognostic biomarkers and elucidate the underlying molecular mechanisms of OS.
Long noncoding RNAs (lncRNAs) are a class of noncoding RNA with length >200 nucleotides that play vital roles in epigenetic regulation, cell differentiation, etc. lncRNA dysfunction is closely related to the pathogenesis of multiple human diseases including cancer.3–6 More specifically, lncRNAs have been identified as oncogenes and as antioncogenes in OS, for instance, PCAT1, 91H, SPRY4-IT1, MALAT1, and FGFR3-AS1.7–11 The lncRNA antisense noncoding RNA gene at the INK4 locus (ANRIL) was transcribed from the INK4b–ARF–INK4a gene cluster,12 which has been proven to be upregulated in multiple cancers, such as breast cancer, cervical cancer, nasopharyngeal carcinoma, and thyroid cancer.13–16 However, the impact of ANRIL on OS biological behavior has not been studied in depth; therefore, our study aimed to explore the roles and molecular mechanisms of ANRIL in OS.
Here, we demonstrated that ANRIL acts as a tumor promoter while further showing that knockdown of ANRIL inhibited OS cell proliferation, invasion, and migration and promoted apoptosis through regulating Bcl-2, caspase-3, MTA1, TIMP-2, and E-cadherin. This study provided data on the crucial roles of ANRIL in OS growth and metastasis, potentially leading to the use of ANRIL as an oncotherapeutic molecular target.
Materials and methods
Tissue sample
The 30 OS tissues and paired adjacent normal tissues were obtained from patients who never received therapy prior to surgery between January 2015 and September 2016 at the First Affiliated Hospital of Zhengzhou University. The specimens were immediately frozen in liquid nitrogen and stored at −80°C until use. Diagnosis was confirmed by histopathological analysis of the tissues. Each patient signed an informed consent form, and the research was conducted with the approval of the Ethics Committee of the First Affiliated Hospital of Zhengzhou University.
Quantitative real-time PCR (qRT-PCR)
RNAiso Plus (TaKaRa, Dalian, People’s Republic of China) was used to extract RNA from tissues and cells, and then a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA) to determine RNA concentration. Then, 1,000 ng total RNA was reverse transcribed for qRT-PCR, which was performed with an ABI 7500 system using SYBR Green Premix Ex Taq (Takara). The primer sequences used were as follows: 5′-CCACATCGCTCAGACACCAT-3′ (forward) and 5′-ACCAGGCGCCCAATACG-3′ (reverse) for glyceraldehyde phosphate dehydrogenase (GAPDH); and 5′-GGGCCTCAGTGGCACATACC-3′ (forward) and 5′-TGCTCTATCCGCCAATCAGG-3′ (reverse) for ANRIL. The sequences of the siRNA targeting the ANRIL coding sequence (si-ANRIL) were as follows: 5′-GGUCAUCUCAUUGCUCUAUTT-3′ (forward) and 5′-AUAGAGCAAUGAGAUGACCTT-3′ (reverse). The nontargeting siRNA (si-NC) sequences were as follows: 5′-CUCCGAACGUGUCACGUT-3′ (forward) and 5′-CGUGACACGUUCGGAGAAT-3′ (reverse). The 2−ΔΔCt method was used to calculate the relative expression of ANRIL, which was normalized to GAPDH expression. All reactions were executed in triplicate.
Cell cultures and transfection
Human osteoblast hFOB1.19 cells and 4 OS cell lines (HOS, U-2OS, MG63, and SAOS-2) were purchased from the Chinese Academy of Medical Sciences (Beijing, People’s Republic of China). hFOB1.19 cells were cultured in Dulbecco’s Modified Eagle’s Medium at 35°C in 5% CO2, and osteosarcoma cell lines were cultured at 37°C in 5% CO2 in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA). MG63 cells were subcultured in 6-well plates and were transiently transfected with si-ANRIL or si-NC using Lipofectamine 2000 (Thermo Fisher Scientific). Transfection efficiency was evaluated with real-time PCR 24 h after transfection. The transfected cells were cultured and cell behavior was examined.
Cell proliferation assay
Transfected MG63 cells were reseeded in 96-well plates and then cultured as previously described. Subsequently, 10 μL of cell counting kit-8 solution (Beyotime, Jiangsu, People’s Republic of China) was added to each well and the cells were incubated at 37°C for 2 h. We then used a microplate reader (Molecular Devices, San Jose, CA, USA) to detect the absorbance at 450 nm at different time points (24, 48, 72, and 96 h). Five replicate wells were prepared for each experimental group.
Flow cytometric analysis of apoptosis
Apoptosis of MG63 cells was detected with the Annexin V–FITC/PI apoptosis detection kit (KeyGEN Biotech, Nanjing, People’s Republic of China). The resulting mixture was then used for flow cytometric analysis. Briefly, MG63 cells were harvested 48 h after transfection, resuspended with 400 μL of 1× binding buffer, and then double-stained with 5 μL Annexin V–FITC and 10 μL PI. Lastly, a flow cytometer (FACScan; BD Biosciences, Franklin Lakes, NJ, USA) analysis of the sample was performed for 30 min.
Caspase-3 activity assay
A caspase activity assay kit (Beyotime) was used to measure cellular caspase activity. Briefly, MG63 cells were harvested 48 h after treatment with si-NC or si-ANRIL, washed with PBS, and then resuspended in cold lysis buffer. Caspase-3 substrate (5 μL) was added to the supernatant after centrifugation and the mixture was incubated at 37°C in the dark for 4 h. Finally, a microplate reader (Infinite M200, Tecan, Männedorf, Switzerland) was used to measure absorbance values and analyze apoptotic ability.
Wound-healing assay
Twenty-four hours after transfection, cells were reseeded into 6-well plates and cultured with serum-free medium. After cells reached 90% confluence, wounds were made with a pipette tip by scratching of the cell monolayer. PBS was used to wash the wounded monolayers and remove cell debris. At 0 and 12 h after wounding, the distance between the 2 edges of wound was measured with micrographs.
Transwell assay
Twenty-four hours post-transfection, the cells were reseeded into a Matrigel-coated upper chamber (8 μm pore size) of a Transwell assay system (Corning, NY, USA). After the cells were cultured for 24 h, we used a cotton swab to scrape off the noninvading cells on the upper surface of the membrane and stained the invading cells with 0.1% crystal violet. Twenty minutes after staining, cells were counted and imaged with a microscope.
Western blot
In Western blot analysis, protein was extracted with RIPA radio immunoprecipitation assay (RIPA) protein extraction reagent (Beyotime) containing phenylmethanesulfonyl fluoride (PMSF), and the protein concentrations were detected using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Proteins were separated by sulfate–polyacrylamide gel lectrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). The membranes were blocked in tris-buffered saline Tween-20 (TBST) with 5% nonfat milk for 2 h at room temperature and then incubated with primary antibodies against Bcl-2 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), caspase-3 (1:800, Santa Cruz Biotechnology), MTA1 (1:200, Santa Cruz Biotechnology), TIMP-2 (1:800, Santa Cruz Biotechnology), and E-cadherin (1:500, Santa Cruz Biotechnology) at 4°C overnight. The next day, membranes were washed 3 times with 1× TBST buffer and then incubated with the corresponding secondary antibody at 37°C for 2 h. After being washed 3 times, the membranes were visualized using an ECL Plus Detection Kit (Pierce, Rockford, IL, USA). GAPDH served as the internal control.
Statistical analysis
SPSS 21.0 for windows (IBM Corporation, Armonk, NY, USA) was used to analyze all data, which are presented as mean ± SD. Each experiment was repeated at least 3 times, and differences between groups were evaluated by the Student’s t-test or one-way analysis of variance. Values of P<0.05 were considered to be significant.
Results
ANRIL was upregulated in OS tissues and cells
The expression of ANRIL in human OS tissues and cell lines were detected by qRT-PCR and the results revealed that relative ANRIL levels in OS tissues were higher than those in adjacent normal tissues (Figure 1A). Additioally, ANRIL expression was elevated in OS cell lines (HOS, U-2OS, MG63 and SAOS-2) than in hFOB1.19 osteoblast cell lines (Figure 1B).
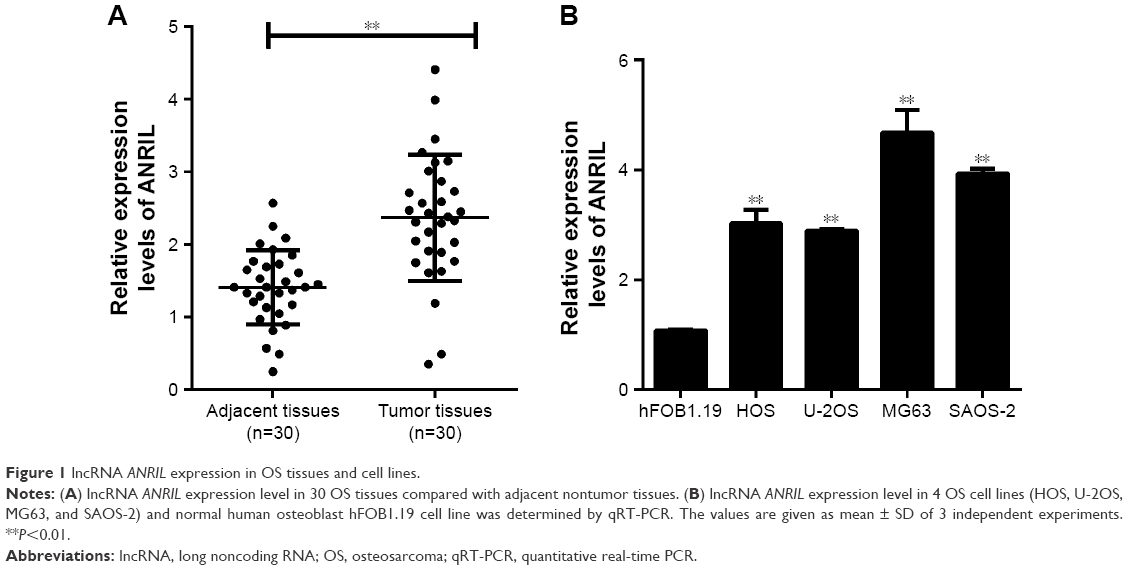 | Figure 1 lncRNA ANRIL expression in OS tissues and cell lines. |
Suppression of ANRIL expression inhibited cell proliferation
Transfection efficiency was determined with qRT-PCR, and the results demonstrated that the expression of ANRIL was decreased in MG63 cells after treatment with siRNA-ANRIL (Figure 2A). Then, cell counting kit-8 assay was used to observe the effect of decreased ANRIL on MG63 cell proliferation, revealing that inhibition of ANRIL suppressed cell proliferation (Figure 2B).
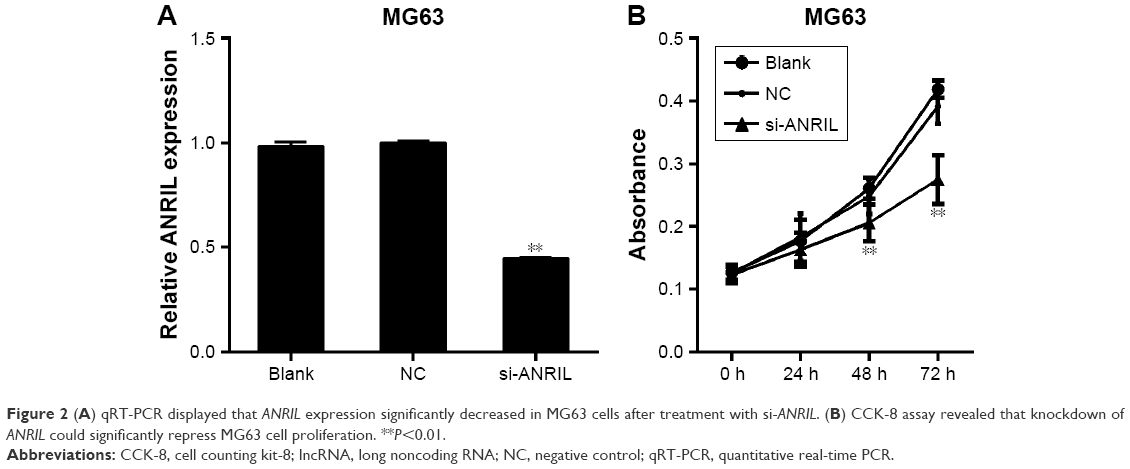 | Figure 2 (A) qRT-PCR displayed that ANRIL expression significantly decreased in MG63 cells after treatment with si-ANRIL. (B) CCK-8 assay revealed that knockdown of ANRIL could significantly repress MG63 cell proliferation. **P<0.01. |
Suppression of ANRIL expression promoted cell apoptosis
In order to investigate the effect of decreased ANRIL levels on cell apoptosis, we carried out flow cytometric analysis and caspase-3 activity assays. The results showed that knockdown of ANRIL in MG63 cells resulted in a marked increase in their capacity for cell apoptosis (Figure 3A–C). Furthermore, Bcl-2 expression was significantly decreased, while caspase-3 expression was increased in ANRIL knockdown cells compared with control cells (Figure 3D and E).
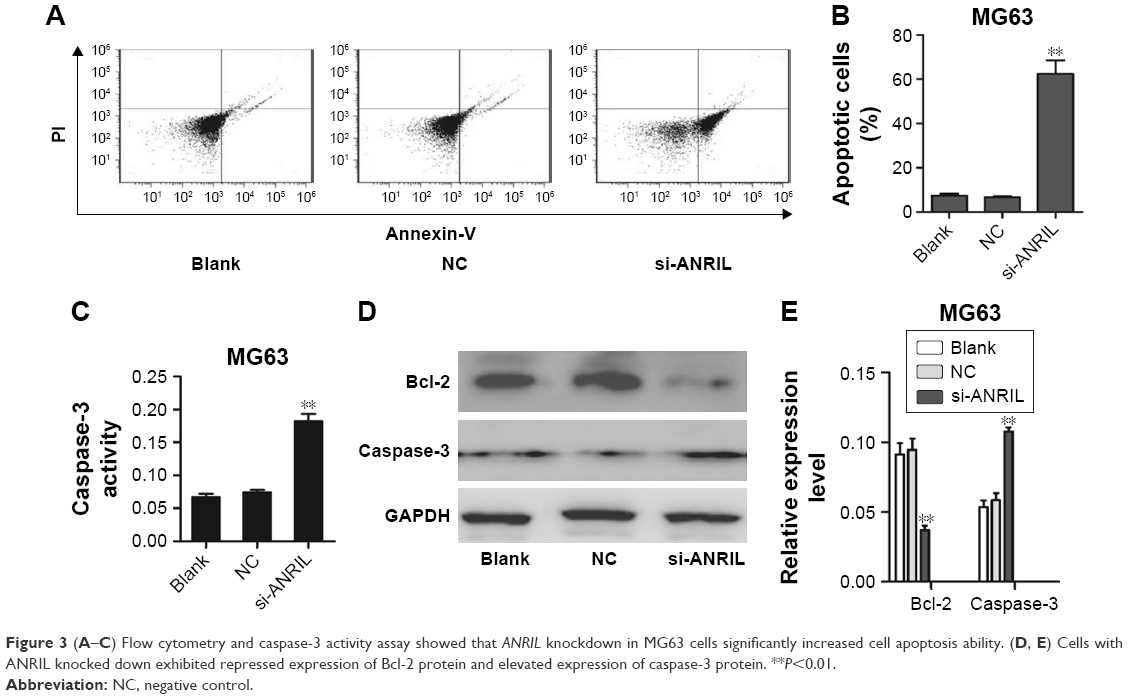 | Figure 3 (A–C) Flow cytometry and caspase-3 activity assay showed that ANRIL knockdown in MG63 cells significantly increased cell apoptosis ability. (D, E) Cells with ANRIL knocked down exhibited repressed expression of Bcl-2 protein and elevated expression of caspase-3 protein. **P<0.01. |
Suppression of ANRIL expression inhibited cell migration and invasion
In order to examine the effect of decreased ANRIL on OS cell migration and invasion, we performed wound-healing and Transwell assays. The wound-healing assay uncovered that the migratory activity of MG63 cells transfected with si-ANRIL was decreased (Figure 4A). In addition, the Transwell assay revealed that the invasive activity of MG63 cells transfected with si-ANRIL was reduced compared with that of cells transfected with siRNA-NC and untransfected cells (Figure 4B and C). Furthermore, the expression of the tumor metastasis-related protein MTA1 was significantly decreased, and TIMP-2 as well as E-cadherin levels were increased in cells with ANRIL knocked down, compared with control cells (Figure 4D and E).
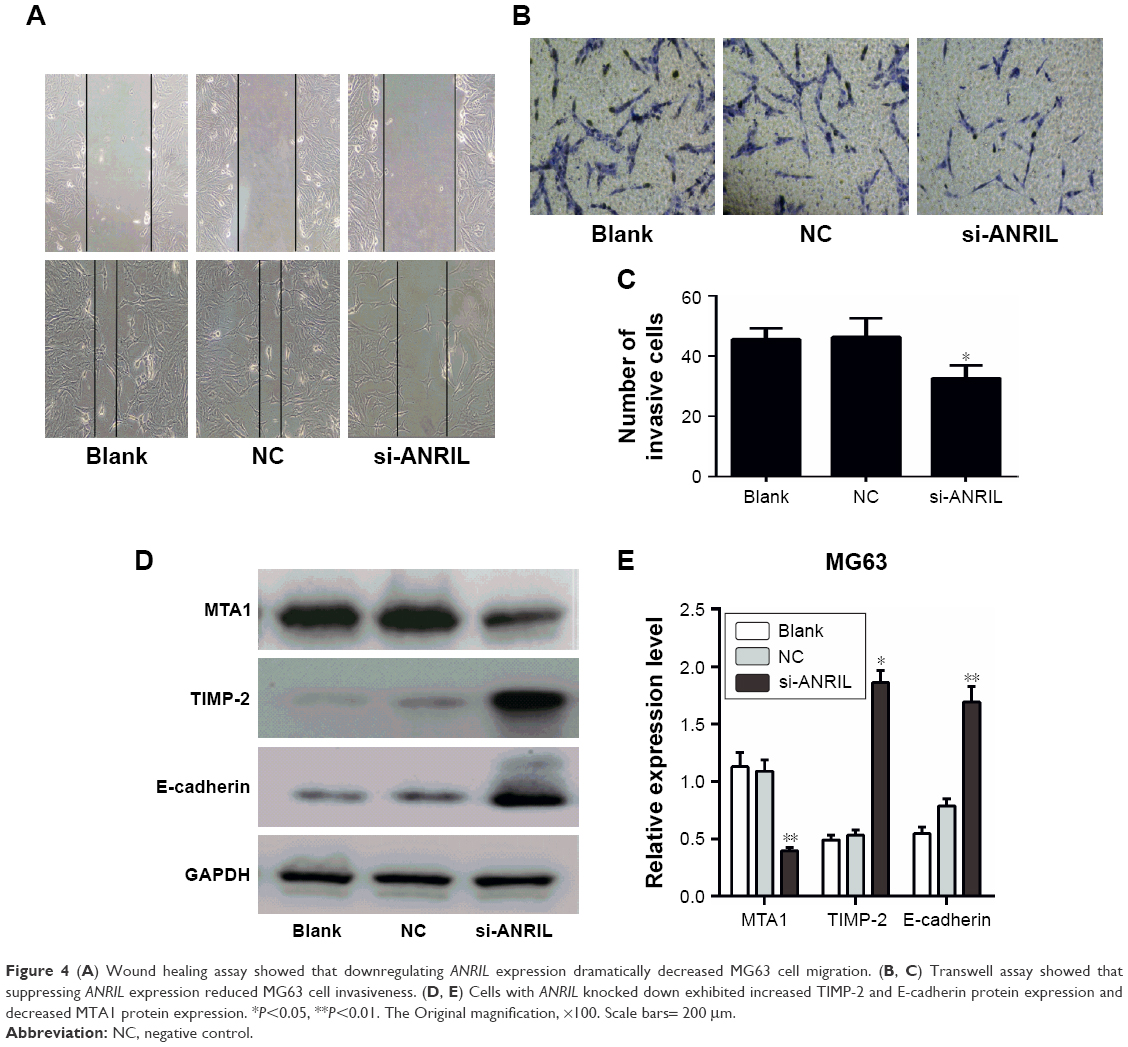 | Figure 4 (A) Wound healing assay showed that downregulating ANRIL expression dramatically decreased MG63 cell migration. (B, C) Transwell assay showed that suppressing ANRIL expression reduced MG63 cell invasiveness. (D, E) Cells with ANRIL knocked down exhibited increased TIMP-2 and E-cadherin protein expression and decreased MTA1 protein expression. *P<0.05, **P<0.01. The Original magnification, ×100. Scale bars= 200 μm. |
Discussion
An increasing number of studies have implicated that lncRNAs act as fundamental regulators in diverse biological processes, where aberrant expression was found to be strongly linked to the tumorigenesis and progression of human cancers, including in OS. For example, ZEB1-AS1 was confirmed to be associated with tumor size, Enneking stage, tumor metastasis, and recurrence. Functional experiments showed that ZEB1-AS1 promoted OS proliferation and migration by activating ZEB1 transcription.17 Elevated expression of HULC was correlated with shorter overall survival of patients with OS, and its suppression inhibited OS growth.18 Additional similar functional studies have demonstrated potent pro- and antitumorigenic activity of lncRNAs in cancer. Thus, identifying and elucidating the biological function and underlying the mechanisms of lncRNAs in OS may help us to fully understand the pathogenesis of this malignancy.
The ANRIL gene is located at chromosome 9p21, with a size of 126.3 kb consisting of 19 exons, and encodes a 3834-nucleotide lncRNA.19 ANRIL has been reported to be overexpressed and to exert protumorigenic roles in bladder cancer, lung cancer, cervical cancer, esophageal squamous cell carcinoma, gastric cancer, and hepatocellular carcinoma.20–26 Zhao et al16 reported that in thyroid cancer cells, ANRIL was significantly upregulated and promoted invasion as well as metastasis by attenuating the transforming growth factor β (TGF-β)/Smad signaling pathway. Additionally, ANRIL was discovered to be upregulated in colorectal cancer tissues, and knockdown of it could inhibit cell proliferation, migration, and invasion.27 All of these discoveries suggest that ANRIL can function as an oncogene. However, to the best of our knowledge, the roles of ANRIL in OS, in particular, have not been previously examined.
Here, our study demonstrated that ANRIL expression was markedly higher in human OS tissues and cell lines. Next, we analyzed the fundamental function of ANRIL in regulating the malignant biological behaviors of OS. To do this, MG63 cells were transfected with siRNA-ANRIL. ANRIL expression was confirmed to be significantly decreased in the siRNA-ANRIL-transfected cells, compared to ANRIL expression in siRNA-NC-transfected or nontransfected cells where there was no change. Additionally, we found that downregulation of ANRIL expression could restrain OS cell proliferation and facilitate cell apoptosis. Previous studies have exhibited that silencing of ANRIL could decrease Bcl-2 expression and increase caspase-3 expression in bladder cancer.22 In line with these findings, mechanistic investigations in present study suggested that the antigrowth and proapoptotic effects of ANRIL occurred via increased Bcl-2 expression and decreased caspase-3 expression. Wound healing and Transwell assays demonstrated that downregulation of ANRIL expression attenuated the migration and invasion ability of MG63 cells. A review of the literature have revealed several genes were dysregulated after ANRIL silencing in ovarian cancer cells, including MTA1, TIMP-2, and E-cadherin.23 To investigate the mechanisms by which ANRIL affected migration and invasion of OS cells, we conducted an initial, tentative analysis of these genes. Surprisingly, we discovered that MTA1 protein expression was significantly decreased in OS cells with ANRIL knocked down, while TIMP-2 and E-cadherin levels were increased, suggesting that ANRIL promoted OS cell migration and invasion maybe partly through upregulating MTA1 and downregulating TIMP-2 and E-cadherin.
Conclusion
Taken together, we have shown that a significant upregulation of ANRIL in OS tissues and cells acts as a tumor promoter. For the first time, we have provided evidence that ANRIL influences OS cell proliferation and apoptosis through the regulation of Bcl-2 and caspase-3, as well as affecting cell migration and invasion through regulation of MTA1, TIMP-2, and E-cadherin. These findings improve our understanding of OS pathogenesis and may provide a novel promising therapeutic target for OS. Further studies are needed to investigate the detailed molecular mechanisms by which ANRIL regulates OS.
Disclosure
The authors report no conflicts of interest in this work.
References
Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125(1):229–234. | ||
Bousquet M, Noirot C, Accadbled F, et al. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann Oncol. 2016;27(4):738–744. | ||
Lorenzen JM, Thum T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol. 2016;12(6):360–373. | ||
Wan P, Su W, Zhuo Y. The role of long noncoding RNAs in neurodegenerative diseases. Mol Neurobiol. 2017;54(3):2012–2021. | ||
Luo Q, Chen Y. Long noncoding RNAs and Alzheimer’s disease. Clin Interv Aging. 2016;11:867–872. | ||
Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. J Clin Invest. 2016;126(8):2775–2782. | ||
Zhang X, Zhang Y, Mao Y, Ma X. The lncRNA PCAT1 is correlated with poor prognosis and promotes cell proliferation, invasion, migration and EMT in osteosarcoma. Onco Targets Ther. 2018;11:629–638. | ||
Xia WK, Lin QF, Shen D, Liu ZL, Su J, Mao WD. Clinical implication of long noncoding RNA 91H expression profile in osteosarcoma patients. Onco Targets Ther. 2016;9:4645–4652. | ||
Xu J, Ding R, Xu Y. Effects of long non-coding RNA SPRY4-IT1 on osteosarcoma cell biological behavior. Am J Transl Res. 2016;8(12):5330–5337. | ||
Luo W, He H, Xiao W, et al. MALAT1 promotes osteosarcoma development by targeting TGFA via MIR376A. Oncotarget. 2016;7(34):54733–54743. | ||
Sun J, Wang X, Fu C, et al. Long noncoding RNA FGFR3-AS1 promotes osteosarcoma growth through regulating its natural antisense transcript FGFR3. Mol Biol Rep. 2016;43(5):427–436. | ||
Li CH, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol. 2013;45(8):1895–1910. | ||
Meseure D, Vacher S, Alsibai KD, et al. Expression of ANRIL-polycomb complexes-CDKN2A/B/ARF genes in breast tumors: identification of a two-gene (EZH2/CBX7) signature with independent prognostic value. Mol Cancer Res. 2016;14(7):623–633. | ||
Naemura M, Murasaki C, Inoue Y, Okamoto H, Kotake Y. Long noncoding RNA ANRIL regulates proliferation of non-small cell lung cancer and cervical cancer cells. Anticancer Res. 2015;35(10):5377–5382. | ||
Zou ZW, Ma C, Medoro L, et al. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing side-population stem-like cancer cells. Oncotarget. 2016;7(38):61741–61754. | ||
Zhao JJ, Hao S, Wang LL, et al. Long non-coding RNA ANRIL promotes the invasion and metastasis of thyroid cancer cells through TGF-β/Smad signaling pathway. Oncotarget. 2016;7(36):57903–57918. | ||
Liu C, Lin J. Long noncoding RNA ZEB1-AS1 acts as an oncogene in osteosarcoma by epigenetically activating ZEB1. Am J Transl Res. 2016;8(10):4095–4105. | ||
Sun XH, Yang LB, Geng XL, Wang R, Zhang ZC. Increased expression of lncRNA HULC indicates a poor prognosis and promotes cell metastasis in osteosarcoma. Int J Clin Exp Pathol. 2015;8(3):2994–3000. | ||
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6(12):e1001233. | ||
Zhu H, Li X, Song Y, Zhang P, Xiao Y, Xing Y. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochem Biophys Res Commun. 2015;467(2):223–228. | ||
Lu Y, Zhou X, Xu L, Rong C, Shen C, Bian W. Long noncoding RNA ANRIL could be transactivated by c-Myc and promote tumor progression of non-small-cell lung cancer. Onco Targets Ther. 2016;9:3077–3084. | ||
Nie FQ, Sun M, Yang JS, et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14(1):268–277. | ||
Qiu JJ, Lin YY, Ding JX, Feng WW, Jin HY, Hua KQ. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int J Oncol. 2015;46(6):2497–2505. | ||
Chen D, Zhang Z, Mao C, et al. ANRIL inhibits p15(INK4b) through the TGFβ1 signaling pathway in human esophageal squamous cell carcinoma. Cell Immunol. 2014;289(1–2):91–96. | ||
Zhang EB, Kong R, Yin DD, et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget. 2014;5(8):2276–2292. | ||
Hua L, Wang CY, Yao KH, Chen JT, Zhang JJ, Ma WL. High expression of long non-coding RNA ANRIL is associated with poor prognosis in hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8(3):3076–3082. | ||
Sun Y, Zheng ZP, Li H, Zhang HQ, Ma FQ. ANRIL is associated with the survival rate of patients with colorectal cancer, and affects cell migration and invasion in vitro. Mol Med Rep. 2016;14(2):1714–1720. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.