")
Back to Journals » OncoTargets and Therapy » Volume 12
Downregulation of KLF13 through DNMT1-mediated hypermethylation promotes glioma cell proliferation and invasion
Authors Wu R, Yun Q , Zhang JP, Bao J
Received 21 September 2018
Accepted for publication 1 December 2018
Published 22 February 2019 Volume 2019:12 Pages 1509—1520
DOI https://doi.org/10.2147/OTT.S188270
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Rile Wu, Qiang Yun, Jianping Zhang, Jingang Bao
Department of Neurosurgery, Inner Mongolia People’s Hospital, Hohhot 010017, China
Background: Recent evidence indicates that Kruppel-like factor 13 (KLF13) has critical roles in regulating cell differentiation, proliferation and may function as a tumor suppressor. However, its role in glioma progression is poorly understood.
Methods: Public database was used to explore the expression and prognostic value of KLF13 in glioma. Cell proliferation and invasion assays were used to explore the role of KLF13. Bisulfite sequencing and ChIP assay were used to determine the methylation of KLF13 promoter in glioma and the regulation of KLF13 by DNMT1.
Results: We found that KLF13 inhibited glioma cell proliferation and invasion, which could be reversed by AKT activation. DNMT1-mediated hypermethylation was responsible for downregulation of KLF13. Knocking down of DNMT1 restored KFL13 expression and inhibited cell proliferation and invasion as well. Patients with high expression of KLF13 might have a better prognosis.
Conclusion: KLF13 suppressed glioma aggressiveness and the regulation of KLF13 could be a potential therapeutic target.
Keywords: KLF13, DNMT1, AKT, glioma, methylation
Introduction
Glioma is the most common brain cancer and the seventh leading cause of cancer-related death worldwide.1 Despite the advances in the diagnosis and treatment of glioma in the last few decades, the overall survival is still short due to the high resistance to traditional chemo- or radiotherapy and metastasis. A number of oncogenes and tumor suppressor genes were investigated, regulating glioma initiation and aggressiveness, such as inducing cell cycle, cancer cell stemness and epithelial–mesenchymal transition.2 However, the management of glioma remains problematic. Hence, there is an urgent need to further investigate the molecular mechanisms underlying glioma progression and identify new therapeutic targets.
Kruppel-like factor 13 (KLF13) belongs to the KLF family and has been involved in various biological processes, such as cell proliferation and differentiation. KLF13 is predominantly expressed in heart, interacting with GATA-4 and regulating the early stage of cardiogenesis.3 In the mice model, KLF13 deficiency impaired the generation of memory-like CD8+ T cells and erythropoiesis.4,5 Moreover, KLF13 promoted porcine adipocyte differentiation through PPARγ activation.6 KLF13 is also found to have diverse roles in cancer. Wang et al7 reported that KLF13 was notably decreased and overexpression of KLF13 inhibited prostate cancer cell proliferation by suppressing AKT activation. A functional screening suggested that KLF13 induced apoptosis in pancreatic cancer cells.8 However, overexpression of KLF13 and FGFR3 was found in oral cancer and is responsible for cell proliferation and resistance to ionizing radiation.9 Also, KLF13 is critical for human papillomavirus (HPV) life cycle and cervical cancer.10 Thus, its roles in glioma need to be further elucidated.
Emerging evidence has demonstrated that epigenetic modification played pivotal roles in cancer.11–13 DNMT1 is a member of DNA methyl transferase family, along with DNMT2, DNMT3A, DNMT3B, DNMT3L, regulating target gene expression by the CpG island methylation.14 In glioma, DNMT1 was reported to epigenetically repress the expression of miRNAs and lncRNAs to promote cell proliferation, stem-cell-like phenotype and chemoresistance.15–18
In this study, we demonstrated that KLF13 expression was significantly downregulated in glioma tissues and overexpression of KLF13 inhibited cell proliferation and invasion, which could be reversed by AKT activation. Mechanically, DNMT1 was responsible for the hypermethylation of the KLF13 promoter in glioma tissues and cell lines. Knocking down of DNMT1 could restore the expression of KLF13 and suppress cell proliferation and invasion. Moreover, we found negative correlation between DNMT1 and KLF13 in both cell lines and glioma tissues. Therefore, upregulating KLF13 or inhibiting DNMT1 might serve as a new approach for glioma.
Materials and methods
Tissue samples
Three glioma tissues and paired adjacent normal tissues were collected from patients undergoing surgical resection between 2012 and 2015. Written informed consent was obtained from all the patients, and this study was conducted in accordance with the Declaration of Helsinki. The tissues were stored at −80°C for further investigation.
Vector construction
The vectors were purchased from GeneChem (Shanghai, China). Briefly, N-terminal Flag-tagged KLF13 was constructed by PCR and cloned into pLVX-TetOne vector. Myr-AKT was constructed by adding the src myristoylation signal sequence19 to the 5′-end of AKT cDNA, generating the constitute activation form. pCDNA3-Myr-AKT-ER was generated by cloning Myr-AKT without the stop codon into pCDNA3 containing the estrogen receptor (ER) ligand-binding domain as described previously.20,21 The Myr-AKT-ER fusion protein can be specifically activated by 4-hydroxytamoxifen (4-OHT; Calbiochem, La Jolla, CA, USA).
Cell culturing and lentivirus infection
Glioma cell lines U251, U87MG and A172 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were maintained in DMEM supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL) at 37°C in a humidified chamber with 5% CO2.
Lentiviral packaging was performed in 293T cells by following the lentivirus packaging protocol using pLVX-TetOne-KLF13, psPAX2 and pMD2.G vectors (Addgene, Cambridge, MA, USA). Glioma cells were infected with lentivirus at a multiplicity of infection of 10 with polybrene and selected with 2 μg/mL puromycin for stable cell lines.
siRNA transfection
For knocking down DNMT1, DNMT3A and DNMT3B, we transfected siRNAs targeting the three genes into glioma cells using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s protocol. siRNAs were purchased from Genepharma (Shanghai, China). The sequences of siRNAs were as follows: si-dnmt1, 5′-UUGUUAAUAGGGAUGGCGGUU-3′; si-dnmt3a, 5′-CAACAUCGAAUCCAUGAAAUU-3′; and si-dnmt3b, 5′-ACGCACAGCUGACGACUCAUU-3′.
Quantitative real-time PCR (qRT-PCR)
RNAs from cells were isolated using Trizol reagent (Thermo Fisher Scientific) as described previously. The first-strand cDNA was synthesized using PrimeScript RT Master Kit (Takara, Kusatsu, Japan). The qRT-PCR was performed using SYBR Green methods on ABI7500 System (Thermo Fisher Scientific). The relative expressions of genes were calculated by the 2−ΔΔCt method, and β-actin was used as internal control. The primers used were as follows: KLF13, 5′-CGGCCTCAGACAAAGGGTC-3′, 5′-TTCCCGTAAACTTTCTCGCAG-3′; DNMT1, 5′-AGGCGGCTCAAAGATTTGGAA-3′, 5′-GCAGAAATTCGTGCAAGAGATTC-3′; DNMT3A, 5′-AGTACGACGACGACGGCTA-3′, 5′-CACACTCCACGCAAAAGCAC-3′; DNMT3B, 5′-AGGGAAGACTCGATCCTCGTC-3′, 5′-GTGTGTAGCTTAGCAGACTGG-3′; and β-actin, 5′-CATGTACGTTGCTATCCAGGC-3′, 5′-CTCCTTAATGTCACGCACGAT-3′.
Western blot
Total protein was extracted using RIPA buffer containing protease and phosphatase inhibitors (Hoffman-La Roche Ltd., Basel, Switzerland). Western blot was performed as described previously. The bands were visualized using electrogenerated chemiluminescence methods. The primary antibodies used were as follows: anti-KLF13 (18352-1-AP), anti-Flag (66008-2-Ig), anti-β-actin (60008-1-Ig) obtained from ProteinTech (Wuhan, China); anti-AKT (4691), anti-phospho-S6 (4858), and anti-S6 (2317) obtained from Cell Signaling Technology (Cambridge, MA, USA); anti-DNMT1 (ab13537), anti-DNMT3A (ab2850), and anti-DNMT3B (ab2851) obtained from Abcam (Cambridge, UK).
Cell proliferation
Cells were plated into 96-well plates at a density of 1,500/well. For cell proliferation assay, 10 μL Cell Counting Kit-8 (CCK-8) reagent was added, incubated for 2 hours, and the absorbance at 450 nm was measured.
Transwell assay
Cell invasion ability was measured using 24-well BD Matrigel (BD Biosciences, San Jose, CA, USA) invasion chamber as described previously. Cells were pretreated with DMSO or doxycycline or 4-OHT and then seeded into the upper chamber without FBS, at the density of 5×105 cells/well. The lower chamber had medium with 20% FBS. After 48-hour culturing, noninvaded cells were removed, and invaded cells were stained by crystal violet and calculated.
DNA extraction and bisulfite sequencing PCR
Genomic DNA from tissues and cells was extracted using TIANamp Genomic DNA kit (Tiangen, Beijing, China), followed by the treatment of sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo, Orange, CA, USA). The treated DNA was amplified by PCR using paired primers to recognize the region (−984 to −697 before the transcriptional start site) of the KLF13 promoter. The primers used were as follows: 5′-GAGGGGCAGCCTAGAAGAGTTGGCA-3′ and 5′- CAACTCCCACAAAAAAAACAAACTC-3′.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously.22 In brief, 107 cells were fixed in 1% formaldehyde and sonicated until the average length of DNA fragment was about 250 bp. Equal amount of DNA was used as “Input” in each group. Then, the anti-DNMT1, anti-DNMT3A and anti-DNMT3B were added to immunoprecipitate the DNA–protein overnight at 4°C. The DNA–protein was digested with proteinase K, and DNA fragment was purified and used for PCR. The primers used were as follows: 5′-CTGGCACGTAGTTCGCTTCTGGAG-3′ and 5′-GAGGGACAGACTCGGGCTCGTTC-3′. To calculate the amount of immunoprecipitated DNA, % Input =2(−ΔCt [normalized ChIP]); ΔCt [normalized ChIP] = (Ct [ChIP] − (Ct [Input] − Log2 (input dilution factor))).
Public data acquisition
The Oncomine database (https://www.oncomine.org/resource/login.html) was used to explore the expression of KLF13 in glioma. GEO data sets GSE4290 (23 nontumor brain tissues and 77 glioma tissues in grade 4 and four glioma tissues of unknown grade) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE4290) and GSE4058 (three nontumor tissues and 30 glioblastoma multiforme tissues of unknown grade) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE4058) were used to measure the expression values of KLF13 and DNMT1 in glioma tissues. The data from the PROGgeneV2 Prognostic database23 (http://watson.compbio.iupui.edu/chirayu/proggene/database/index.php) were used to explore the association between KLF13 expression and glioma patients’ survival according to the data in GSE4271 containing 77 primary high-grade tumors and 23 matched recurrences.
Statistical analysis
The statistical analysis was performed using SPSS 21.0 (IBM Corporation, Armonk, NY, USA). Data were presented as mean ± SEM from at least three independent experiments. Student’s t-test or ANOVA test was used to measure the difference between groups. The Mann–Whitney test was used to measure the expression of KLF13 or DNMT1 in glioma tissues and normal tissues. Pearson’s test was used to measure the correlation between KLF13 and DNMT1. Survival was estimated by the Kaplan–Meier method with the log-rank test. A P-value of <0.05 was considered statistically significant.
Ethics statement
Ethical approval for this study was obtained from the ethics committee of Inner Mongolia People’s Hospital.
Results
KLF13 is downregulated in glioma tissues and associated with better survival
To explore the role of KLF13 in the progression of glioma, the expression of KLF13 was evaluated in clinical glioma tissues and normal tissues according to the Oncomine database. As shown in Figure 1A and B, KLF13 was significantly downregulated in glioma tissues compared to that in normal tissues. More importantly, we noticed that patients with high expression of KLF13 had a longer survival by analyzing the PROGgene V2 database. The 3- and 5-year survival rates of the patients with high expression of KLF13 were about 45% and 33%, respectively. However, in patients with low expression of KLF13, the rates were only about 20% and <10%, respectively (Figure 1C). Altogether, these results indicated that KLF13 might play a tumor suppressor role in glioma.
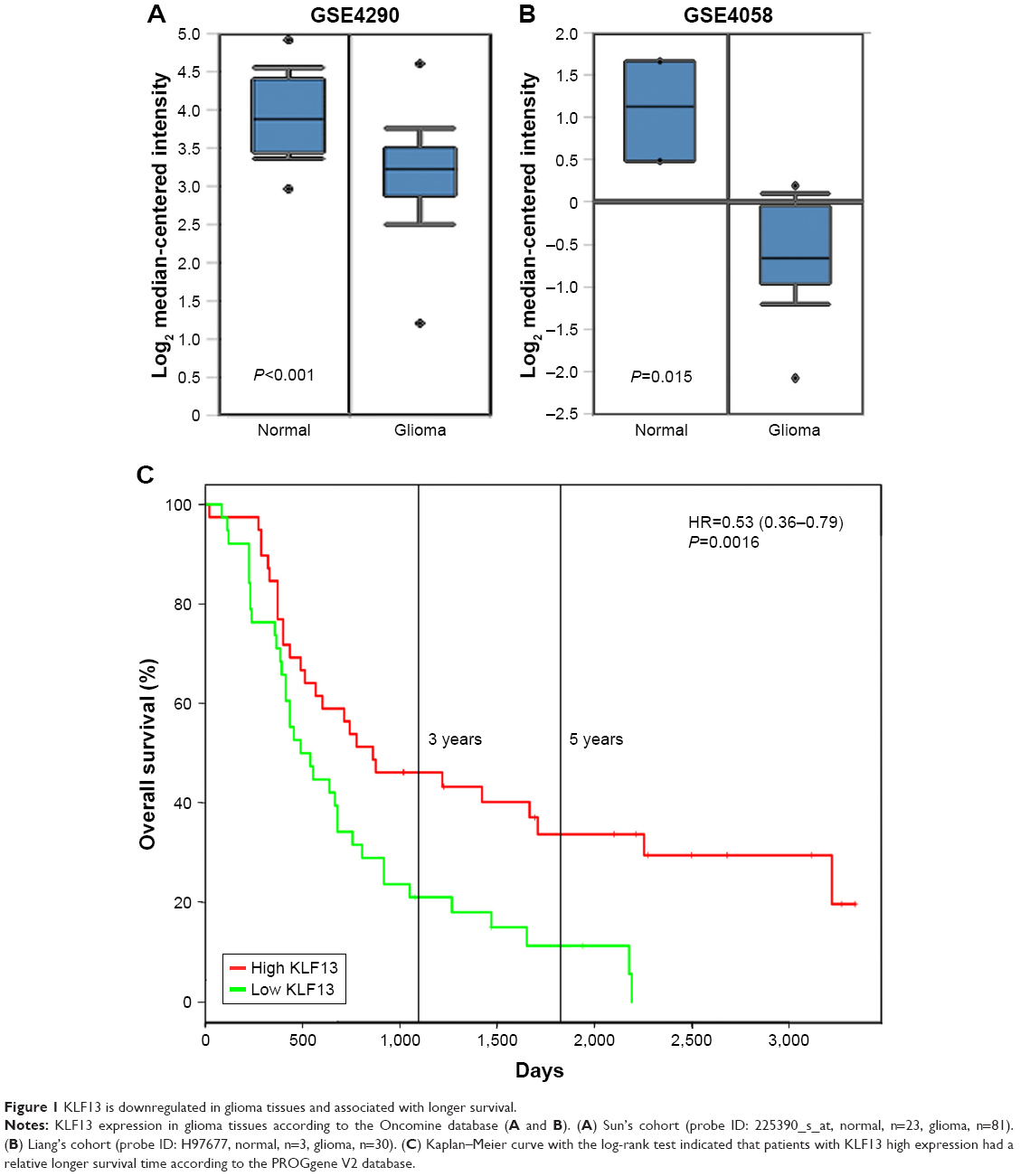 | Figure 1 KLF13 is downregulated in glioma tissues and associated with longer survival. |
KLF13 inhibits cell proliferation and invasion
To further explore the function of KLF13 in glioma, we performed an inducible overexpression of KLF13 in A172 and U87MG cells. By doxycycline treatment, KLF13 was effectively expressed, as evidenced by Western blot using anti-KLF13 antibody (Figure 2A and B) and qRT-PCR (Figure S1). The results from CCK-8 assay indicated that the proliferation was significantly retarded when KLF13 was overexpressed compared to control groups (Figure 2C and D). Moreover, the cell invasion ability was suppressed as well after KLF13 overexpression (Figure 2E and F). Altogether, these results indicated that KLF13 could inhibit cell proliferation and invasion.
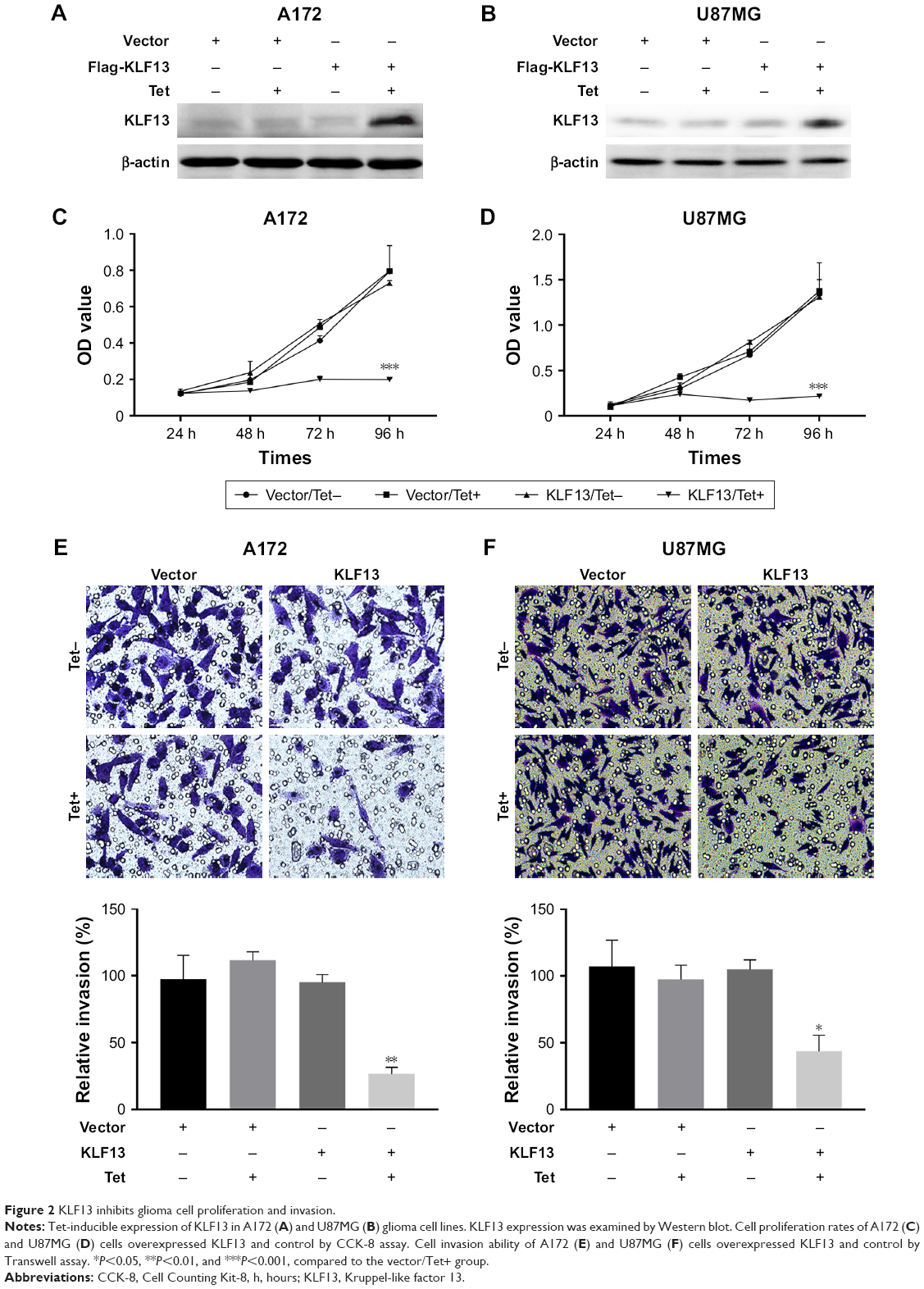 | Figure 2 KLF13 inhibits glioma cell proliferation and invasion. |
KLF13-mediated suppression effects can be reversed by AKT activation
Previous study suggested that KLF13 inhibited AKT activation, thereby, impaired cell proliferation. So, we used ER-inducible expression system of constitute-activated AKT (caAKT) to assess whether KLF13-mediated tumor suppression could be reversed by AKT signaling. We transfected Myr-AKT into A172 and U87MG cells and induced the expression of AKT by 4-OHT. The Western blot results demonstrated that caAKT could significantly upregulate pS6 expression, which was suppressed by KLF13 overexpression (Figure 3A and B). Consistently, the cell proliferation rate was significantly elevated when caAKT was overexpressed, regardless of the expression of KLF13, compared to the control group (Figure 3C and D). We noticed that there was no significant difference between KLF13−/AKT+ and KLF13+/AKT+ groups. Also, the cell invasion ability was enhanced after caAKT overexpression (Figure 3E and F). Altogether, these results suggested that AKT activation could abrogate the suppression function of KLF13 in glioma cells.
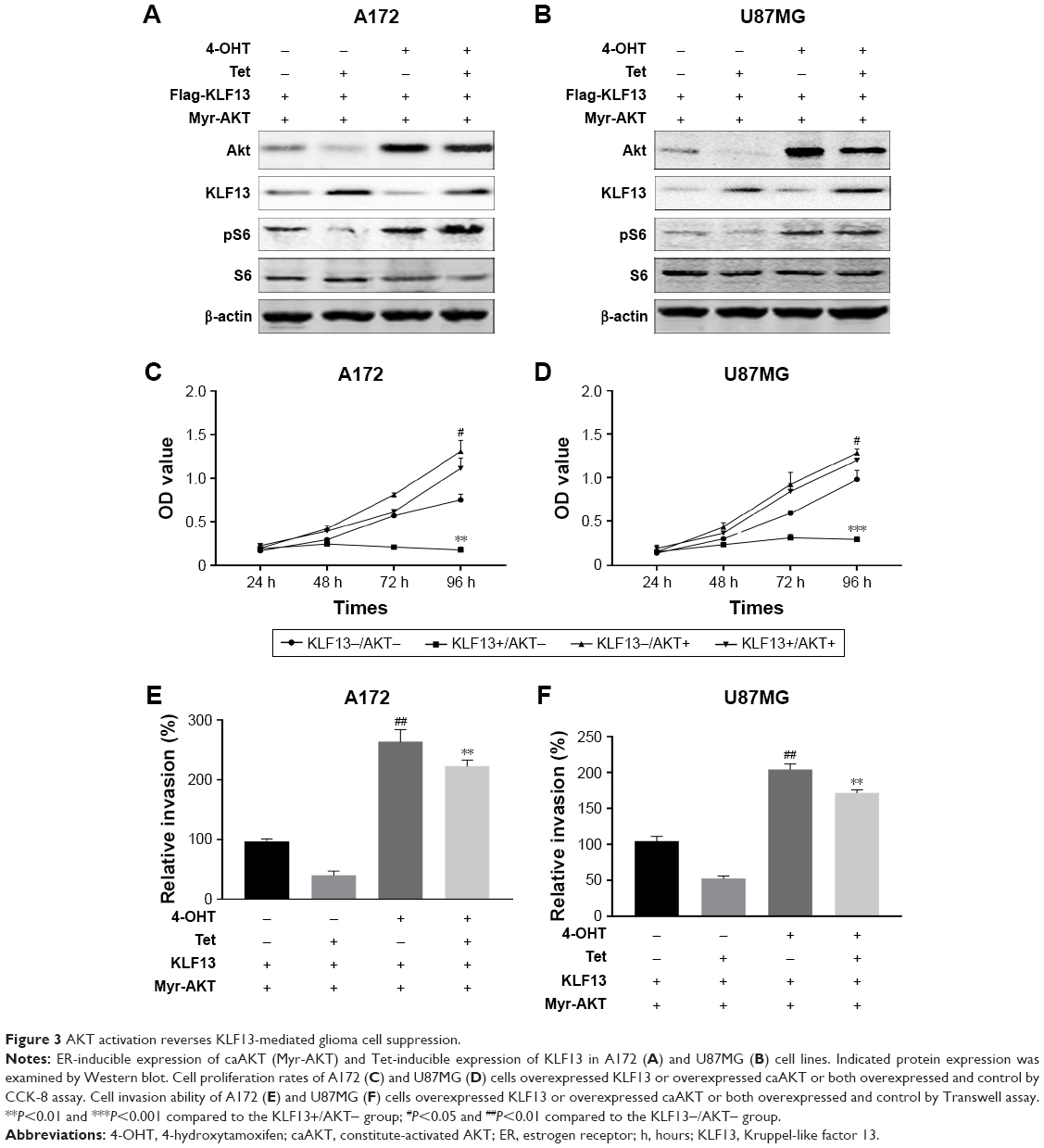 | Figure 3 AKT activation reverses KLF13-mediated glioma cell suppression. |
DNMT1 regulates methylation of KLF13 promoter
To investigate the mechanism of KLF13 repression in glioma, we hypothesized that KLF13 was regulated by DNA methylation in the promoter region. We first searched for CpG islands in the promoter of KLF13 and found 28 CpG dinucleotides predicted to be methylated. Then, we performed bisulfite sequencing in three glioma tissues and paired adjacent normal tissues and three glioma cell lines. The results showed that most of the CpG islands were hypermethylated in glioma tissue samples and cell lines compared to that in normal tissues (Figure 4A). Then, we determined whether the DNMT family was involved in the hypermethylation of KLF13 promoter in glioma. As shown in Figure 4B, we performed quantitative ChIP assay and found that DNMT1 bound to the KLF13 promoter instead of DNMT3A and DNMT3B (Figure 4B). Furthermore, we used siRNAs to knock down the expression of DNMT1, DNMT3A and DNMT3B, respectively, and found that DNMT1 was responsible for suppressing KLF13 expression, as knocking down of DNMT1 upregulated KLF13 expression in both mRNA and protein levels (Figures 4C, D and S2).
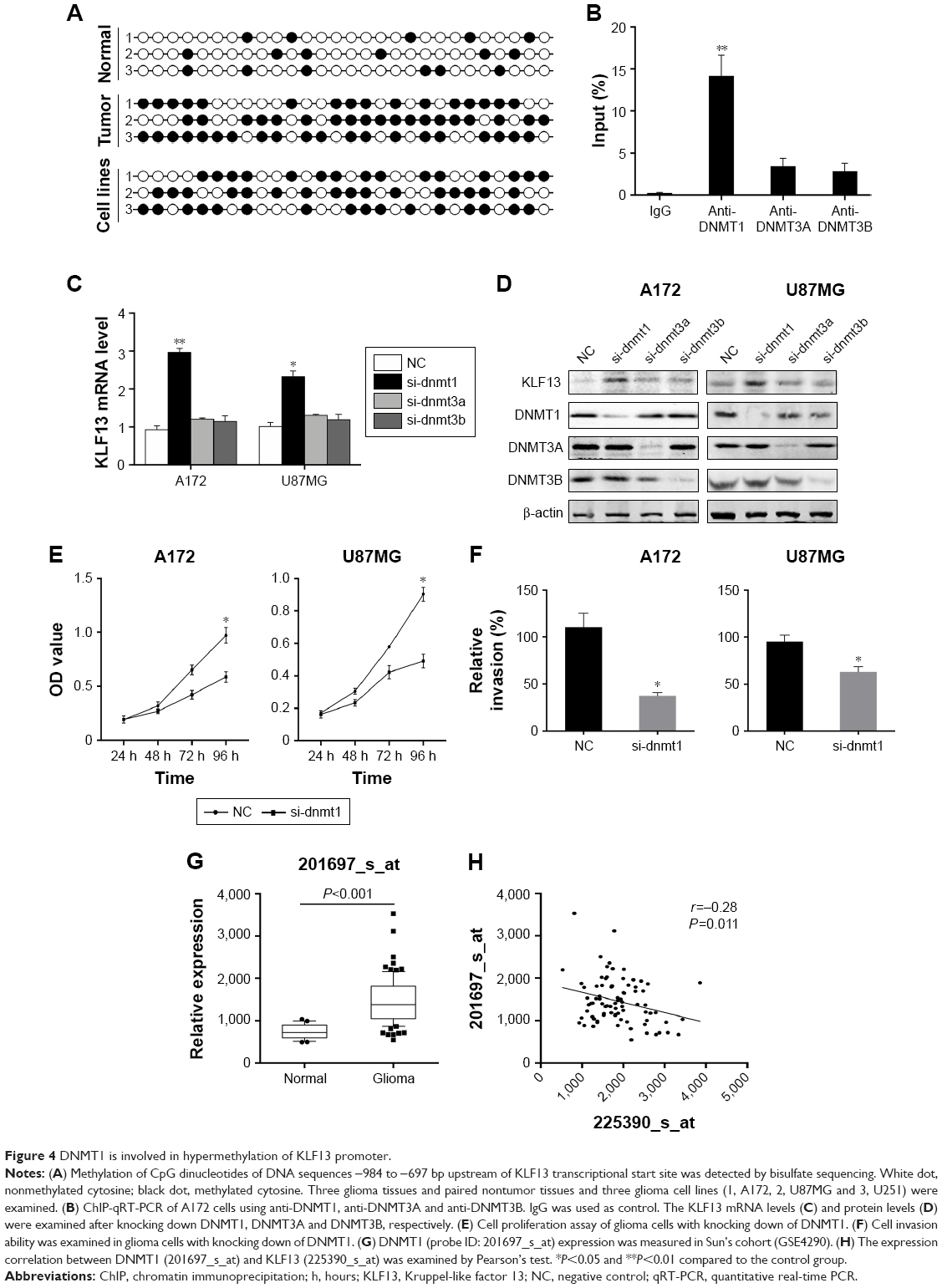 | Figure 4 DNMT1 is involved in hypermethylation of KLF13 promoter. |
Additionally, we knocked down DNMT1 and found that cell proliferation and invasion were significantly decreased in A172 and U87MG cells (Figure 4E and F). To verify the association between KLF13 and DNMT1, we explored the expression values of DNMT1 in GSE4290 and found that the expression value of DNMT1 was significantly higher in glioma tissues compared to that in normal tissues (Figure 4G). Moreover, the expressions of DNMT1 (probe ID: 201697_s_at) and KLF13 (probe ID: 225390_s_at) were negatively correlated in glioma samples (Figures 4H and S3). Taken together, the results demonstrated that KLF13 could be epigenetically silenced by DNMT1 in glioma.
Discussion
Previous studies implicated that several KLFs had tumor suppression roles, such as KLF9 in hepatocellular carcinoma, KLF4 in bladder cancer and KLF2 in non-small-cell lung cancer.24–26 However, given the tissue abundance diversity of KLF13, the role of KLF13 in different tumor types needs to be further addressed, especially in glioma, which is characterized by its high aggressiveness, metastasis and stemness. This study suggested that KLF13 downregulation was critical for glioma cell proliferation and invasion. More importantly, we indicated that patients with high expression of KLF13 had a relative longer survival time according to the public database.
Consistent with previous study that KLF13 inhibited AKT activation to suppress tumor cell proliferation, we found that KLF13 indeed decreased pS6 expression. However, AKT constitutive activation abrogated the KLF13-induced tumor suppression effects. Our hypothesis was that overexpression of AKT could abrogate the suppression effects caused by KLF13, and the results were consistent with our hypothesis that overexpression of AKT could significantly enhance the proliferation and invasion abilities regardless of the status of KLF13, as evidenced by the fact that there was no significant difference between KLF13−/AKT+ and KLF13+/AKT+ groups. Numerous studies have reported that AKT signaling activation was critical for tumor stem-like phenotype, chemoresistance and metastasis in glioma. For example, AKT induced ABCG2 expression and regulated side population phenotype.27 TRIM24 could bind to the PI3KCA promoter and enhance Akt activation to induce glioma chemoresistance.28 These results could partially explain that some cases of glioma might be still aggressive even if KLF13 is present and functional, because of the AKT activation.
Aberrant epigenetic modulation has been demonstrated in various types of cancers, including colorectal carcinoma, breast cancer and glioma.29–31 The KLF family was reported to be regulated by epigenetic silencing. Among that, KLF4 was methylated in a subset of tumors and cell lines.32 KLF2 methylation was associated with non-small cell lung carcinoma progression.33 In this study, we noticed that KLF13 promoter was frequently hypermethylated in glioma tissues and cell lines, compared to normal tissues, which explained the downregulation of KLF13 in glioma tissues and cell lines. Mechanically, the DNMT1, instead of DNMT3A and DNMT3B, was responsible for the hypermethylation of KLF13 promoter. DNMT1 was found to bind to the rich CpG dinucleotides regions according to our ChIP data. Interestingly, AKT activation was reported to increase DNMT1 stability and nuclear translocation.34,35 Consequently, the KLF13 was downregulated by AKT/DNMT1 axis in glioma. Nevertheless, KLF13 could also be regulated by miRNAs. miR-147b was reported to downregulate KLF13 and inhibit cell viability of rat cardiomyocytes.36 miR-125a and miR-125b could regulate KLF13 in systemic lupus erythematosus and acute myocardial infarction, respectively.37,38 Moreover, KLF13 could also be regulated by posttranslational mechanisms. For instance, its activity was reported to be regulated by CBP and PCAF acetylation.39 Fbw7γ was involved in the degradation of KLF13.40 So, the modulation of KLF13 needs to be further investigated.
Conclusion
Our current findings demonstrated a role for DNMT1-mediated epigenetic silencing of KLF13 in the regulation of glioma cell proliferation and invasion and its clinical prognostic values. Loss of KLF13 in glioma is predominant and correlated with poor prognosis. Thus, upregulation of KLF13 might be employed as a new approach for the management of glioma patients.
Acknowledgment
This study was supported by grants from Fund for Less Developed Regions of the National Natural Science Foundation of China (81360187).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Wang J, Su H-Kai, Zhao H-Fu, et al. Progress in the application of molecular biomarkers in gliomas. Biochem Biophys Res Commun. 2015;465(1):1–4. | ||
Lavallée G, Andelfinger G, Nadeau M, et al. The Kruppel-like transcription factor KLF13 is a novel regulator of heart development. EMBO J. 2006;25(21):5201–5213. | ||
Lai D, Zhu J, Wang T, et al. KLF13 sustains thymic memory-like CD8+ T cells in BALB/c mice by regulating IL-4-generating invariant natural killer T cells. J Exp Med. 2011;208(5):1093–1103. | ||
Gordon AR, Outram SV, Keramatipour M, et al. Splenomegaly and modified erythropoiesis in KLF13-/-mice. J Biol Chem. 2008;283(18):11897–11904. | ||
Jiang S, Wei H, Song T, et al. KLF13 promotes porcine adipocyte differentiation through PPARγ activation. Cell Biosci. 2015;5(1):28. | ||
Wang Q, Peng R, Wang B, et al. Transcription factor KLF13 inhibits AKT activation and suppresses the growth of prostate carcinoma cells. Cancer Biomark. 2018;22(3):533–541. | ||
Fernandez-Zapico ME, Lomberk GA, Tsuji S, et al. A functional family-wide screening of SP/KLF proteins identifies a subset of suppressors of KRAS-mediated cell growth. Biochem J. 2011;435(2):529–537. | ||
Henson BJ, Gollin SM. Overexpression of KLF13 and FGFR3 in oral cancer cells. Cytogenet Genome Res. 2010;128(4):192–198. | ||
Zhang W, Hong S, Maniar KP, et al. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene. 2016;35(42):5565–5575. | ||
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. | ||
Baylin SB, Ohm JE. Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. | ||
Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. | ||
Turek-Plewa J, Jagodziński PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10(4):631–647. | ||
Yao J, Zhou B, Zhang J, et al. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumour Biol. 2014;35(8):7935–7944. | ||
Li J, Bian EB, He XJ, et al. Epigenetic repression of long non-coding RNA MEG3 mediated by DNMT1 represses the p53 pathway in gliomas. Int J Oncol. 2016;48(2):723–733. | ||
Zhou D, Wan Y, Xie D, et al. DNMT1 mediates chemosensitivity by reducing methylation of miRNA-20a promoter in glioma cells. Exp Mol Med. 2015;47(9):e182. | ||
Sun J, Tian X, Zhang J, et al. Regulation of human glioma cell apoptosis and invasion by miR-152-3p through targeting DNMT1 and regulating NF2: MiR-152-3p regulate glioma cell apoptosis and invasion. J Exp Clin Cancer Res. 2017;36(1):100. | ||
Cross FR, Garber EA, Pellman D, Hanafusa H. A short sequence in the p60src N terminus is required for p60src myristylation and membrane association and for cell transformation. Mol Cell Biol. 1984;4(9):1834–1842. | ||
Southgate RJ, Neill B, Prelovsek O, et al. FOXO1 regulates the expression of 4E-BP1 and inhibits mTOR signaling in mammalian skeletal muscle. J Biol Chem. 2007;282(29):21176–21186. | ||
Xie L, Ushmorov A, Leithäuser F, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119(15):3503–3511. | ||
Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3(4):698–709. | ||
Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014;14(1):970. | ||
Sun J, Wang B, Liu Y, et al. Transcription factor KLF9 suppresses the growth of hepatocellular carcinoma cells in vivo and positively regulates p53 expression. Cancer Lett. 2014;355(1):25–33. | ||
Ohnishi S, Ohnami S, Laub F, et al. Downregulation and growth inhibitory effect of epithelial-type Krüppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. 2003;308(2):251–256. | ||
Nie FQ, Sun M, Yang JS, et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14(1):268–277. | ||
Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. | ||
Zhang LH, Yin AA, Cheng JX, et al. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene. 2015;34(5):600–610. | ||
Vaiopoulos AG, Athanasoula KCH, Papavassiliou AG. Epigenetic modifications in colorectal cancer: molecular insights and therapeutic challenges. Biochim Biophys Acta. 2014;1842(7):971–980. | ||
Toska E, Osmanbeyoglu HU, Castel P, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355(6331):1324–1330. | ||
Kreth S, Thon N, Kreth FW. Epigenetics in human gliomas. Cancer Lett. 2014;342(2):185–192. | ||
Bureau C, Hanoun N, Torrisani J, Vinel JP, Buscail L, Cordelier P. Expression and function of Kruppel Like-Factors (KLF) in Carcinogenesis. Curr Genomics. 2009;10(5):353–360. | ||
Jiang W, Xu X, Deng S, et al. Methylation of kruppel-like factor 2 (KLF2) associates with its expression and non-small cell lung cancer progression. Am J Transl Res. 2017;9(4):2024–2037. | ||
Spangle JM, Roberts TM, Zhao JJ. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim Biophys Acta Rev Cancer. 2017;1868(1):123–131. | ||
Hodge DR, Cho E, Copeland TD, et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics. 2007;4(6):387–398. | ||
Gu M, Wang J, Wang Y, et al. MiR-147b inhibits cell viability and promotes apoptosis of rat H9c2 cardiomyocytes via down-regulating KLF13 expression. Acta Biochim Biophys Sin. 2018;50(3):288–297. | ||
Zhao X, Tang Y, Qu B, et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010;62(11):3425–3435. | ||
Bayoumi AS, Park KM, Wang Y, et al. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2018;114:72–82. | ||
Song CZ, Keller K, Chen Y, Stamatoyannopoulos G. Functional interplay between CBP and PCAF in acetylation and regulation of transcription factor KLF13 activity. J Mol Biol. 2003;329(2):207–215. | ||
Kim DS, Zhang W, Millman SE, et al. Fbw7γ-mediated degradation of KLF13 prevents RANTES expression in resting human but not murine T lymphocytes. Blood. 2012;120(8):1658–1667. |
Supplementary materials
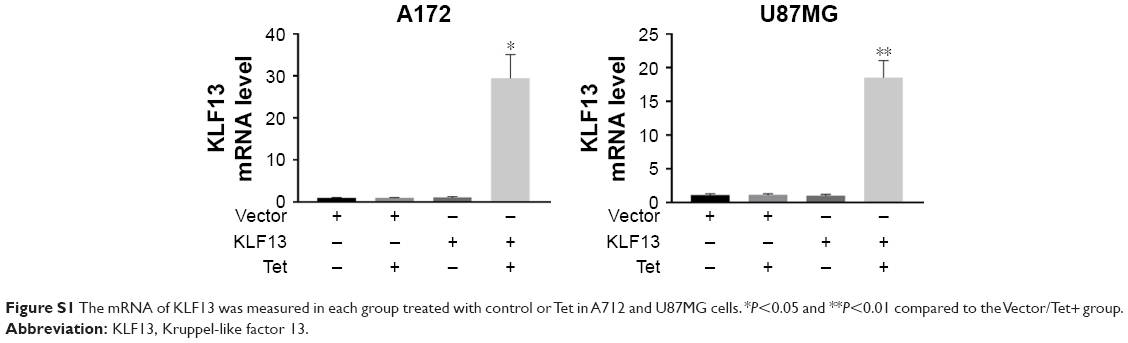 | Figure S1 The mRNA of KLF13 was measured in each group treated with control or Tet in A712 and U87MG cells. *P<0.05 and **P<0.01 compared to the Vector/Tet+ group. |
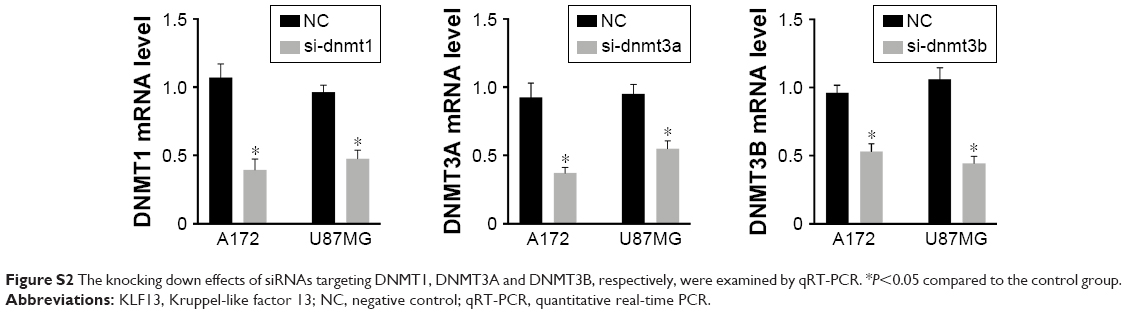 | Figure S2 The knocking down effects of siRNAs targeting DNMT1, DNMT3A and DNMT3B, respectively, were examined by qRT-PCR. *P<0.05 compared to the control group. |
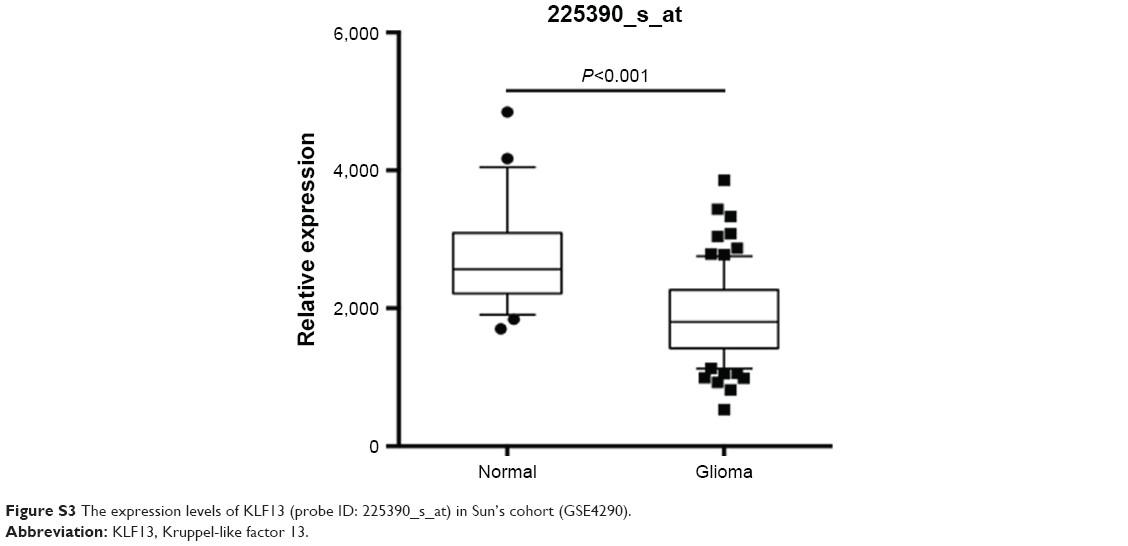 | Figure S3 The expression levels of KLF13 (probe ID: 225390_s_at) in Sun’s cohort (GSE4290). |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.