")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Downregulation of IRF2 Alleviates Sepsis-Related Acute Kidney Injury in vitro and in vivo
Authors Zhang Y, Zhang Y, Yang A, Xia F
Received 16 August 2021
Accepted for publication 25 November 2021
Published 22 December 2021 Volume 2021:15 Pages 5123—5132
DOI https://doi.org/10.2147/DDDT.S334518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yanyan Zhang, Yun Zhang, Aixiang Yang, Fei Xia
Department of Critical Care Medicine, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, 215008, Jiangsu, People’s Republic of China
Correspondence: Fei Xia
Department of Critical Care Medicine, The Affiliated Suzhou Hospital of Nanjing Medical University, No. 242, Guangji Road, Suzhou, 215008, Jiangsu, People’s Republic of China
Email [email protected]
Objective: We investigated the roles and mechanisms of IRF2 in sepsis-related acute kidney injury (S-AKI) in a lipopolysaccharide (LPS)-induced HK-2 cell line and caecal ligation and puncture (CLP)-induced IRF2−/− mouse model.
Methods: Quantitative real-time polymerase chain reaction assay was used to detect IRF2 in the serum of S-AKI patients and LPS-induced HK-2 cells. Cell proliferation, death, and apoptosis were analysed by CCK-8, lactate dehydrogenase release, and flow cytometry assays, respectively. The levels of interleukin (IL)-1β, IL-18, IL-6, tumour necrosis factor (TNF)-α, non-canonical inflammasomes, including caspase-4 and gasdermin-D (GSDMD), and canonical inflammasomes, such as caspase-1, NLR family pyrin domain containing 3 (NLRP3), and apoptosis-associated speck-like protein (ASC) in S-AKI cells or animal models were analysed by enzyme-linked immunosorbent assay or Western blotting.
Results: IRF2 was upregulated in the serum of S-AKI patients and LPS-induced HK-2 cells. IRF2 downregulation promoted cell proliferation and inhibited cell death and apoptosis, respectively. IRF2 inhibition reduced the levels of IL-1β, IL-18, IL-6, and TNF-α in S-AKI cells and animal models. IRF2 knockdown inhibited LPS-treated HK-2 cell pyroptosis by decreasing the expression of caspase-4 and GSDMD, instead of affecting caspase-1, NLRP3, and ASC. An elevated survival rate and alleviated pathological features and scores were observed in the CLP-induced IRF2−/− animal models. IRF2 deficiency also suppressed inflammation and pyroptosis by inhibiting non-canonical inflammasomes as indicated by the decreased expression of caspase-11 and GSDMD.
Conclusion: Our findings suggest that IRF2 downregulation protects against S-AKI in vitro and in vivo.
Keywords: sepsis acute kidney injury, IRF2, inflammation, pyroptosis
Introduction
Sepsis is characterised by diffuse inflammation, which is generally caused by the fungal, viral, or bacterial infections. It is the most common factor in the development of acute kidney injury (AKI), and AKI of any origin is associated with a high risk of sepsis.1 Sepsis-related AKI (S-AKI) is a common complication in critically ill patients and is associated with high morbidity and mortality.2 It is the 7th and 8th leading cause of the global mortality of 1–4-year-old children and 65–75-year-old adults, respectively.3 S-AKI is accompanied by abnormal changes in the expression and function of various genes, RNAs, and proteins, which can be ameliorated by effectively regulating the expression of these abnormal molecules. Sepsis is the primary cause of AKI in critically ill patients,4 however, the pathophysiological mechanisms of S-AKI remain unclear. Thus, more mechanistic studies are required to deeply understand the complicated pathophysiology of S-AKI and to apply these results into potential treatment strategies for pharmacological approaches and clinical trials.
Studies have indicated that inflammation plays a critical role in the pathogenesis of S-AKI.5 Inflammation is triggered by pattern recognition receptors, which sense external stimuli to activate canonical inflammasomes, resulting in the production of pro-inflammatory cytokines, including tumour necrosis factor-α (TNF-α), interleukin (IL)-18, and IL-1β, or leading to caspase-1 activation, thereby causing gasdermin-D (GSDMD)-mediated pyroptosis.6,7 Pyroptosis differs from other forms of cell death in morphology and mechanism, and it is characterised by rapid rupture of the plasma membrane and the release of pro-inflammatory factors that recruit a greater participation of inflammatory cells, thereby expanding the inflammatory response.8–10 Both pyrolysis and apoptosis are programmed mechanisms of cell death, however, each process relies on different caspases. Caspase-1 and caspase-11 (caspase-4 in humans) are activated by canonical and non-canonical inflammasomes in various sterile inflammatory and infectious states, such as septic shock.11,12 Activated caspase-1, caspase-11, and caspase-4 cleave GSDMD results cytokine release and rupture of the cytolytic membrane (pyroptosis).7 Distinct from apoptosis, pyrolysis leads to cell lysis and pro-inflammatory cytokine release into the extracellular space, including IL-1β and IL-18.13,14 This inflammatory pathway is found in a variety of cells, such as macrophages, monocytes, and epithelial cells.15–17 The pathological characteristics of S-AKI are renal tubular sublethal and lethal damage, leading to apoptosis, necrosis, and pyrolysis.18
Interferon regulatory factor 2 (IRF2) is a transcription factor located on chromosome 4, which was originally identified as a regulator of the type I interferon system.19 Emerging evidence has shown that IRF2 plays an important role in the inflammatory response.20 IRF2 is important for GSDMD transcriptional activation.21 Destruction of a single IRF2 binding site eliminates signal transduction in canonical and non-canonical inflammasomes.21 Apolipoprotein L1 (APOL1) risk variants considerably elevate the risk of kidney disease in African Americans, and IRF2 knockdown markedly decreases APOL1 expression in the unstimulated state.22 However, the roles and mechanisms of IRF2 in S-AKI have not been elucidated.
In this study, we explored the effects and potential mechanisms of IRF2 regulation on S-AKI using a lipopolysaccharide (LPS)-induced in vitro model and caecal ligation and puncture (CLP)-induced in vivo model. We found that IRF2 was upregulated in the serum of S-AKI patients and LPS-treated HK2 cells. IRF2 downregulation could alleviate sepsis-induced renal injury by inhibiting inflammation and pyroptosis in vivo and in vitro. Our results provide a new potential therapeutic target for S-AKI treatment.
Materials and Methods
Serum Samples
The serum samples were collected and then refrigerated at −80 °C. All serum samples were obtained with informed consent from patients. This study was approved by the Ethics Committee of the Affiliated Suzhou Hospital of Nanjing Medical University (KL907125), and was conducted in accordance with the Declaration of Helsinki.
Cell Culture, Induction, and Transfection
The human renal tubular epithelial cell line (HK-2) was purchased from Procell (Wuhan, China). HK-2 cells were cultured in Dulbecco’s Modified Eagle Medium (Invitrogen, MA, USA) containing 10% foetal bovine serum (Invitrogen) in an incubator at 37 °C with 5% CO2. To establish the S-AKI model in vitro, HK-2 cells were treated with 1 μg/mL LPS (Sigma-Aldrich, MO, USA) for 24 h. The LPS-treated HK-2 cells were seed into 60 mm culture plates at a density of 1×106 cells/mL. Si-RNAs (JTS Scientific, Wuhan, China) were transfected into HK-2 cells using Lipofectamine 3000 reagent (Invitrogen) following the manufacturer’s instructions.
Experimental Animals
Male 8–12-week-old C57BL/6N mice and IRF2 knockout (IRF2−/−) mice (20–25 g body weight) were purchased from Vital River (Beijing, China). A sepsis in vivo model was established using CLP surgery, as previously described.23 The survival rate of each mouse was determined starting 24 h after CLP treatment for 10 days. The renal tissue of each mouse was collected 24 h later.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from cells and tissues using TRIzol Reagent (TIANGEN, Beijing, China). cDNA was synthesized from RNA by TIANSeq M-MLV reverse transcriptase (TIANGEN, China). Subsequently, qRT-PCR was performed. The primer sequences used in this section are as follows: IRF2, Forward (F): 5ʹ-AGTGTGGCCAGTGATGAAGA-3ʹ, Reverse (R): 5ʹ-GAGCTGTTGTAAGGCATCGG-3ʹ; GAPDH, F: 5ʹ- ACATGGCCTCCAAGGAGTAAGAA-3ʹ, R:5ʹ-GGGATAGGGCCTCTCTTGCT-3ʹ. The results of this assay were calculated using 2−ΔΔCT method.
Lactate Dehydrogenase (LDH) Release, Enzyme-Linked Immunosorbent Assay (ELISA), and Serum Biochemical Indicators Assays
Cytotoxicity was assessed using the LDH release assay according to the manufacturer’s instructions (Clontech, CA, USA). The levels of TNF-α, IL-6, IL-1β, and IL-18 were tested using ELISA kits (Beyotime, Jiangsu, China) following the manufacturer’s instructions. The levels of serum creatinine (SCR) and blood urea nitrogen (BUN) were detected using the automatic biochemical analyzer (Hitachi 7170A, Japan).
Flow Cytometry
HK-2 cells cell apoptosis was detected using an Annexin V-FITC/Propidium Iodide Apoptosis Detection Kit (Abcam, Cambridge, UK) following the manufacturer’s instructions. The cells were fixed in FACS fixing buffer containing 1% paraformaldehyde. Flow cytometry (FACS Calibre, BD Biosciences, NJ, USA) was used to perform the assay. The positive cells were analysed using the FlowJo software (Ashland, DE, USA).
Cell Proliferation Assay
Cells were seeded onto 96-well culture plates and grown in an incubator for 48 h. Cell Counting Kit 8 (CCK-8) reagent (Beyotime) was added to each well, and the cells were incubated for 1 h. The optical density value of each well at 450 nm was measured using a microplate reader (ELX-800, BioTek, VT, USA).
Haematoxylin and Eosin (H&E)
For H&E staining, tissues were fixed in 4% formaldehyde for 24 h and then dehydrated with ethanol in a gradient manner. The samples were embedded in paraffin and cut into 5 μm-thick sections. The sections were stained with haematoxylin for 5 min and stained with eosin for 30 s. The stained sections were scored in a blinded manner, and tubular damage, including tubular necrosis, apoptosis, dilatation, and cast formation was recorded as follows: 0 (none), 1 (1–10%), 2 (11–25%), 3 (26–45%), and 4 (46–75%).24
Western Blot
HK-2 cells and animal tissues were lysed using RIPA lysis buffer (Thermo Fisher Scientific, MA, USA). Each protein sample was electrophoresed by 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Sigma-Aldrich). Next, the membranes were blocked with 5% bovine serum albumin (BSA) at room temperature for 2 h and incubated with the following primary antibodies: anti-IRF2 (ab124744, 1:1000, Abcam); anti-Bax (ab32503, 1:1000, Abcam); anti-Bcl-2 (ab196495, 1:1000, Abcam); anti-caspase-1 (ab62698, ab138483, 1:1000, Abcam); anti-caspase-4 (4450, 1:1000, Cell Signaling Technology, MA, USA), anti-caspase-11 (ab246496, 1:1000, Abcam); anti- apoptosis-associated speck-like protein (ASC; 13833, 67824, 1:1000, Cell Signaling Technology); anti-TNF-α (3707, 1:500, Cell Signaling Technology); anti-IL-1β (12242, 1:500, Cell Signaling Technology); anti-IL-18 (54943, 57058, 1:500, Cell Signaling Technology); anti-IL-6 (ab259341, 1:500, Abcam); anti-GSDMD (39754, 1:1000, Cell Signaling Technology); anti-NLRP3 (13158, 15101, 1:1000, Cell Signaling Technology); and GAPDH (ab8245, 1:3000, Abcam) overnight at 4 °C. Each membrane was incubated with corresponding secondary antibodies (7233, 7076, 1:5000, Cell Signaling Technology) for 1 h at room temperature. Protein expression was detected using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific).
Statistics
Statistical analysis was carried out using GraphPad Prism software (version 8.0). Data are presented as mean ± standard deviation (SD). Comparisons among groups were performed using t-test or one-way ANOVA. Differences were considered statistically significant at P<0.05 and P<0.01.
Results
IRF2 is Upregulated in the Serum of S-AKI Patients and LPS-Induced HK-2 Cells
To investigate IRF2 in expression S-AKI, we recruited 27 S-AKI patients and 18 healthy volunteers. Using qRT-PCR, we found that IRF2 expression was significantly upregulated in the serum of S-AKI patients compared to that in normal serum (Figure 1A). IRF2 expression in the LPS-induced S-AKI in vitro model was quantified; likewise, IRF2 expression was markedly elevated in LPS-treated HK-2 cells (Figure 1B). These results suggest that IRF2 may be involved in the development of S-AKI.
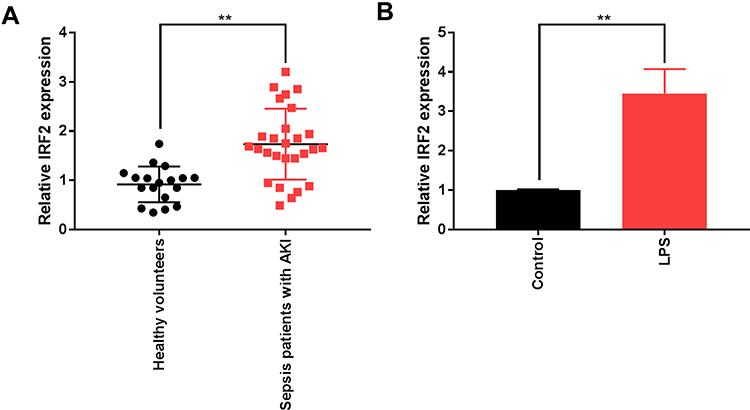 |
Figure 1 IRF2 expression is upregulated in the serum of S-AKI patients and cells. (A) The mRNA level of IRF2 in the serum of of S-AKI patients and healthy individuals was measured by qRT-PCR. (B) The mRNA level of IRF2 in LPS-induced HK-2 cells was detected by qRT-PCR. Data were expressed as mean + SD. **p<0.01. Abbreviations: IRF2, interferon regulatory factor 2; S-AKI, sepsis-related acute kidney injury; mRNA, messenger RNA; qRT-PCR, quantitative real-time polymerase chain reaction; LPS, lipopolysaccharide; HK-2, human renal tubular epithelial cell line; SD, standard deviation. |
IRF2 Knockdown Promotes Proliferation and Inhibits Cell Death and Apoptosis in LPS-Induced HK-2 Cells
We transfected the negative control siRNA or IRF2 siRNA into HK-2 cells treated with LPS to explore the role of IRF2 in S-AKI. We found that IRF2 inhibition notably reduced IRF2 mRNA expression (Figure 2A) and protein levels (Figure 2B) in S-AKI cells. Additionally, CCK-8 assay results revealed that IRF2 inhibition promoted the viability of S-AKI cells (Figure 2C). The results of LDH release and flow cytometry assays indicated that IRF2 downregulation remarkably suppressed cell death (Figure 2D) and apoptosis (Figure 2E) in the S-AKI in vitro model. Western blotting results showed that IRF2 inhibition notably reduced the protein expression of Bax, whereas it induced the protein expression of Bcl-2 (Figure 2F). Taken together, IRF2 knockdown attenuated LPS-induced injury in HK-2 cells.
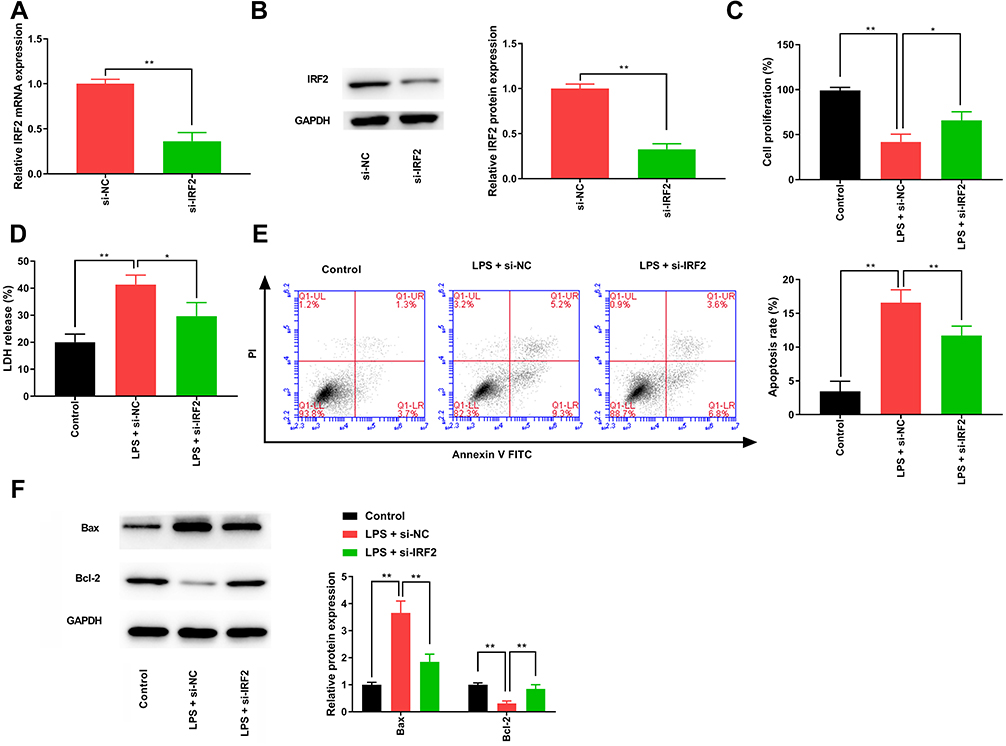 |
Figure 2 IRF2 knockdown attenuates the LPS-induced injury in HK-2 cells. (A) qRT-PCR was performed to measure the level of IRF2 in LPS-induced HK-2 cells. (B) The protein level of IRF2 was detected in LPS-induced HK-2 cells by Western blotting. (C) CCK-8 assay was used to determine the changes in cell viability. (D) LDH release assay was used to detect cell death. (E) The apoptotic cells were quantified using flow cytometry. (F) Western blotting was used to detect the protein expression levels of Bax and Bcl-2. Data were expressed as mean + SD. *p<0.05, **p<0.01. Abbreviations: IRF2, interferon regulatory factor 2; LPS, lipopolysaccharide; HK-2, human renal tubular epithelial cell line; qRT-PCR, quantitative real-time polymerase chain reaction; CCK-8, cell counting kit 8; LDH, lactate dehydrogenase; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated X; SD, standard deviation. |
IRF2 Knockdown Inhibits LPS-Induced Inflammatory Response in HK-2 Cells
We measured the concentrations of certain inflammatory cytokines by ELISA to investigate the effects of IRF2 on LPS-induced inflammatory response in S-AKI cells. LPS treatment significantly increased the levels of IL-1β, IL-18, IL-6, and TNF-α in HK-2 cells, while these inflammatory factors were notably reduced following IRF2 inhibition (Figure 3A–D). Similarly, lower protein expression levels of IL-1β, IL-18, IL-6, and TNF-α were observed in S-AKI cells after suppressing IRF2 (Figure 3E). These results indicate that IRF2 knockdown inhibited the LPS-induced inflammatory response in HK-2 cells.
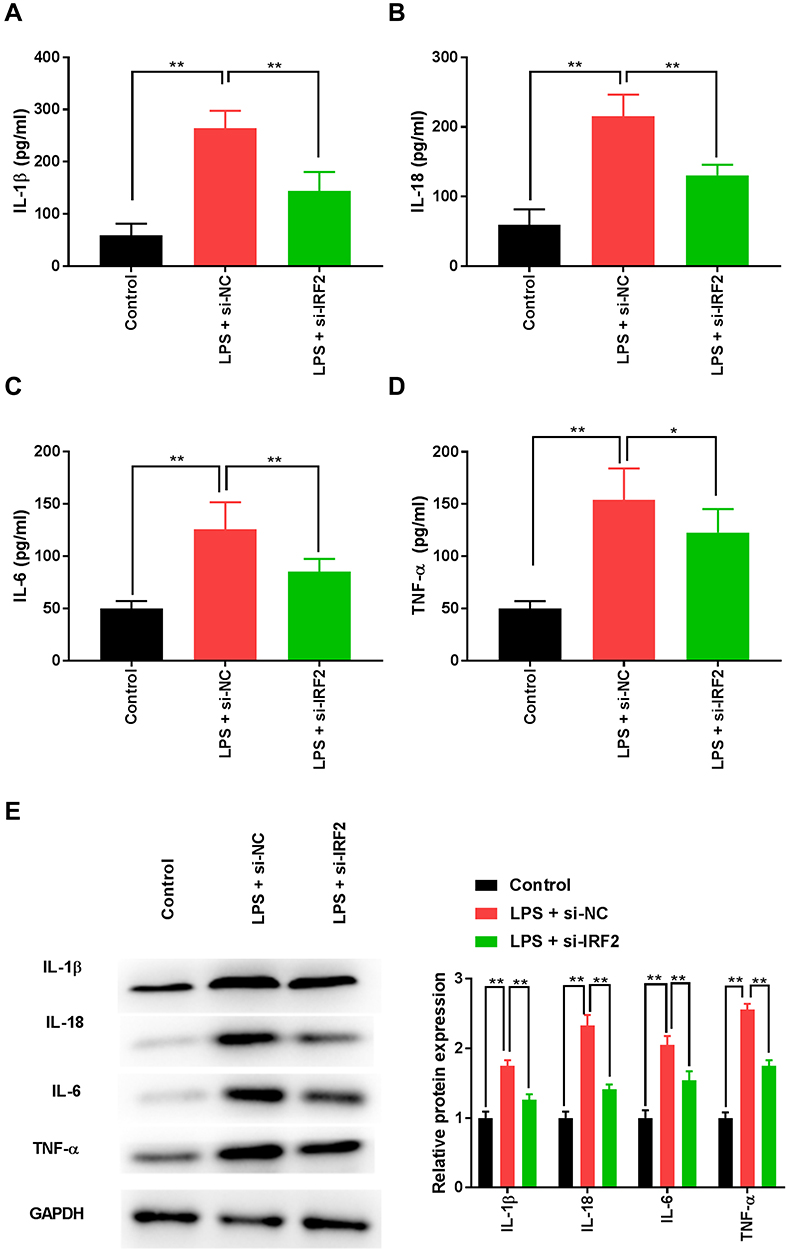 |
Figure 3 IRF2 knockdown inhibits LPS-induced inflammatory cytokines in HK-2 cells. (A–D) ELISA was performed to compare the levels of IL-1β, IL-18, IL-6, and TNF-α in LPS-induced HK-2 cells. (E) Western blotting was used to detect the protein expression levels of IL-1β, IL-18, IL-6, and TNF-α in LPS-induced HK-2 cells. Data were expressed as mean + SD. *p<0.05, **p<0.01. Abbreviations: IRF2, interferon regulatory factor 2; LPS, lipopolysaccharide; HK-2, human renal tubular epithelial cell line; ELISA, enzyme-linked immunosorbent assay; IL-1β, interleukin-1β; TNF-α, tumour necrosis factor-α; SD, standard deviation. |
IRF2 Knockdown Inhibits Non-Canonical Inflammasomes and Pyroptosis
Infection and pathological stimuli exacerbate inflammation and frequently trigger pyroptosis.25 The activation of caspase-1, caspase-4, NLRP3, ASC, and GSDMD might be involved in the process of pyroptosis in S-AKI.26 The results of Western blotting showed that LPS treatment significantly enhanced caspase-4, caspase-1, NLRP3, ASC, and GSDMD expression (Figure 4A and B), indicating that canonical and non-canonical inflammasomes were activated. IRF2 downregulation dramatically reduced the expression levels of caspase-4 and GSDMD, however, it had no significant effect on caspase-1, NLRP3, and ASC expression (Figure 4A and B), suggesting that the non-canonical pathway of pyroptosis was suppressed. Collectively, IRF2 knockdown inhibited non-canonical inflammasomes, thereby ameliorating pyroptosis.
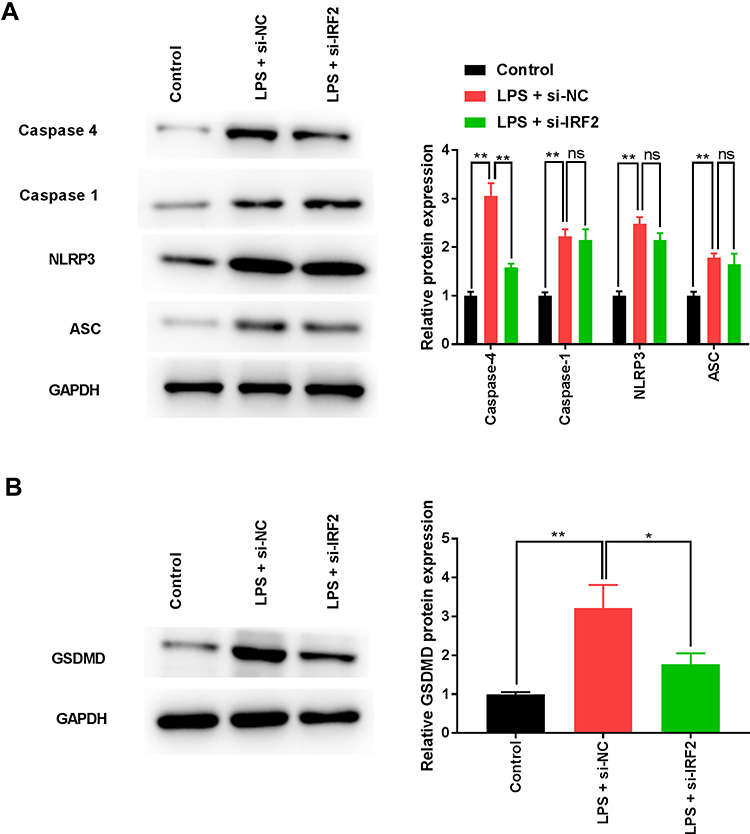 |
Figure 4 IRF2 knockdown inhibits LPS-induced pyroptosis. (A) The expression of caspase-1, caspase-4, NLRP3, and ASC was measured by Western blotting. (B) The expression of GSDMD was detected by Western blotting. Data were expressed as mean + SD. *p<0.05, **p<0.01. Abbreviations: IRF2, interferon regulatory factor 2; LPS, lipopolysaccharide; NLRP3, NLR family pyrin domain containing 3; ASC, apoptosis-associated speck-like protein; GSDMD, gasdermin-D; SD, standard deviation. |
IRF2 Knockout Alleviates the Inflammation and Pyroptosis of S-AKI in vivo
We determined the effects of IRF2 on CLP-induced renal injury in vivo to validate the protective activity of IRF2 inhibition. Figure 5A confirmed that IRF2 was not expressed in CLP-induced IRF2−/− mice. IRF2 knockout remarkably increased the survival rate of CLP-induced mice (Figure 5B). SCR and BUN levels were significantly higher in mice injected with LPS. In contrast, IRF2 knockout decreased the elevation of the levels of SCR and BUN (Figure 5C and D). Histological evaluation revealed that IRF2 knockout reduced inflammatory cell infiltration in the renal tissues of S-AKI mice, and reduced the scores of pathological sections in S-AKI mice (Figure 5E). IRF2 knockout notably decreased the levels of inflammatory cytokines IL-1β, TNF-α, IL-18, and IL-6 in the renal tissues of mice (Figure 5F). The protein expression of caspase 1, NLRP3, and ASC was unchanged in the renal tissues of mice by IRF2 knockout (Figure 5G). Western blot assay showed that the expression of pyroptosis pathway-associated proteins, caspase-11 and GSDMD, was markedly reduced (Figure 5H). Taken together, our results demonstrated that IRF2 knockout alleviated renal damage of S-AKI mice through a non-canonical inflammatory pathway.
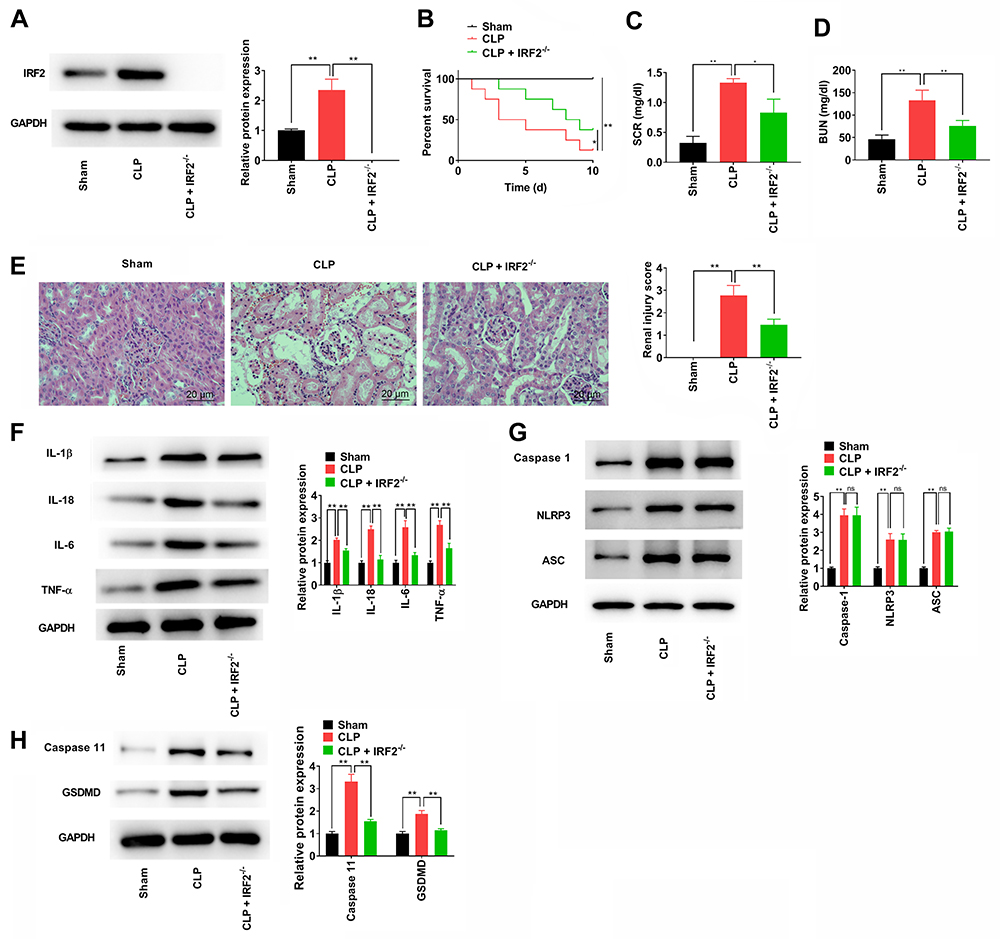 |
Figure 5 IRF2 knockout alleviates the renal damage of CLP-induced mice. (A) The IRF2 expression in renal tissues of CLP-induced IRF2−/− mice was detected by Western blotting. (B) The survival rates of mice from each group with the indicated condition were recorded for 10 days. (C and D) The content of SCR and BUN among different mice groups. (E) H&E staining and pathological scores of S-AKI mice were assessed. (F) The levels of the inflammatory cytokines, IL-1β, TNF-α, IL-18, and IL-6 in renal tissues collected were measured by Western blotting. (G) The protein expression of caspase-1, NLRP3, and ASC in renal tissues was determined by Western blotting. (H) The protein expression of caspase-11 and GSDMD in renal tissues was determined by Western blotting. Data were expressed as mean + SD. *p<0.05, **p<0.01. Abbreviations: IRF2, interferon regulatory factor 2; CLP, caecal ligation and puncture; SCR, serum creatinine; BUN, blood urea nitrogen; S-AKI, sepsis-related acute kidney injury; IL-1β, interleukin-1β; TNF-α, tumour necrosis factor-α; NLRP3, NLR family pyrin domain containing 3; ASC, apoptosis-associated speck-like protein; GSDMD, gasdermin-D; SD, standard deviation. |
Discussion
Sepsis is the leading cause of AKI in intensive care units. Furthermore, S-AKI is associated with a high mortality rate.27 Therefore, new therapeutic interventions are required to reduce S-AKI.
In this study, we collected serum samples from S-AKI patients and healthy individuals and established an S-AKI model in vitro. Our data demonstrated that IRF2 expression was upregulated in the serum of S-AKI patients and LPS-induced HK-2 cells. Moreover, IRF2 inhibition significantly increased S-AKI cell viability. Apoptosis and necrosis are the major pathways of cell death in S-AKI28,29. In the present study, we found that IRF2 downregulation dramatically reduced cell death and apoptosis in S-AKI. Increasing evidence has shown that sepsis can promote inflammatory factor release in kidney tissues, thereby causing severe renal cell apoptosis, leading to AKI.30 Thus, we explored the effect of IRF2 on inflammation in S-AKI by measuring the levels of several inflammatory cytokines. IL-1β is an important inflammatory factor that participates in the defence mechanism of hosts against pathogens.31 IL-18 is a pluripotent cytokine primarily produced by activated mononuclear macrophages and mediates ischaemic AKI.32 IL-1β and IL-18 can also elevate the expression of other inflammatory cytokines, such as TNF-α and IL-6, and facilitate the exudation of inflammatory cells.33 Our data showed that LPS treatment increased the levels of IL-1β, IL-18, IL-6, and TNF-α in HK-2 cells, thereby inducing an inflammatory response resulting in S-AKI. However, IRF2 knockdown notably reduced the levels of these factors to inhibit inflammation in S-AKI.
Apoptosis is characterised by cell shrinkage, membrane blebbing, phosphatidylserine externalization, nuclear DNA fragmentation, and nuclear externalisation.34 Apoptosis is considered to be immunologically silent and even anti-inflammatory, resulting in cell clearance in the absence of explicit activation of the immune system.35 Our data showed that LPS treatment increased LDH release, cell apoptosis, and the protein expression of Bax, and decreased the protein expression of Bcl-2 in an in vitro S-AKI model. IRF2 knockdown attenuated LPS-induced apoptosis in HK-2 cells. In recent years, increasing evidence has shown that, in addition to necrosis and apoptosis, pyroptosis also plays an important role in the pathogenesis of S-AKI.33 NLRP3 is predominantly distributed in the intercellular substance and cell membrane, and plays crucial roles in AKI by regulating inflammation and pyroptosis.6 It is responsible for caspase-1 activation by interacting with ASC to trigger the production of mature IL-1β and IL-18.36,37 NLRP3 is activated in an LPS-induced AKI cell model, leading to a decrease in cell viability,38 which is consistent with our results. GSDMD has been identified as a critical mediator of pore formation in cells undergoing pyroptosis.39 The tetramer active forms of caspase-1, caspase-4, and caspase-11 were found to be capable of cleaving purified recombinant GSDMD.7 GSDMD is a component of the inflammasome that is responsible for the implementation of pyroptosis and the secretion of mature IL-1β.40 Moreover, IRF2 binds to a unique site within the promoter of GSDMD and drives the GSDMD transcription to perform pyroptosis.21 Our results showed that inhibiting IRF2 does not alleviate the pyrolysis of S-AKI in vitro by affecting the expression of NLRP3, ASC, and caspase-1 (canonical inflammasomes), but by reducing the expression of caspase-4 and GSDMD (non-canonical inflammasomes).
In mice, caspase-11 is associated with non-canonical inflammasomes activated by multiple infections, resulting in cell death by pyroptosis.41 GSDMD is a crucial target of caspase-11, and its cleavage and resultant N-terminal fragments were identified as the executioner of pyroptosis.42 IL-1β levels in the serum and kidney tissues of caspase-11−/− mice remained low after LPS challenge.41 In the present study, IRF2 knockout dramatically ameliorated CLP-induced renal injury in vivo. Meanwhile, we found that IRF2 knockout could inhibit the CLP-induced inflammatory response and pyroptosis in vivo by suppressing non-canonical inflammasomes. Our study provides a new insight into the pathogenesis and treatment of S-AKI.
Conclusion
In conclusion, this study revealed that IRF2 is upregulated in the serum of S-AKI patients and LPS-induced HK-2 cells. IRF2 downregulation showed protective effects in LPS-induced HK-2 cells and CLP-induced mice. IRF2 inhibition can reduce inflammation and pyroptosis in vitro and in vivo by regulating non-canonical inflammasomes, showing that IRF2 is an effective drug target for S-AKI treatment.
Data Sharing Statement
All data of this manuscript used to support the findings of this study may be released upon application to the correspondence author.
Ethics Approval and Consent to Participate
The protocol of this research has been approved by the Ethics Committee of the Affiliated Suzhou Hospital of Nanjing Medical University (KL907125). All patients have signed written informed consent. The experimental protocol of our study was performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Affiliated Suzhou Hospital of Nanjing Medical University. (KL907125-DW).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no potential conflicts of interest.
References
1. Mehta RL, Bouchard J, Soroko SB, et al. Sepsis as a cause and consequence of acute kidney injury: program to improve care in acute renal disease. Intensive Care Med. 2011;37(2):241–248. doi:10.1007/s00134-010-2089-9
2. Bagshaw SM, Uchino S, Bellomo R, et al. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007;2(3):431–439. doi:10.2215/CJN.03681106
3. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–150. doi:10.1056/NEJMra021333
4. Hoste EA, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi:10.1007/s00134-015-3934-7
5. Alobaidi R, Basu RK, Goldstein SL, Bagshaw SM. Sepsis-associated acute kidney injury. Semin Nephrol. 2015;35(1):2–11. doi:10.1016/j.semnephrol.2015.01.002
6. Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol. 2019;15(8):501–520. doi:10.1038/s41581-019-0158-z
7. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
8. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi:10.1038/nature10759
9. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28(1):137–161. doi:10.1146/annurev-cellbio-101011-155745
10. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916. doi:10.1128/IAI.73.4.1907-1916.2005
11. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
12. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
13. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–164. doi:10.1038/nri.2016.147
14. Yuan J, Najafov A, Py BF. Roles of caspases in necrotic cell death. Cell. 2016;167(7):1693–1704. doi:10.1016/j.cell.2016.11.047
15. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. 2017;152(2):207–217. doi:10.1111/imm.12787
16. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
17. Wang Y, Zhang M, Sun Y, et al. Role of short-wavelength blue light in the formation of cataracts and the expression of caspase-1, caspase-11, Gasdermin D in rat lens epithelial cells: insights into a novel pathogenic mechanism of cataracts. BMC Ophthalmol. 2020;20(1):020–01565. doi:10.1186/s12886-020-01565-z
18. Levey AS, James MT. Acute kidney injury. Ann Intern Med. 2017;167(9):ITC66. doi:10.7326/AITC201711070
19. Harada H, Takahashi E, Itoh S, Harada K, Hori TA, Taniguchi T. Structure and regulation of the human interferon regulatory factor 1 (IRF-1) and IRF-2 genes: implications for a gene network in the interferon system. Mol Cell Biol. 1994;14(2):1500–1509. doi:10.1128/mcb.14.2.1500-1509.1994
20. Cui H, Banerjee S, Guo S, Xie N, Liu G. IFN regulatory factor 2 inhibits expression of glycolytic genes and lipopolysaccharide-induced proinflammatory responses in macrophages. J Immunol. 2018;200(9):3218–3230. doi:10.4049/jimmunol.1701571
21. Kayagaki N, Lee BL, Stowe IB, et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal. 2019;12(582):582. doi:10.1126/scisignal.aax4917
22. Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int. 2015;87(2):332–342. doi:10.1038/ki.2014.270
23. Wilson RL, Selvaraju V, Lakshmanan R, et al. Thioredoxin-1 attenuates sepsis-induced cardiomyopathy after cecal ligation and puncture in mice. J Surg Res. 2017;220:68–78. doi:10.1016/j.jss.2017.06.062
24. Wu Y, Wang L, Meng L, Cao GK, Zhao YL, Zhang Y. Biological effects of autophagy in mice with sepsis-induced acute kidney injury. Exp Ther Med. 2019;17(1):316–322. doi:10.3892/etm.2018.6899
25. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070
26. Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22(6):1007–1018. doi:10.1681/ASN.2010080798
27. Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;9:364.
28. Pickkers P, Ostermann M, Joannidis M, et al. The intensive care medicine agenda on acute kidney injury. Intensive Care Med. 2017;43(9):1198–1209. doi:10.1007/s00134-017-4687-2
29. Holthoff JH, Wang Z, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012;81(4):370–378.
30. Shum HP, Kong HH, Chan KC, Yan WW, Chan TM. Septic acute kidney injury in critically ill patients - a single-center study on its incidence, clinical characteristics, and outcome predictors. Ren Fail. 2016;38(5):706–716. doi:10.3109/0886022X.2016.1157749
31. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214. doi:10.1111/j.1600-065X.2011.01044.x
32. Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046–3052. doi:10.1681/ASN.2005030236
33. Yamauchi K, Choi IJ, Lu H, Ogiwara H, Graham DY, Yamaoka Y. Regulation of IL-18 in Helicobacter pylori infection. J Immunol. 2008;180(2):1207–1216. doi:10.4049/jimmunol.180.2.1207
34. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
35. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17(5):333–340. doi:10.1038/nri.2016.153
36. Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transplant. 2014;29(1):41–48. doi:10.1093/ndt/gft332
37. Hutton HL, Ooi JD, Holdsworth SR, Kitching AR. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology. 2016;21(9):736–744. doi:10.1111/nep.12785
38. Shen J, Wang L, Jiang N, et al. NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci Rep. 2016;6(34682):1.
39. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
40. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi:10.1038/nature13683
41. Cheng KT, Xiong S, Ye Z, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124–4135. doi:10.1172/JCI94495
42. Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. doi:10.1038/nature15541
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.