")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Downregulation of DOCK1 sensitizes bladder cancer cells to cisplatin through preventing epithelial–mesenchymal transition
Authors Chen D, Chen W, Jiang H, Yang H, Wang Y, Chen J
Received 7 December 2015
Accepted for publication 4 February 2016
Published 8 September 2016 Volume 2016:10 Pages 2845—2853
DOI https://doi.org/10.2147/DDDT.S101998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Da-jin Chen,1–3,* Wei Chen,4,* Hong Jiang,1–3 Hao Yang,1–3 Yu-cheng Wang,1–3 Jiang-hua Chen1–3
1Kidney Disease Center, The First Affiliated Hospital, Zhejiang University School of Medicine, 2Key Laboratory of Nephropathy, 3Kidney Disease Immunology Laboratory, The Third Grade Laboratory, State Administration of Traditional Chinese Medicine, 4Department of Hepatobiliary Surgery, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Abstract: During the past several decades, resistance to single or multiple anticancer agents has posed a great challenge in cancer therapy. Dedicator of cytokinesis 1 (DOCK1), the first identified member in DOCK family, plays diverse roles in cellular processes, including tumorigenesis. In this study, we explored the biological role of DOCK1 in the chemotherapeutic resistance in bladder cancer and its underlying mechanism. Our results showed that the bladder cancer cell lines UM-UC-3 and J82 with higher DOCK1 are more resistant to cisplatin, whereas B87 cells with the lowest expression of DOCK1 exhibited the highest sensitivity to cisplatin. Downregulation of DOCK1 with small interfering RNA (siRNA) increased the cisplatin sensitivity in bladder cancer cells. Moreover, treatment with cisplatin induced epithelial–mesenchymal transition (EMT), while transfection with Twist siRNA restored the chemosensitivity to cisplatin. In addition, we found that downregulation of DOCK1 reversed EMT program in bladder cancer cells. However, cotransfection with DOCK1 siRNA could not further enhance the cisplatin sensitivity and cellular phenotypic changes in tumor cells. Taken together, these results demonstrate that downregulation of DOCK1 could increase the chemosensitivity in bladder cancer cells via preventing cisplatin-induced EMT, suggesting that DOCK1 may serve as a potential therapeutic target in bladder cancer.
Keywords: bladder cancer, epithelial–mesenchymal transition, drug resistance, DOCK1
Introduction
Bladder cancer is the most common urinary tract malignancy and remains one of the costliest diseases to manage from diagnosis to death.1 In accordance with the invasion degree, patients with bladder cancer can be diagnosed as nonmuscle-invasive or muscle-invasive tumor.2 In addition to the radical cystectomy, cisplatin-based neoadjuvant chemotherapy serves as an important therapy option and shows increased response rate and prolonged survival in patients with bladder cancer.3 However, cisplatin resistance in the past several decades has become a great challenge for clinical treatment of bladder cancer, highlighting the urgency to investigate the molecular mechanism underlying the drug resistance.4
The dedicator of cytokinesis (DOCK) family with eleven mammalian members belongs to the atypical Rho guanine nucleotide exchange factors. Emerging evidences reported the critical roles of DOCK in various cellular processes such as actin cytoskeleton and cell adhesion and migration.5,6 As the first identified member of DOCK protein family, DOCK1 plays crucial roles in cytoskeletal organization, myoblast fusion, and phagocytosis via Rac1 activation in Caenorhabditis elegans and Drosophila.7–9 The expression of DOCK1 is upregulated in ovarian cancer correlated with aggressive phenotype and poor patient survival.10 Moreover, engulfment and cell motility 1 (Elmo1) synergistically with DOCK180 promote cell motility in ovarian carcinoma.11 Functional studies reveal that DOCK1 is associated with tumorigenesis, growth, and invasion of glioma, suggesting that DOCK1 could be a potential therapeutic target for malignant brain tumor.12–14 However, the biological function of DOCK1 in bladder cancer has not been investigated. Thus, this study aimed to explore the role of DOCK1 in the chemotherapeutic resistance in bladder cancer and its underlying mechanism.
Materials and methods
Cell culture
Three bladder cancer cell lines (UM-UC-3, J82, and B87) were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were maintained at 37°C in 5% CO2 incubator. All experiments included in this research were in compliance with the defined policies and protocols, and the Ethical Committee of the First Affiliated Hospital of Zhejiang University approved the research protocol. Cisplatin was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). The DOCK1 small interfering RNA (siRNA) and Twist siRNA were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
CCK-8 assay
Cells at 3.0×103 cells/well were seeded into 96-well plates. Then, 10 μL/well Cell Counting Kit-8 (CCK-8) solution (Dojindo, Kumamoto, Japan) was added, the plates were incubated for 3 hours, and absorbance was measured at 450 nm using an MRX II microplate reader (Dynex, Chantilly, VA, USA).
EdU incorporation assay
DNA incorporation synthesis was calculated using EdU (5-ethynyl-2-deoxyuridine) incorporation assay. Measurement of inhibitive rate of cell proliferation was carried out using a Click-iT EdU Imaging Kit (Thermo Fisher Scientific) according to the procedure described previously.15
siRNA transfection
Bladder cancer cells were transfected with DOCK1 siRNA and Twist siRNA using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instruction. The transfection medium (Opti-MEM; Thermo Fisher Scientific) was replaced with complete medium 12 hours after transfection, and the cells were incubated for the indicated times.
Immunofluorescence
Bladder cancer cells were seeded into 48-well plates at 6.0×103 cells/well. Slides were blocked with bovine serum albumin and incubated with mouse anti-human vimentin or anti-human E-cadherin primary antibodies (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight. After washing, the slides were incubated with goat anti-mouse fluorescein isothiocyanate-conjugated secondary antibody (Abcam, Cambridge, UK). After incubation with 4’,6-diamidino-2-phenylindole (DAPI), cells were observed with a fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Western blotting
Cells were lysed, and protein concentration was determined using a BCA kit (Beyotime, Nanjing, People’s Republic of China). Afterward, protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were incubated with anti-DOCK1, anti-E-cadherin, anti-vimentin, and anti-Twist (Cell Signaling Technology) antibodies at 4°C overnight. The membranes were washed three times with Tris-buffered saline–Tween 20 and then incubated with the appropriate horseradish peroxide-conjugated secondary antibodies. The protein bands were detected by chemiluminescence (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
Statistical analysis
Each experiment was performed in triplicate and repeated at least three times. Experimental data were presented as mean ± standard deviation and treated for statistics analysis by SPSS program. Comparison between groups was made using analysis of variance, and statistically significant difference was defined as P<0.05.
Results
Different cisplatin chemosensitivity in bladder cancer cells
First, we determined the protein expression of DOCK1 in three bladder cancer cell lines, including UM-UC-3, J82, and B87, using Western blotting. The protein levels varied in these three bladder cancer cell lines (Figure 1A). Interestingly, CCK-8 assay (Figure 1B) and EdU incorporation assay (Figure 1C) showed that UM-UC-3 and J82 cells with higher DOCK1 are more resistant to cisplatin, whereas B87 cells with the lowest expression of DOCK1 exhibited the highest sensitivity to cisplatin. These results implied that DOCK1 may be involved in the chemoresistance to cisplatin in bladder cancer.
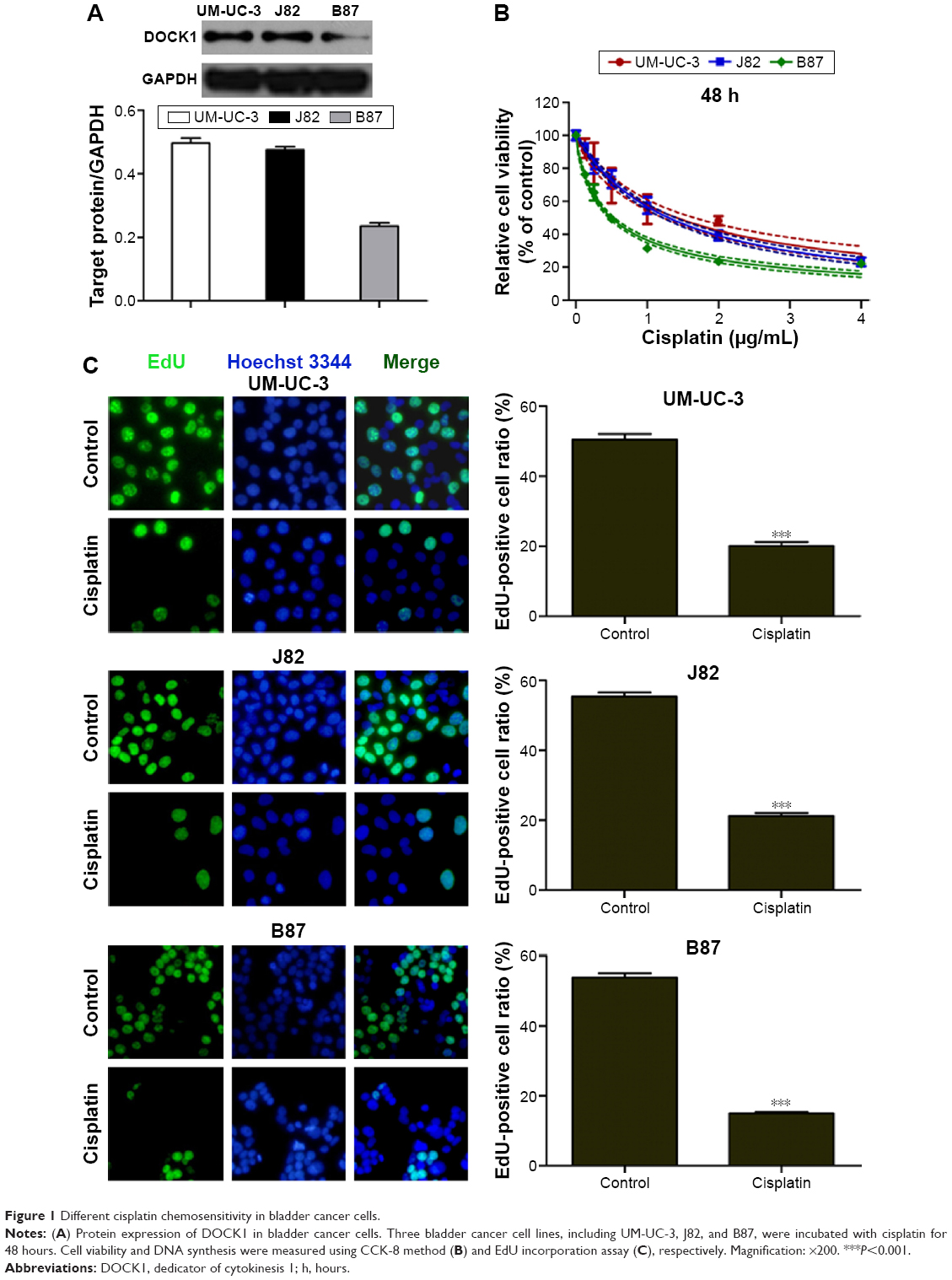 | Figure 1 Different cisplatin chemosensitivity in bladder cancer cells. |
Downregulation of DOCK1 sensitized bladder cancer cells to cisplatin
UM-UC-3, J82, and B87 cells were transiently transfected with DOCK1 siRNA or control siRNA. After 48 hours, the expression of DOCK1 was obviously suppressed in these three cancer cell lines (Figure 2D). As a result, we found that downregulation of DOCK1 increased the cisplatin sensitivity in bladder cancer cells (Figure 2A–C). These data demonstrated that decreased expression of DOCK1 sensitized bladder cancer cells to chemotherapy agents.
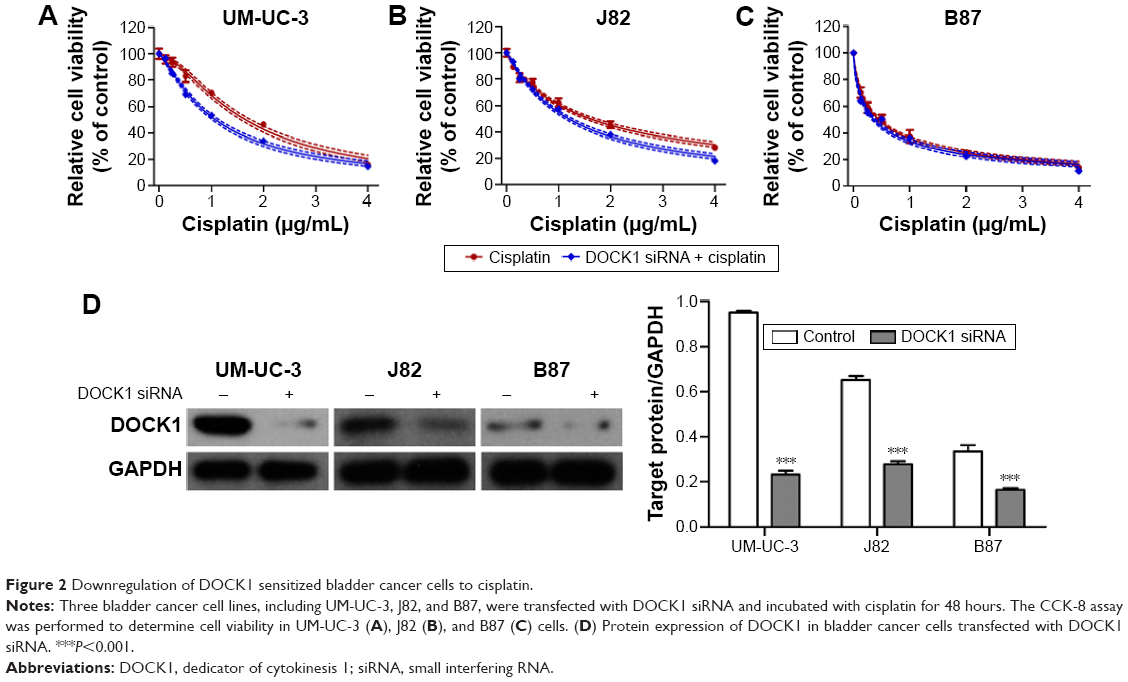 | Figure 2 Downregulation of DOCK1 sensitized bladder cancer cells to cisplatin. |
DOCK1 regulated EMT in bladder cancer cells
EMT program is involved in various tumor behaviors, such as growth, invasion, metastasis, and drug resistance.16,17 Immunostaining and Western blot assay showed that cisplatin treatment inhibited the expression of E-cadherin (epithelial marker) and promoted the expression of vimentin (mesenchymal marker) in bladder cancer cells (Figure 3A). In order to block EMT in bladder cancer cells, Twist level was suppressed through transfection of Twist siRNA into tumor cells (Figure 3B). Consequently, the cisplatin sensitivity was remarkably enhanced after EMT blockage in bladder cancer cells (Figure 3C).
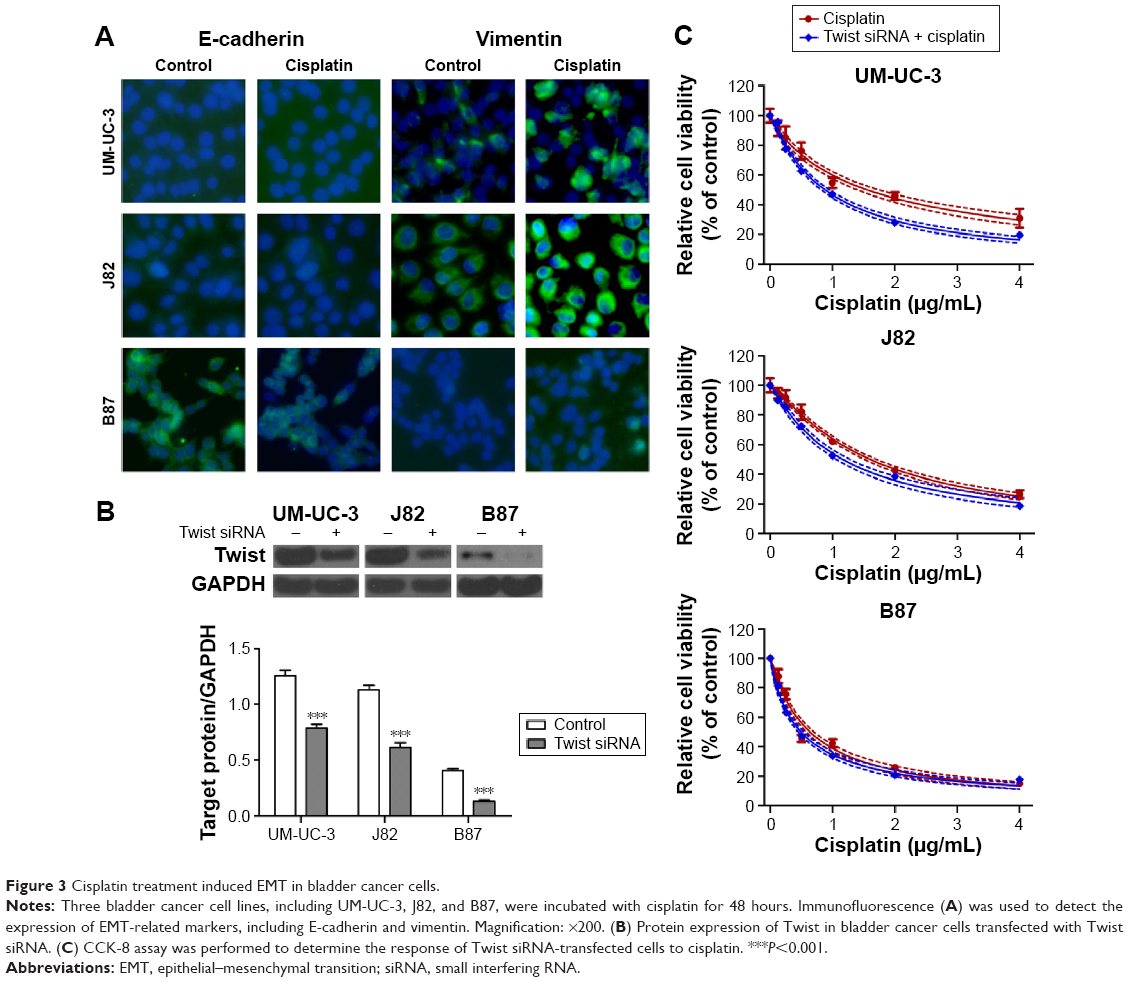 | Figure 3 Cisplatin treatment induced EMT in bladder cancer cells. |
Next, we tested the hypothesis that DOCK1 regulated EMT in bladder cancer cells. Western blot analysis showed that EMT-related markers varied in different cell lines (Figure 4A). Interestingly, we observed that B87 cells with lower DOCK1 exhibited a significant increase of E-cadherin protein and reduced expression of vimentin. In addition, we found that downregulation of DOCK1 via siRNA transfection reversed EMT as shown by upregulation of E-cadherin and downregulation of vimentin in bladder cancer cells (Figure 4B). Based on these findings, we concluded that DOCK1 could regulate EMT in bladder cancer cells.
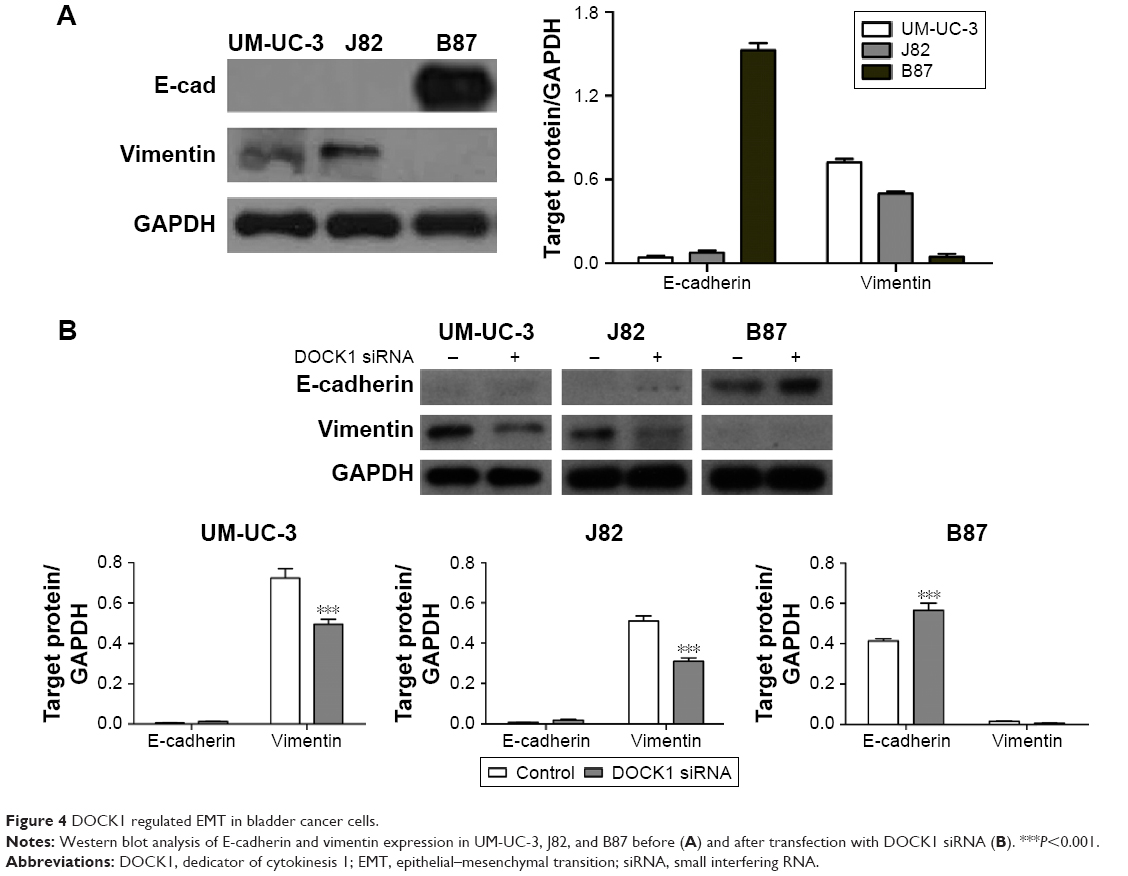 | Figure 4 DOCK1 regulated EMT in bladder cancer cells. |
DOCK1 inhibition enhanced cisplatin sensitivity via EMT
To further define the role of DOCK1 in cisplatin-induced EMT, we detected the expression level of DOCK1 in cisplatin-treated cells. Western blot analysis revealed that cisplatin treatment promoted the expression of DOCK1 in UM-UC-3, J82, and B87 cells (Figure 5A). However, cisplatin addition failed to induce EMT in DOCK1-knockdown cells (Figure 5B), suggesting that EMT induction by cisplatin was dependent on DOCK1 in bladder cancer cells. To further elucidate the effects of DOCK1-regulated EMT on cisplatin sensitivity, bladder cancer cells were transfected with Twist siRNA alone or in combination with DOCK1 siRNA. CCK-8 assay showed that DOCK1 inhibition could not further sensitize tumor cells to cisplatin after transfection with Twist siRNA (Figure 6A–C). On the molecular level, Western blot analysis showed that Twist knockdown led to a reduction of the mesenchymal marker vimentin in bladder cancer cells. However, cotransfection with DOCK1 siRNA had no obvious effects on the expression of cellular phenotype markers in Twist siRNA-transfected bladder cancer cells (Figure 6D). Taken together, these results confirmed that downregulation of DOCK1 sensitized bladder cancer cells to cisplatin via regulation of EMT.
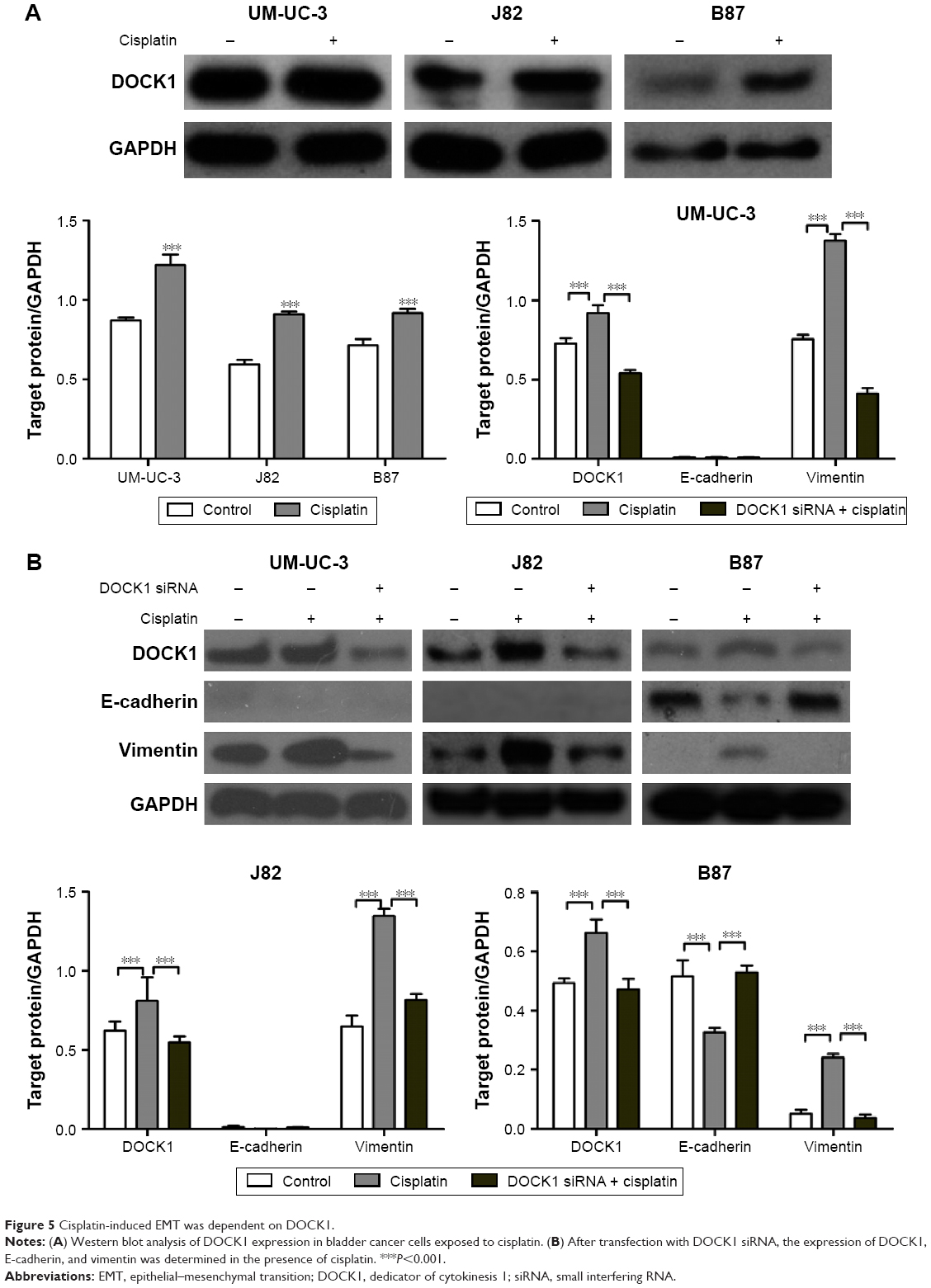 | Figure 5 Cisplatin-induced EMT was dependent on DOCK1. |
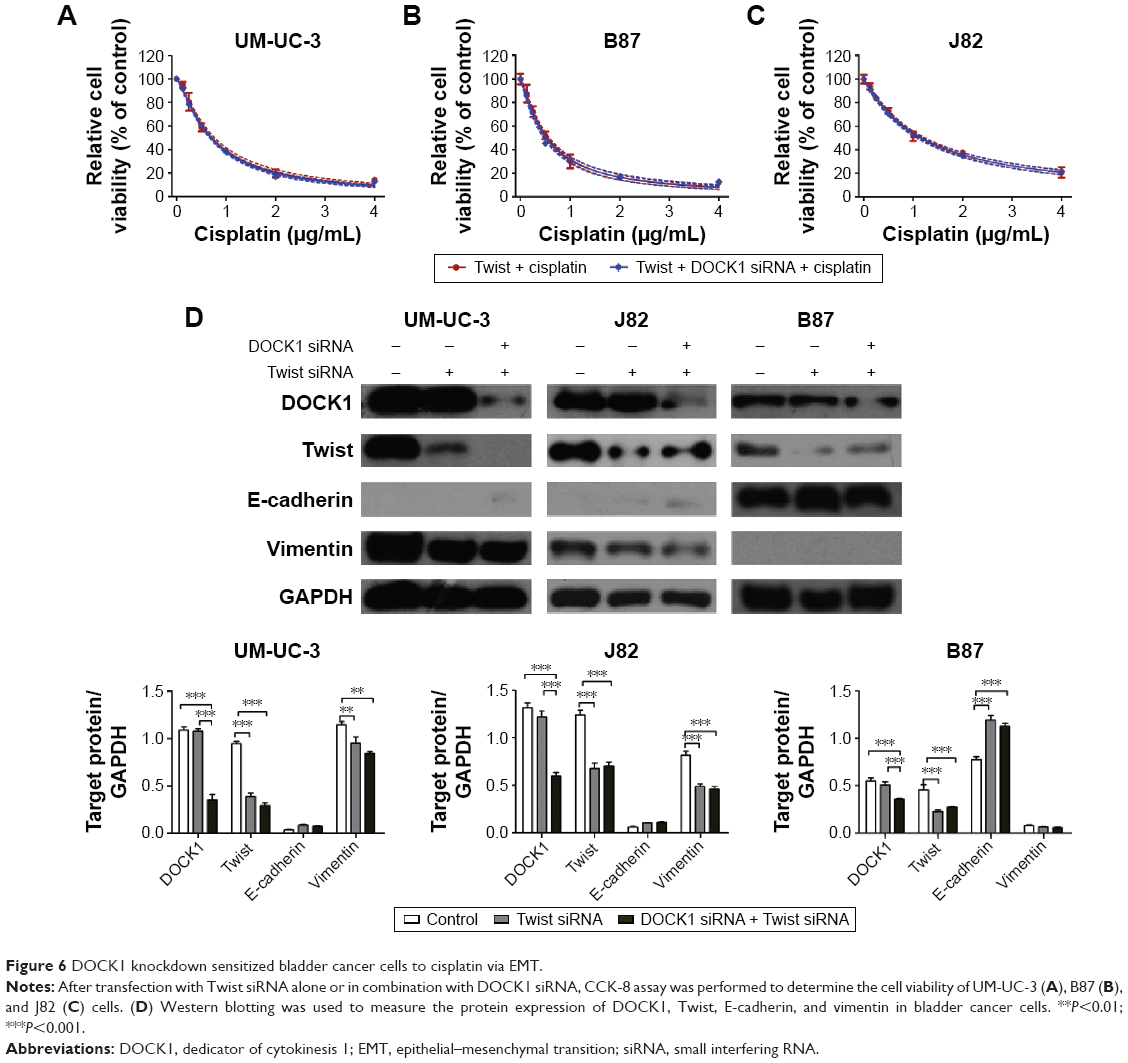 | Figure 6 DOCK1 knockdown sensitized bladder cancer cells to cisplatin via EMT. |
Discussion
Emerging evidences point that a wide range of pro-oncogene and antioncogene alterations are involved in the pathogenesis of bladder cancer.18 Despite significant progress in diagnosis and treatment, the molecular mechanism of bladder carcinogenesis still remains elucidated, and specific therapeutic targets for bladder cancer are currently limited. In this study, we demonstrated that downregulation of DOCK1 increased cisplatin sensitivity via inhibition of EMT in bladder cancer cells.
To our knowledge, this study for the first time demonstrated the regulatory effect of DOCK1 on cisplatin sensitivity in bladder cancer cells, which we think is of biological importance given its potential therapeutic intervention for patients with bladder cancer resistant to chemotherapeutic agents.
Functioning as a guanine nucleotide exchange factor, DOCK1 activates Rac1 and controls multiple cellular behaviors, including cell growth, motility, and survival, in both normal and pathological conditions.19,20 Previous exploration in gliomas demonstrated that DOCK1 is required for platelet-derived growth factor receptor α-stimulated glioma cell migration and survival in vitro and tumor growth, survival, and invasion in the brain.13 A recent study has revealed that DOCK1 is also critical for epidermal growth factor receptor (EGFR) vIII-driven glioma growth and invasion via EGFRvIII-PKA-DOCK180-Rac1 signaling axis, providing a novel target to develop potential therapeutic strategies for malignant gliomas.12 Immunohistochemistry analysis shows the upregulation of DOCK1 in ovarian cancer, which is correlated with aggressive phenotype and poor patient survival.10 Documented biological role of DOCK1 in breast cancers demonstrates that DOCK1-Rac signaling is essential for HER2-mediated breast cancer progression to metastasis and offers a therapeutic opportunity to limit cancer spread.21 Moreover, the low molecular weight heparin enoxaparin sensitizes human lung cancer cells to gefitinib through inhibition of DOCK1 expression.22 All these findings suggest the oncogenic property of DOCK1 in human cancer. In our study, we found that the expression of DOCK1 varied in three bladder cancer cell lines, and its downregulation enhanced the cisplatin sensitivity, suggesting that DOCK1 is involved in the chemoresistance in bladder cancer.
A large number of studies demonstrate that EMT is of paramount importance in a plethora of cancer-related events, such as cancer metastasis, invasion, and refractory responses to chemotherapy.23–27 During EMT, epithelial markers such as E-cadherin decrease, while mesenchymal markers such as vimentin increase.28 In our study, we found that cisplatin treatment induced EMT in bladder cancer cells, while transfection with Twist siRNA restored the chemosensitivity to cisplatin. Moreover, the expression of EMT-related markers E-cadherin (epithelial) and vimentin (mesenchymal) varied in UM-UC-3, J82, and B87 cells. Interestingly, B87 cells with low-level DOCK1 exhibited a significant increase of E-cadherin and reduced vimentin. Based on these findings, we hypothesized that DOCK1 may regulate EMT in bladder cancer cells. Unsurprisingly, our results showed that DOCK1 knockdown reversed EMT program after DOCK1 siRNA transfection into bladder cancer cells.
Drug resistance in bladder cancer involves a complex interplay of various factors, including the accumulation of oncogenes and loss of tumor repressors.29–31 Serving as a potential oncogene, the expression of DOCK1 was increased in bladder cancer cells exposed to cisplatin. However, downregulation of DOCK1 prevented cisplatin-induced EMT in DOCK siRNA-transfected cells. Moreover, downregulation of DOCK1 failed to further enhance the cisplatin sensitivity in bladder cancer cells transfected with Twist siRNA. We also found that Twist knockdown attenuated the mesenchymal phenotype of bladder cancer cells. However, DOCK1 downregulation resulted in no further changes in cellular phenotype in Twist siRNA-transfected cells. Taken together, these results confirmed that DOCK1 knockdown sensitized bladder cancer cells to cisplatin via EMT.
Conclusion
In conclusion, this study showed that downregulation of DOCK1 enhances the cytotoxicity of cisplatin in bladder cancer cells through preventing cisplatin-induced EMT. Thus, these findings may provide evidences to define DOCK1 as a potential therapeutic target in bladder cancer.
Acknowledgments
This study was supported by Zhejiang Provincial Education Department Program (491010-G21410): Biological role and mechanism investigation of DOCK1 in chemoresistance in bladder cancer, and Zhejiang Medical Science and Technology Program (2014KYA057): Biological effect and mechanism investigation of Th17 cells, the new T cell subsets, on the early acute renal transplant rejection
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Mertens LS, Neuzillet Y, Horenblas S, van Rhijn BW. Landmarks in non-muscle-invasive bladder cancer. Nat Rev Urol. 2014;11(8):476–480. | ||
Moon DG, Lee SE, Oh MM, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor synergistically potentiates the antitumor effects of cisplatin in bladder cancer cells. Int J Oncol. 2014;45(3):1027–1035. | ||
Stenzl A, Cowan NC, De Santis M, et al. Treatment of muscle-invasive and metastatic bladder cancer: update of the EAU guidelines. Eur Urol. 2011;59(6):1009–1018. | ||
Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014;93(10–12):466–477. | ||
Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24(1):203–216. | ||
Erickson MR, Galletta BJ, Abmayr SM. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol. 1997;138(3):589–603. | ||
Wu YC, Horvitz HR. C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature. 1998;392(6675):501–504. | ||
Jarzynka MJ, Hu B, Hui KM, et al. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 2007;67(15):7203–7211. | ||
Zhao F, Siu MK, Jiang L, et al. Overexpression of dedicator of cytokinesis I (Dock180) in ovarian cancer correlated with aggressive phenotype and poor patient survival. Histopathology. 2011;59(6):1163–1172. | ||
Wang J, Dai JM, Che YL, et al. Elmo1 helps dock180 to regulate Rac1 activity and cell migration of ovarian cancer. Int J Gynecol Cancer. 2014;24(5):844–850. | ||
Feng H, Hu B, Vuori K, et al. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene. 2014;33(19):2504–2512. | ||
Feng H, Hu B, Liu KW, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest. 2011;121(12):4670–4684. | ||
Feng H, Hu B, Jarzynka MJ, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci U S A. 2012;109(8):3018–3023. | ||
Chehrehasa F, Meedeniya AC, Dwyer P, Abrahamsen G, Mackay-Sim A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J Neurosci Methods. 2009;177(1):122–130. | ||
Jing Y, Cui D, Guo W, et al. Activated androgen receptor promotes bladder cancer metastasis via Slug mediated epithelial-mesenchymal transition. Cancer Lett. 2014;348(1–2):135–145. | ||
Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66(23):11271–11278. | ||
Costello JC, Theodorescu D. Decade in review-bladder cancer: international progress: from cytology to genomics. Nat Rev Urol. 2014;11(11):609–610. | ||
Laurin M, Dumouchel A, Fukui Y, Cote JF. The Rac-specific exchange factors Dock1 and Dock5 are dispensable for the establishment of the glomerular filtration barrier in vivo. Small GTPases. 2013;4(4):221–230. | ||
Laurin M, Fradet N, Blangy A, Hall A, Vuori K, Côté JF. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc Natl Acad Sci U S A. 2008;105(40):15446–15451. | ||
Laurin M, Huber J, Pelletier A, et al. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci U S A. 2013;110(18):7434–7439. | ||
Pan Y, Li X, Duan J, et al. Enoxaparin sensitizes human non-small-cell lung carcinomas to gefitinib by inhibiting DOCK1 expression, vimentin phosphorylation, and Akt activation. Mol Pharmacol. 2015;87(3):378–390. | ||
Hanze J, Henrici M, Hegele A, Hofmann R, Olbert PJ. Epithelial mesenchymal transition status is associated with anti-cancer responses towards receptor tyrosine-kinase inhibition by dovitinib in human bladder cancer cells. BMC Cancer. 2013;13:589. | ||
Tran MN, Choi W, Wszolek MF, et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem. 2013;288(5):3275–3288. | ||
Li W, Kidiyoor A, Hu Y, et al. Evaluation of transforming growth factor-beta1 suppress Pokemon/epithelial-mesenchymal transition expression in human bladder cancer cells. Tumour Biol. 2015;36(2):1155–1162. | ||
Islam SS, Mokhtari RB, Noman AS, et al. Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial-to-mesenchymal transition (EMT) in bladder cancer. Mol Carcinog. Epub 2015 Mar 1. | ||
Zhang J, Wei J, Lu J, et al. Overexpression of Rab25 contributes to metastasis of bladder cancer through induction of epithelial-mesenchymal transition and activation of Akt/GSK-3beta/Snail signaling. Carcinogenesis. 2013;34(10):2401–2408. | ||
Thomson S, Petti F, Sujka-Kwok I, et al. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis. 2011;28(2):137–155. | ||
Massari F, Santoni M, Ciccarese C, et al. Emerging concepts on drug resistance in bladder cancer: Implications for future strategies. Crit Rev Oncol Hematol. 2015;96(1):81–90. | ||
Tada Y, Wada M, Migita T, et al. Increased expression of multidrug resistance-associated proteins in bladder cancer during clinical course and drug resistance to doxorubicin. Int J Cancer. 2002;98(4):630–635. | ||
Yu S, Meng Q, Hu H, Zhang M. Correlation of ANXA1 expression with drug resistance and relapse in bladder cancer. Int J Clin Exp Pathol. 2014;7(9):5538–5548. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.