")
Back to Journals » OncoTargets and Therapy » Volume 12
Downregulation of CENPK suppresses hepatocellular carcinoma malignant progression through regulating YAP1
Authors Wang J , Li H, Xia C, Yang X, Dai B, Tao K, Dou K
Received 8 October 2018
Accepted for publication 18 December 2018
Published 29 January 2019 Volume 2019:12 Pages 869—882
DOI https://doi.org/10.2147/OTT.S190061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Jianlin Wang,* Haimin Li,* Congcong Xia,* Xisheng Yang, Bin Dai, Kaishan Tao, Kefeng Dou
Department of Hepatobiliary Surgery, Xijing Hospital, The Fourth Military Medical University, Xi’an 710032, China
*These authors contributed equally to this work
Background: Several studies have found that centromere protein K (CENPK) is overexpressed in several tumour types and promotes tumor progression. However, there has been little research on the role of CENPK in the progression of hepatocellular carcinoma (HCC).
Materials and methods: The expression of CENPK in HCC tissues was quantified by Western blot and quantitative real-time PCR. Cells were transfected with lentiviral plasmids containing shRNA sequences targeting CENPK and YAP1 to silence the expression of CENPK and YAP1. Cell Counting Kit-8 assay, colony formation assay, wound healing assay, and transwell invasion assay were performed to evaluate cell growth, migration, and invasion of HCC cells. Tumorigenicity assay was used to detect the effect of CENPK on the growth of HCC cells. Western blot assay was performed to investigate the expression of epithelial–mesenchymal transition (EMT) markers and YAP1.
Results: Compared to that in adjacent non-tumor tissues, CENPK was aberrantly upregulated in HCC tumor tissues. Furthermore, CENPK knockdown significantly inhibited proliferation, migration, invasion, and EMT progression in HCC cells. Mechanistically, we identified that YAP1 was responsible for the tumor-suppressive effects of CENPK knockdown in the HCC cells. The inhibitory effects of CENPK silencing on cell proliferation, migration, invasion, and EMT were partially reversed by the restoration of YAP1 expression.
Conclusion: Our results suggested that the CENPK–YAP1–EMT axis plays a critical role in regulating HCC malignant progression, indicating the role of this axis as a potential therapeutic target for HCC.
Keywords: CENPK, YAP1, proliferation, migration and invasion, EMT, HCC
Corrigendum for this paper has been published.
Introduction
Hepatocellular carcinoma (HCC) is a major histological subtype of liver cancer, accounting for 90% of primary liver cancers, and is the third most frequent cause of cancer-related mortality worldwide.1,2 Genetic and epigenetic alterations, chronic infection with hepatitis B virus or hepatitis C virus, aflatoxin exposure, smoking, obesity, and diabetes are the main risk factors for HCC.2–4 The poor prognosis of HCC is attributed to the high rates of recurrence and metastasis.5–7 At present, transplantation is the most effective treatment for HCC, but either due to tumor burden or poor liver function, more than 70% of cases at advanced stage are unsuitable for transplantation.8 Hence, finding molecular mechanisms underlying HCC tumorigenesis and improving therapeutic strategies for HCC are critically important.
Increasing evidence suggests that kinetochore dysregulation or dysfunction is closely related to the occurrence of cancer.9 Kinetochore is a protein structure on chromatids that consists of at least 80 different proteins and plays an important role in chromosome segregation in all eukaryotes.10 Centromere protein A (CENPA) is one of the first identified kinetochore components in humans and is involved in several human malignancies.11–14 It has been found that several centromere proteins were upregulated in HCC and associated with HCC malignant progression.11,15,16 Centromere protein K (CENPK) is important for proper kinetochore function and mitotic progression.17 Recently, studies have showed that CENPK is specifically upregulated in ovarian cancer, triple-negative breast cancer, and HCC and is associated with malignant progression.18–20 However, the clinical significance and mechanism of CENPK in HCC remain largely unclear, which promoted us to explore the functional roles of CENPK in HCC pathogenesis.
Yes-associated protein (YAP1), the direct downstream effector of the Hippo pathway, controls countless protein targets that influence gene expression and participates in regulating cell proliferation, cell contact, and apoptosis.21–23 YAP1 has been found to be overexpressed in various types of human cancers, and associated with malignant features (eg, high histological grade, late TNM stage, metastasis, poor tumor differentiation) and poor patient outcomes.24 The expression of YAP1 was elevated in HCC tissues and predicted a poor disease-free survival and overall survival in HCC patients.25 Studies have demonstrated that knocking down YAP1 can reduce HCC cell proliferation, migration, and invasion.26–28 Furthermore, YAP1 expression is involved in epithelial–mesenchymal transition (EMT) in many human cancers,29–31 including HCC.28,32 Therefore, YAP1 may have a critical role in the development of hepatocarcinogenesis and targeting YAP1 could be a useful strategy for HCC treatment.
In this study, we sought to investigate the clinicopathological significance of CENPK in HCC and its role in HCC development. Based on our results, compared to that in adjacent non-tumor tissues (ANLTs), CENPK was significantly upregulated in HCC tissues. Knockdown of CENPK significantly inhibited HCC cell proliferation, migration, and invasion, and EMT progression. Moreover, CENPK suppressed the HCC malignant phenotype and EMT progression by regulating YAP1. These data demonstrate that the CENPK–YAP1–EMT axis may play a critical role in HCC development and thus may represent a promising therapeutic target for HCC treatment.
Materials and methods
Cell lines and tissue samples
The HCC cell lines SMMC-7721 and BEL-7404 were obtained from Genechem (Shanghai, China). BEL-7404 and SMMC-7721 cells were maintained in RIPA-1640 and DMEM supplemented with 10% FBS, respectively. The cells were incubated at 37°C in humidified atmosphere containing 5% CO2. Additionally, 30 HCC tissue samples and matched ANLTs were obtained from Xijing Hospital (Shaanxi, China), and all participants provided written informed consent.
Plasmids and cell transfection
Lentiviral plasmids containing shRNA sequences targeting CENPK and YAP1 (shCENPK and shYAP1) or negative control (shNC) were designed and produced by Genechem. BEL-7404 and SMMC-7721 cells were transfected with lentiviral plasmids when the cells were 30% confluent in six-well plates. Then, the culture medium was replaced with transduction-enhancing solution containing lentivirus at 20 multiplicity of infection and 50 μg/mL polybrene. After 12 hours, the medium was replaced with complete medium, and the cells were cultured for 72 hours. The cells were selected with 1 μg/mL puromycin and harvested for subsequent studies. HCC cells were transfected with the pcDNA3.1-YAP1 or pcDNA3.1-YAP1-NC plasmid (Genechem) by using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
RNA extraction and qRT-PCR
Total RNA from HCC tissues, ANLTs, and BEL-7404 and SMMC-7721 cells was isolated with the RNAiso Plus reagent (TaKaRa, Dalian, China) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was carried out using RT primers and the SYBR Premix EX Taq™ II kit purchased from TaKaRa. β-actin was used as a quantitative control to normalize mRNA expression. The amplification of genes was carried out using the primers shown in Table 1, and all primers were designed, synthesized, and validated by Shanghai Genechem.
 | Table 1 Sequences of qRT-PCR primers used for mRNA analysis |
Western blotting
The Western blot assay was performed according to the protocol described in a previous study.33 Antibody list is given in Table 2.
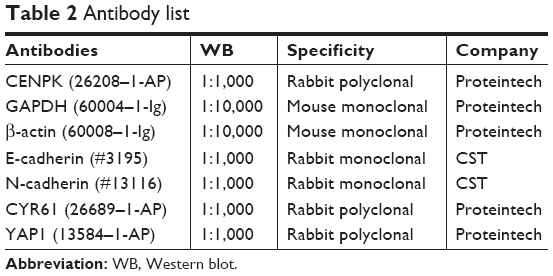 | Table 2 Antibody list |
Immunofluorescence staining
BEL-7404 and SMMC-7721 cells were seeded in 24-well plates for 24 hours. Then, the cells were fixed with 4% paraformaldehyde for 15 minutes and permeabilized with 0.3% Triton X-100 for 10 minutes. After blocking for 1 hour with 5% BSA, the cells were incubated with primary anti-YAP1 antibody (1:100; Proteintech, Chicago, IL, USA) overnight at 4°C, and for 1 hour with tetramethyl-rhodamine isothiocyanate-conjugated anti-rabbit IgG. In addition, the nuclei were stained with DAPI. Images were captured under a fluorescence microscope.
CCK-8 assay
Transfected BEL-7404 and SMMC-7721 cells were plated in 96-well plates at 2,000 cells per well, and cell proliferation was measured at 1, 2, 3, and 4 days after seeding using the Cell Counting Kit (CCK-8; Yiyuan Biotechnologies, Guangzhou, China) assay according to the manufacturer’s instructions.
Colony formation assay
HCC cells were seeded in six-well culture plates at 300 or 500 cells per well. After incubation for 2 weeks, the colonies were washed twice with PBS, fixed with 95% ethanol for 15 minutes, and stained with a 0.5% crystal violet for 15 minutes. The number of colonies was counted using a microscope.
Wound healing assay and invasion assay
HCC cell migration was assessed by the wound healing assay. HCC cells were cultured in a tight cell monolayer in six-well plates, and wounded with 200 μL plastic pipette tips. The migrating distance of cells at the wound front was determined in photographs of microscopic fields at 200× magnification. HCC cell invasion was analyzed with Matrigel-coated transwell cell culture chambers (EMD Millipore, Billerica, MA, USA). BEL-7404 cells (5×104) and SMMC-7721 cells (3×104) were plated in the top chamber onto the Matrigel-coated membrane (24-well insert) and allowed to migrate toward serum-containing medium in the lower chamber. The cells were incubated for 24 hours, then fixed with 4% formaldehyde for 15 minutes and stained with 0.5% crystal violet for 15 minutes. The invaded cells were counted at 200× magnification from five different fields.
Tumorigenicity in nude mice
Xenograft tumors were generated by subcutaneous injection of BEL-7404 cells (5×106), including BEL-7404-shNC and BEL-7404-shCENPK cells, on the right back of 4–6-week-old male athymic nude mice. Tumors were measured with digital calipers at set intervals, and the tumor volume was calculated by the following formula: tumor volume = (length×width2/2). After 24 days, the mice were euthanized via CO2 inhalation according to animal care guidelines, and the tumors were harvested and weighed. Additionally, bioluminescence imaging was used to detect tumor growth. All experimental animals were approved by the Institutional Animal Care and Use Committee of Fourth Military Medical University (Shaanxi, China) in accordance with the principles and procedures outlined in the National Institutes of Health guidelines for the care and use of laboratory animals.
Microarray analysis
The mRNA microarray experiments were performed at Shanghai Genechem Corporation (Shanghai, China). Total RNA was isolated from HCC cell line BEL-7404 transfected with negative control or shCENPK lentiviral plasmids. Total RNA was checked for a RNA integrity number to inspect RNA integrity by an Agilent Bioanalyzer 2,100. Qualified total RNA was further purified by RNeasy micro kit and RNase-Free DNase Set. Biotinylated cRNA was prepared according to the standard Affymetrix protocol from 12 μg of total RNA (Affymetrix, Inc., Santa Clara, CA, USA). Following fragmentation, 10 μg of cRNA was hybridized for 16 hours at 45°C on GeneChip Primeview human Array. GeneChips were washed and stained in the Affymetrix Fluidics Station 450 and scanned using the GeneArray Scanner 3000 7G. Then, the data were analyzed with RMA using Affymetrix default analysis settings and global scaling as normalization method and more details were shown in GSE114983. To assess the molecular signaling pathway change after knockdown of CENPK, data analysis, including canonical pathway and gene network interaction, was performed by the Ingenuity Pathway Analysis (IPA).
Statistical analysis
The data are expressed as the mean ± SD. Two-tailed Student’s t-tests were employed to analyze the differences using SPSS 22.0 software (Chicago, IL, USA). P<0.05 was considered statistically significant.
Ethical approval
The study protocol was approved by the Ethics Committee of Xijing Hospital (Xi’an, Shaanxi, China), and complied with the Declaration of Helsinki. All procedures performed in this study were in accordance with the 1964 Helsinki Declaration and its later amendments.
Results
CENPK is upregulated in HCC tissues
To assess the underlying role of CENPK in HCC, we first determined the expression of CENPK in the Cancer Genome Atlas (TCGA) database, and compared to that in ANLTs; CENPK expression was significantly upregulated in HCC tissues (Figure 1A). Furthermore, the overexpression of CENPK was correlated with histological grade, T stage, and pathologic stage (Table 3). Kaplan–Meier survival analysis revealed that patients with high CENPK expression had poor overall survival in TCGA database (Figure 1B). We further confirmed the upregulation of CENPK in HCC tissues by qRT-PCR and Western blot analysis. Unsurprisingly, compared with those in ANLTs, the mRNA and protein expression levels of CENPK were evidently elevated in HCC tissues (Figure 1C–D). Taken together, these data demonstrated that CENPK mRNA and protein levels were higher in HCC tissues than in ANLTs, suggesting that CENPK may promote HCC progression.
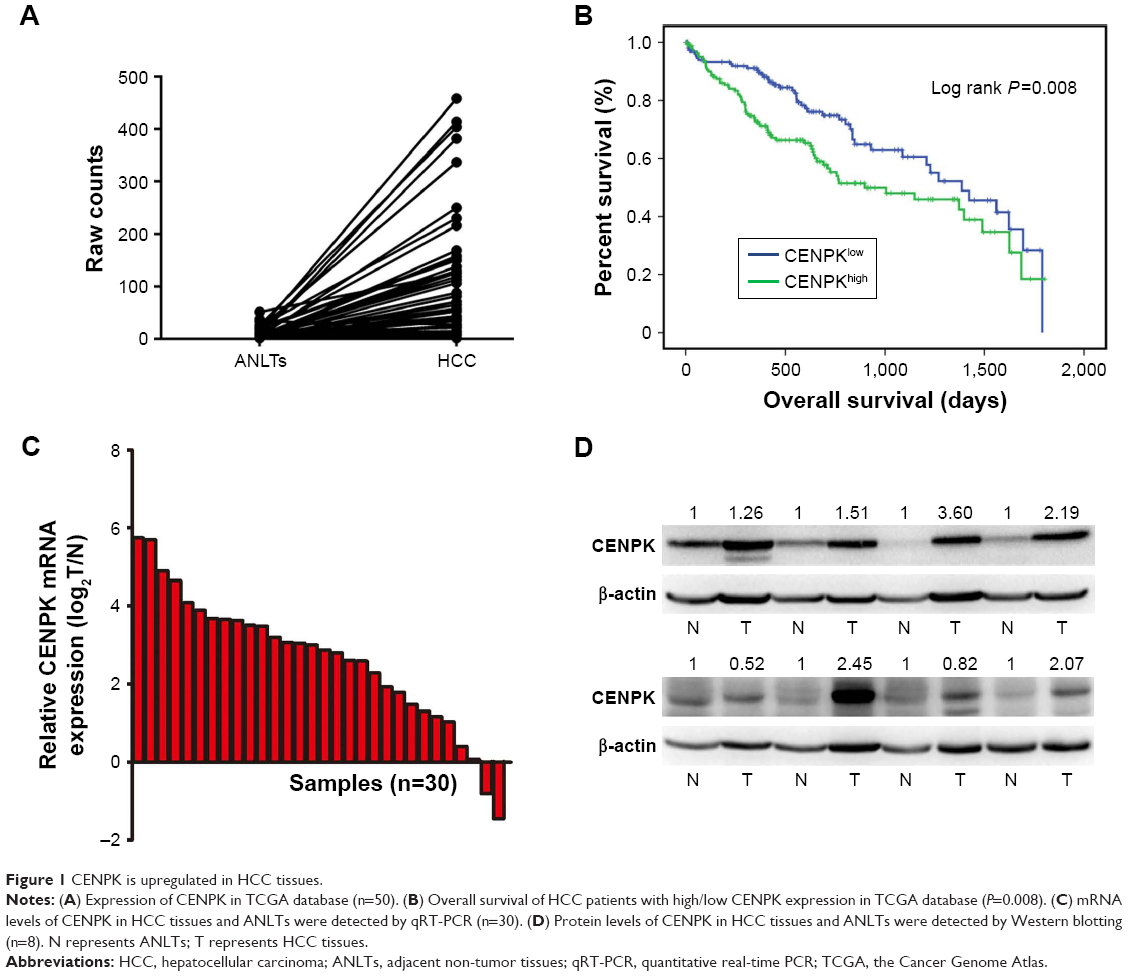 | Figure 1 CENPK is upregulated in HCC tissues. |
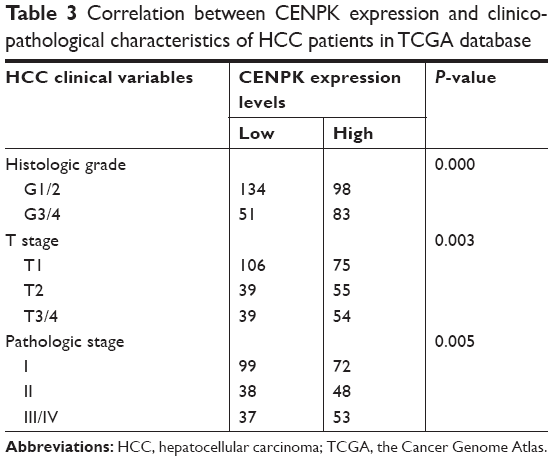 | Table 3 Correlation between CENPK expression and clinicopathological characteristics of HCC patients in TCGA database |
Knockdown of CENPK inhibits HCC cell growth, migration, and invasion in vitro
To investigate the biological function of CENPK in HCC tumorigenesis, we generated lentivirus-mediated shRNA to knockdown the endogenous expression of CENPK in BEL-7404 and SMMC-7721 cells. After transfection with shCENPK-encoding lentivirus, the expression of CENPK was successfully reduced in HCC cells (Figure 2A). We next investigated the effect of CENPK silencing on HCC cell proliferation by the CCK-8 and colony formation assays. Indeed, the downregulation of CENPK significantly inhibited cell viability and reduced the number of colonies formed by BEL-7404 and SMMC-7721 cells (Figure 2B–D). Furthermore, we performed wound healing and transwell assays to evaluate the effect of CENPK silencing on cell migration and invasion, respectively. Indeed, silencing CENPK dramatically decreased the migration and invasion of HCC cells (Figure 2E–G). Collectively, these data suggested that CENPK plays a negative role in HCC cell growth, migration, and invasion.
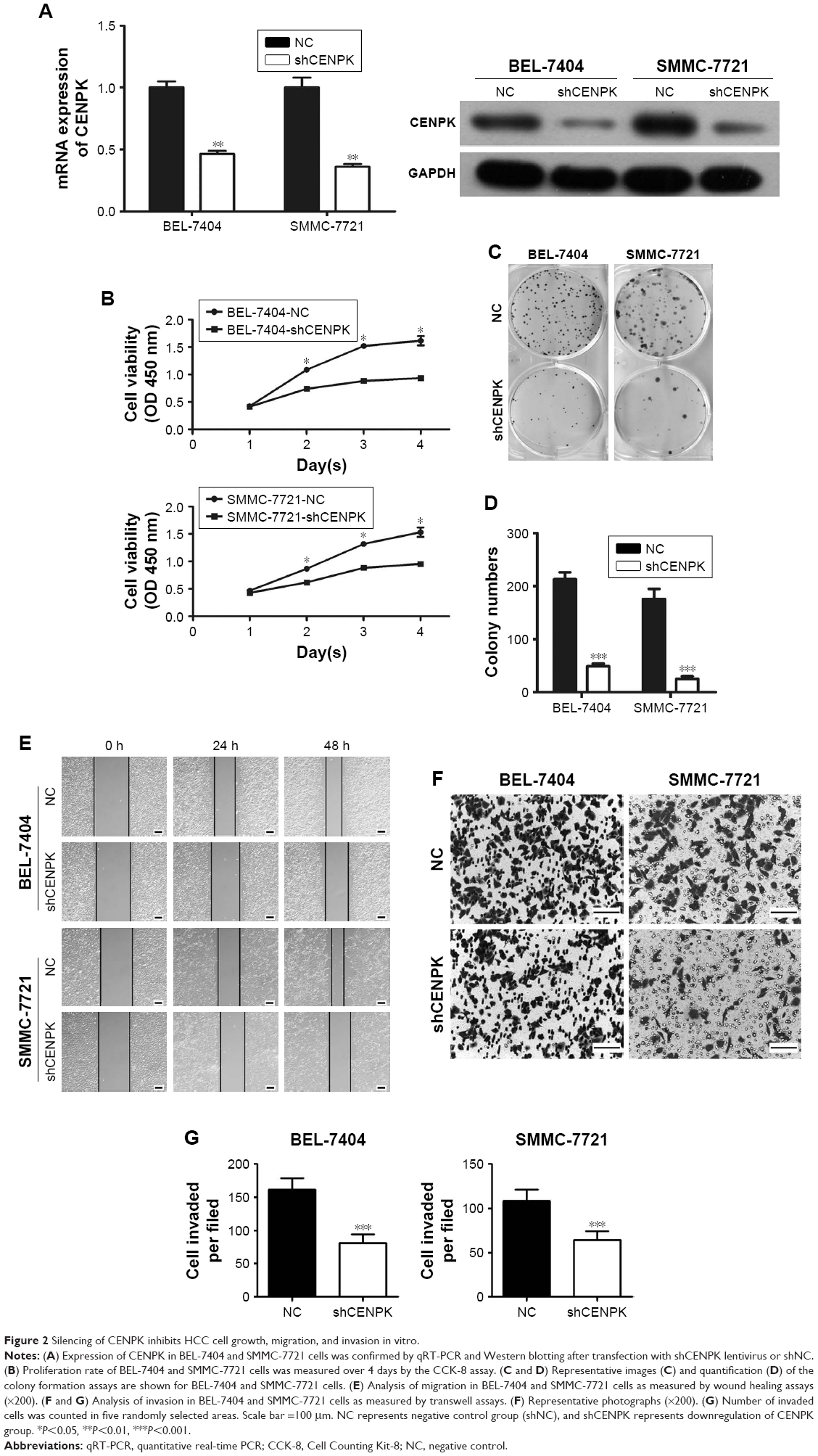 | Figure 2 Silencing of CENPK inhibits HCC cell growth, migration, and invasion in vitro. |
Knockdown of CENPK suppresses HCC tumor growth in vivo
We further detected the effect of CENPK knockdown on the growth of HCC cells by tumorigenicity assay in nude mice. BEL-7404 cells transfected with either negative control shRNA (shNC) or shCENPK were injected into nude mice, and tumor size was measured at set intervals. Twenty-four days later, the fluorescence intensity of green fluorescent protein was weaker in the BEL-7404-shCENPK group than in the BEL-7404-shNC group (Figure 3A–B). As shown in Figure 3C–E, the average tumor volume and weight were lower in the BEL-7404-shCENPK group than in the BEL-7404-shNC group. Throughout the observation period, the tumors formed by BEL-7404-shCENPK cells grew significantly slower than those formed by BEL-7404-shNC cells (Figure 3D). These results indicated that silencing of CENPK decreases the HCC tumor growth in vivo.
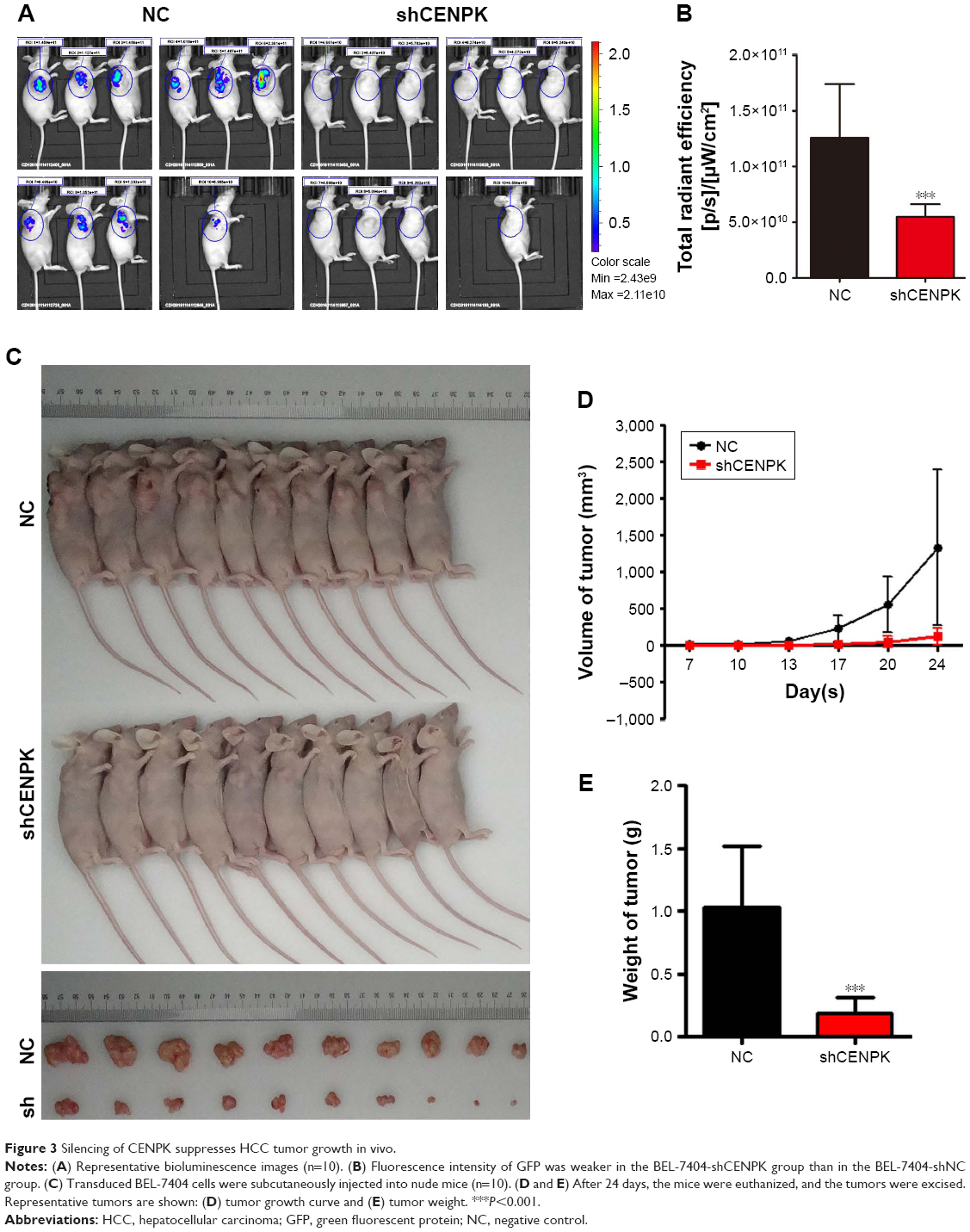 | Figure 3 Silencing of CENPK suppresses HCC tumor growth in vivo. |
Modulation of CENPK expression regulates Yap1 signaling pathway in HCC cells
To further elucidate the underlying mechanisms by which CENPK mediates carcinogenesis in HCC, we analyzed BEL-7404-shCENPK cells and BEL-7404-shNC cells by mRNA microarray. Indeed, silencing CENPK significantly decreased a series of genes involved in malignant solid tumour and cell proliferation and invasion, based on IPA (Figure 4A–B). In addition, silencing of CENPK remarkably repressed YAP1 expression and influenced some YAP1-related genes (Figure 4C). Next, we performed qRT-PCR and Western blot assays to validate the expression of YAP1 and some YAP1-related genes in HCC cells. Based on our data, the mRNA expression of YAP1, NUPR1, CTGF, and CYR61 was significantly downregulated in BEL-7404-shCENPK and SMMC-7721-shCENPK cells (Figure 5A). As shown in Figure 5B, silencing of CENPK decreased the protein expression of YAP1 and CYR61 in HCC cells. Immunofluorescence results also suggested that YAP1 expression was downregulated in CENPK knockdown cells (Figure 5C). These findings indicated that CENPK can regulate YAP1 expression in HCC.
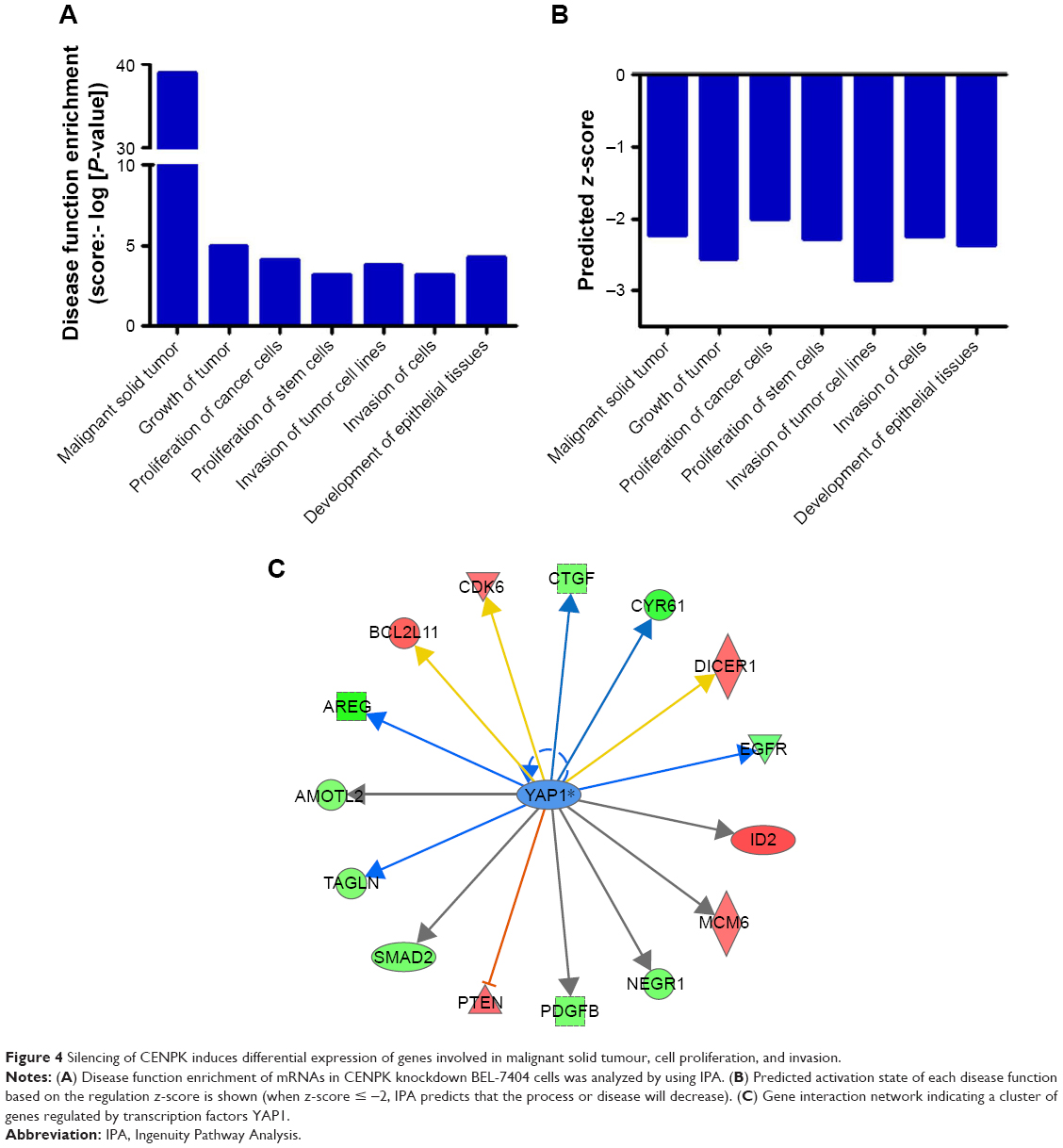 | Figure 4 Silencing of CENPK induces differential expression of genes involved in malignant solid tumour, cell proliferation, and invasion. |
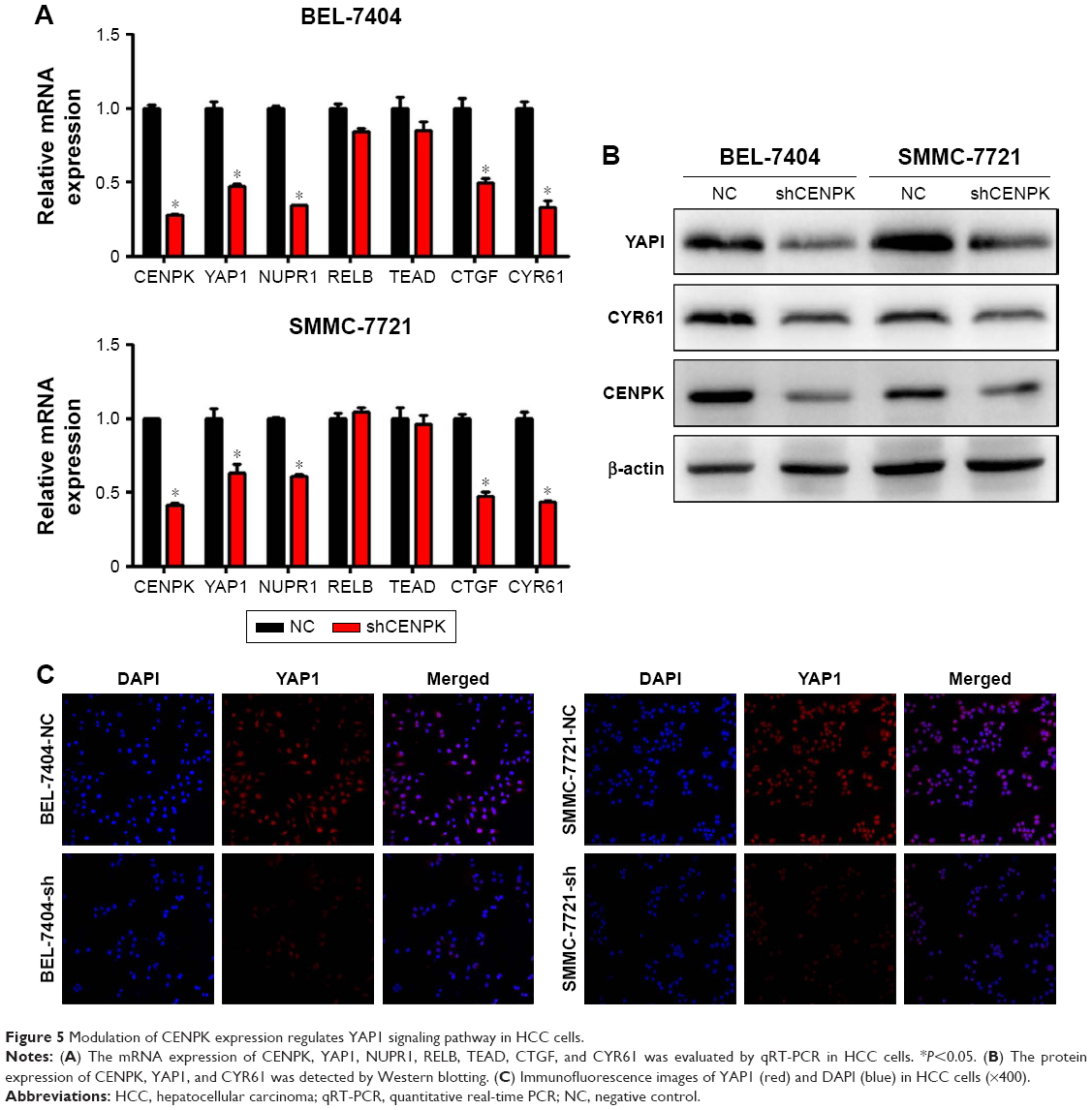 | Figure 5 Modulation of CENPK expression regulates YAP1 signaling pathway in HCC cells. |
Knockdown of CENPK suppresses the proliferation, migration, and invasion of HCC cells by targeting Yap1
Firstly, we evaluated the effect of YAP1 on HCC cell malignant phenotype. Western blot assays showed that the expression of YAP1 and CYR61 was significantly decreased in BEL-7404 and SMMC-7721 cells transfected with lentivirus-mediated shRNA against YAP1 (Figure 6A). Then, the results of CCK-8 and colony formation assays suggested that silencing YAP1 inhibited proliferation in HCC cells (Figure 6B–D). Moreover, migration and invasion were reduced in YAP1 knockdown cells (Figure 6E–G). These data suggested that the inhibitory effects of CENPK knockdown can be incompletely imitated by silencing YAP1 in HCC cells.
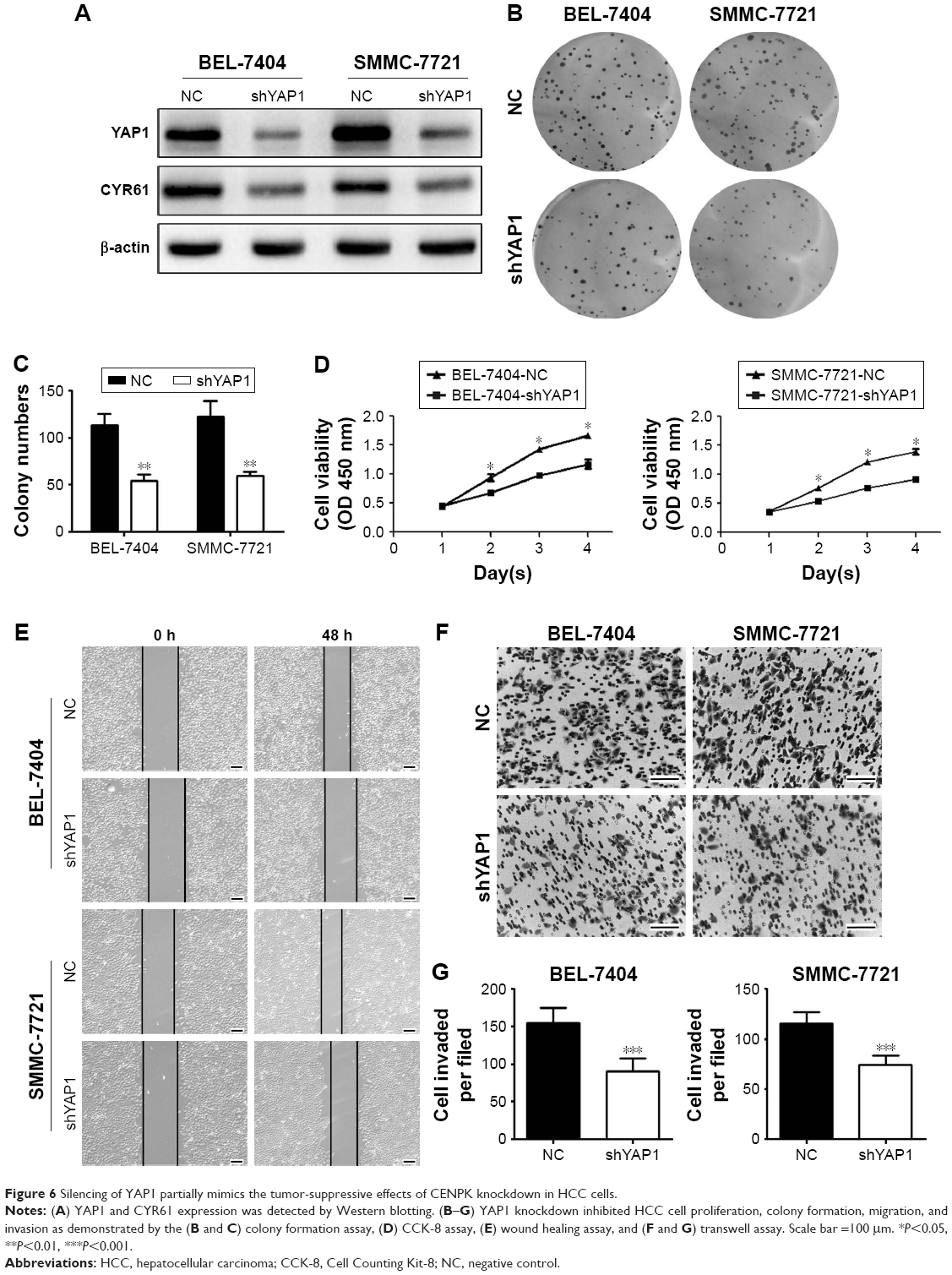 | Figure 6 Silencing of YAP1 partially mimics the tumor-suppressive effects of CENPK knockdown in HCC cells. |
Next, we performed gain- and loss-of-function experiments to investigate whether restoration of YAP1 expression can rescue the biological functions of CENPK knockdown cells. After transfection with full-length YAP1 plasmid or negative control vector, the expression of YAP1 and CYR61 was successfully upregulated in SMMC-7721 cells (Figure 7A). As shown in Figure 7B–E, the inhibitory effects of CENPK knockdown on cell proliferation, migration, and invasion were partially abolished in the YAP1 overexpression group. These results suggested that the inhibition of CENPK can inhibit HCC cell proliferation, migration, and invasion partially through targeting YAP1.
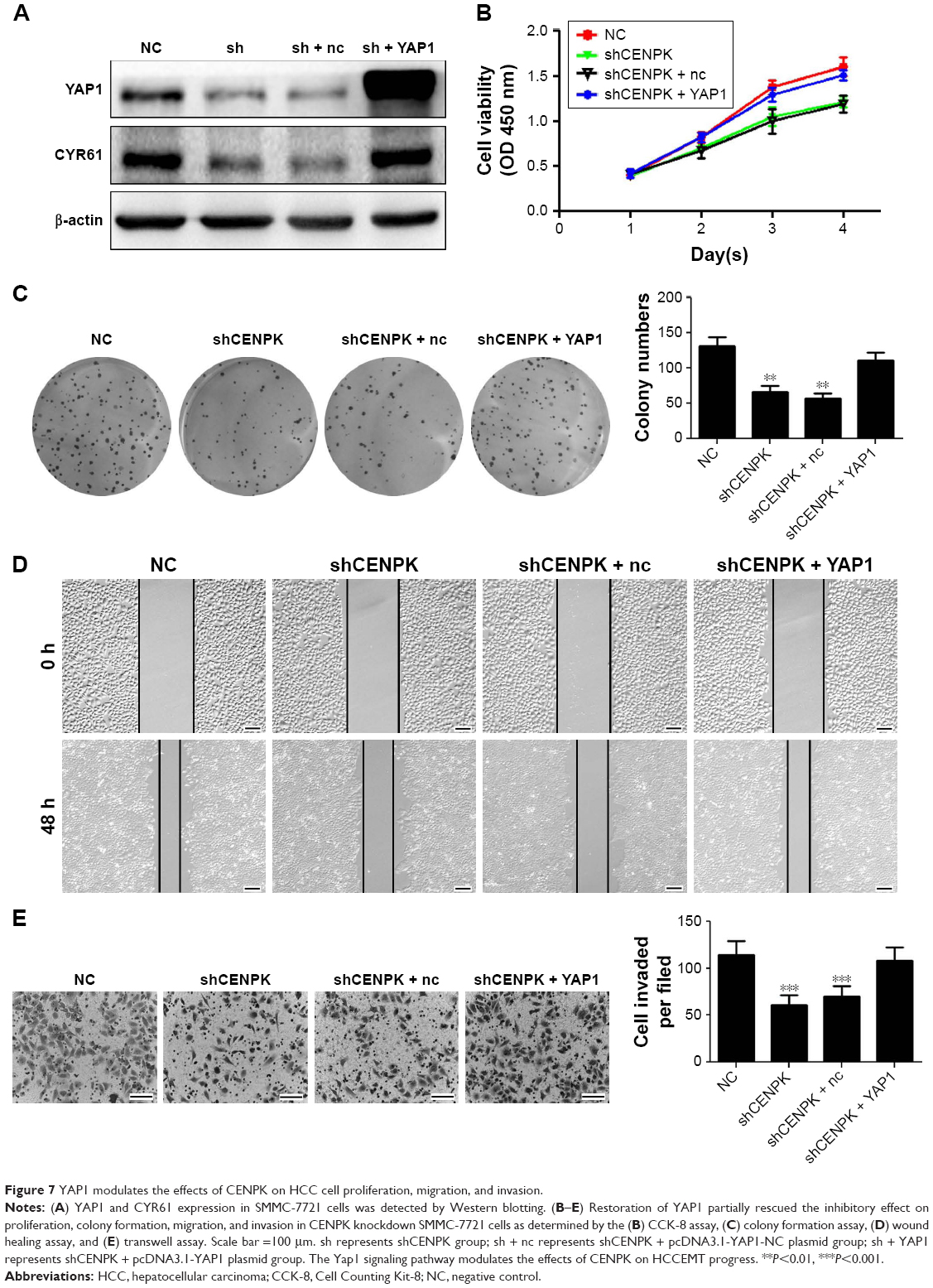 | Figure 7 YAP1 modulates the effects of CENPK on HCC cell proliferation, migration, and invasion. |
Emerging evidence suggests that EMT plays an important role in cancer cell invasion and metastasis,34,35 and YAP1 expression is involved in EMT progression in HCC cells,28,32 so we firstly detected the expression of EMT markers by using Western blot assays. As shown in Figure 8A, silencing of CENPK significantly enhanced the expression of E-cadherin and reduced the expression of N-cadherin in HCC cells. As expected, knockdown of YAP1 blocked EMT progression in HCC cells (Figure 8B). In addition, the EMT blocking effect of CENPK knockdown in HCC cells was partially counteracted by the overexpression of YAP1 (Figure 8C). These results suggested that YAP1 signaling pathway may be the key regulator between CENPK and EMT in HCC.
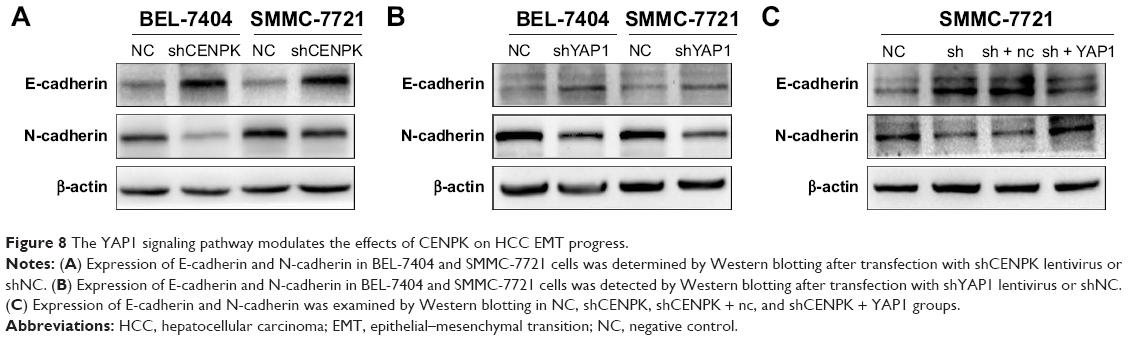 | Figure 8 The YAP1 signaling pathway modulates the effects of CENPK on HCC EMT progress. |
Discussion
HCC is the third leading cause of cancer-related death worldwide and remains a significant clinical challenge with few therapeutic approaches and poor survival.2,36 However, the underlying molecular mechanisms of HCC have not yet been fully clarified. Therefore, identifying potential molecular biomarkers and elucidating the underlying molecular mechanisms of HCC are extremely important. Recently, Wang et al reported that the mRNA expression of CENPK was elevated in HCC tissues and correlated with tumor size and alpha-fetoprotein expression.20 In this study, CENPK was significantly upregulated in HCC tissues, and its upregulation was associated with histological grade, T stage, pathologic stage, and poor overall survival in TCGA database. In addition, qRT-PCR and Western blot assays confirmed that CENPK expression was higher in HCC tissues and in ANLTs collected in our clinical department. Our data were partly consistent with the results of Wang et al.20 Thus, CENPK may act as a pro-tumor molecule in HCC. However, the relationship between CENPK expression and clinicopathologic characteristics of HCC patient needs further exploration.
The overexpression of CENPK in HCC tissues suggests a possible role for this molecule in hepatocarcinogenesis. Consistent with this hypothesis, depletion of CENPK in BEL-7404 and SMMC-7721 cells significantly inhibited cell proliferation, migration, and invasion in vitro. Furthermore, the results of tumorigenicity assay revealed that silencing of CENPK dramatically suppressed tumor growth in vivo. Wang et al demonstrated that the overexpression of CENPK can promote HCC cell proliferation, migration, and invasion by activating the AKT signal pathway.20 However, the mechanism underlying the role of CENPK is still largely unclear. To further evaluate the mechanism by which silencing of CENPK inhibited HCC cell proliferation, migration, and invasion, we performed mRNA microarray analysis in CENPK knockdown cells. According to IPA, many genes involved in malignant solid tumour and cell proliferation and invasion were regulated by CENPK. Moreover, the expression of some downstream genes of YAP1 could be regulated by silencing CENPK. Furthermore, qRT-PCR assay validated that the mRNA expression of YAP1 was downregulated in CENPK knockdown cells. NUPR1,37 CTGF,38 and CYR6139 are downstream of YAP1, and the alterations of these mRNAs were the same as those in YAP1. The protein expression of YAP1 and CYR61 was decreased in CENPK knockdown cells. These data suggest that CEPNK can modulate YAP1 expression at both mRNA and protein levels.
YAP1 is a key component of the Hippo signaling pathway, and has been shown to be essential for the initiation, progression, and metastasis of many types of cancer, including HCC.24,40 Alterations in YAP1 expression can also drive carcinogenesis in animal models.41 Recently, verteporfin, a small molecule inhibitor, has been shown to attenuate YAP activity in murine models,42 and Phase I/II clinical trials in humans have reported that verteporfin therapy is safe and feasible with minimal phototoxicity in patients with locally advanced pancreatic cancer.43 Targeting YAP1 may be a useful method for the treatment of cancer, including HCC.44,45 In this study, the depletion of CENPK significantly decreased the expression of YAP1 and reduced proliferation, migration, and invasion in HCC cells. In addition, restoring the expression of YAP1 partially reversed the inhibitory effects of CENPK knockdown in HCC cells. Taken together, these data suggest that knockdown of CENPK inhibits HCC cell proliferation, migration, and invasion by regulating YAP1.
Emerging studies demonstrate that YAP1 participates in regulating EMT progression in many human cancers, such as pancreatic cancer,46 malignant glioma,47 colorectal cancer,30 and HCC.28 Wang et al reported that silencing of YAP1 markedly prevented EMT progression in HCC cells.28 In this study, the depletion of CENPK or YAP1 markedly upregulated the expression of the epithelial marker E-cadherin and downregulated the expression of the mesenchymal marker N-cadherin in HCC cells, while the blocking effect on EMT was incompletely abolished by the overexpression of YAP1 in CENPK knockdown cells. Taken together, these data suggest that YAP1 is responsible for CENPK-mediated EMT progression in HCC cells. However, the detailed mechanisms underlying CENPK regulating YAP1 need further study.
Conclusion
The present study demonstrates that CENPK is frequently upregulated in HCC and is associated with an aggressive tumor phenotype. We demonstrated that the knockdown of CENPK inhibited proliferation, migration, invasion, and EMT in HCC cells by suppressing YAP1. Therefore, targeting the CENPK–YAP1–EMT axis may be a novel therapeutic approach for HCC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 81672339) and the National High-Tech Research and Development Program of China (grant no. 2016YFC0905902).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. | ||
Wahid B, Ali A, Rafique S, Idrees M. New insights into the epigenetics of hepatocellular carcinoma. Biomed Res Int. 2017;2017(8):1–16. | ||
Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res. 2011;727(3):55–61. | ||
Villanueva A, Hoshida Y, Battiston C, et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology. 2011;140(5):e1502:1501–1512. | ||
Li T, Fan J, Qin LX, et al. Risk factors, prognosis, and management of early and late intrahepatic recurrence after resection of primary clear cell carcinoma of the liver. Ann Surg Oncol. 2011;18(7):1955–1963. | ||
Shao YY, Shau WY, Chan SY, Lu LC, Hsu CH, Cheng AL. Treatment efficacy differences of sorafenib for advanced hepatocellular carcinoma: a meta-analysis of randomized clinical trials. Oncology. 2015;88(6):345–352. | ||
Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology. 2002;35(3):519–524. | ||
Scully R. The spindle-assembly checkpoint, aneuploidy, and gastrointestinal cancer. N Engl J Med. 2010;363(27):2665–2666. | ||
Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112(4):407–421. | ||
Li Y, Zhu Z, Zhang S, et al. ShRNA-targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS One. 2011;6(3):e17794. | ||
Tomonaga T, Matsushita K, Yamaguchi S, et al. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003;63(13):3511–3516. | ||
Wu Q, Qian YM, Zhao XL, et al. Expression and prognostic significance of centromere protein A in human lung adenocarcinoma. Lung Cancer. 2012;77(2):407–414. | ||
Mcgovern SL, Qi Y, Pusztai L, Symmans WF, Buchholz TA. Centromere protein-A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor-positive breast cancer. Breast Cancer Res. 2012;14(3):R72. | ||
Lu G, Hou H, Lu X, et al. CENP-H regulates the cell growth of human hepatocellular carcinoma cells through the mitochondrial apoptotic pathway. Oncol Rep. 2017;37(6):3484–3492. | ||
Dai Y, Liu L, Zeng T, et al. Characterization of the oncogenic function of centromere protein F in hepatocellular carcinoma. Biochem Biophys Res Commun. 2013;436(4):711–718. | ||
Cheeseman IM, Hori T, Fukagawa T, Desai A. KNL1 and the CENP-H/I/K complex coordinately direct kinetochore assembly in vertebrates. Mol Biol Cell. 2008;19(2):587–594. | ||
Lee YC, Huang CC, Lin DY, Chang WC, Lee KH. Overexpression of centromere protein K (CENPK) in ovarian cancer is correlated with poor patient survival and associated with predictive and prognostic relevance. Peer J. 2015;3:e1386. | ||
Komatsu M, Yoshimaru T, Matsuo T, et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int J Oncol. 2013;42(2):478–506. | ||
Wang H, Liu W, Liu L, et al. Overexpression of centromere protein K (CENP-K) gene in hepatocellular carcinoma promote cell proliferation by activating AKT/TP53 signal pathway. Oncotarget. 2017;8(43):73994–74005. | ||
Pan D. The Hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. | ||
Mauviel A, Nallet-Staub F, Varelas X. Integrating developmental signals: a Hippo in the (path)way. Oncogene. 2012;31(14):1743–1756. | ||
Zhao B, Ye X, Yu J, et al. TeaD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. | ||
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783–803. | ||
Xu MZ, Yao TJ, Lee NP, et al. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115(19):4576–4585. | ||
Wang C, Zhu ZM, Liu CL, He XJ, Zhang HY, Dong JH. Knockdown of yes-associated protein inhibits proliferation and downregulates large tumor suppressor 1 expression in MHCC97H human hepatocellular carcinoma cells. Mol Med Rep. 2015;11(6):4101–4108. | ||
Liu AM, Xu MZ, Chen J, Poon RT, Luk JM. Targeting YAP and Hippo signaling pathway in liver cancer. Expert Opin Ther Targets. 2010;14(8):855–868. | ||
Wang S, Li H, Wang G, et al. Yes-associated protein (YAP) expression is involved in epithelial-mesenchymal transition in hepatocellular carcinoma. Clin Transl Oncol. 2016;18(2):172–177. | ||
Shao DD, Xue W, Krall EB, et al. KRAS and Yap1 converge to regulate EMT and tumor survival. Cell. 2014;158(1):171–184. | ||
Ling HH, Kuo CC, Lin BX, Huang YH, Lin CW. Elevation of Yap promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp Cell Res. 2017;350(1):218–225. | ||
Lehmann W, Mossmann D, Kleemann J, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. 2016;7:10498. | ||
Yu S, Jing L, Yin XR, et al. MiR-195 suppresses the metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by inhibiting YAP. Oncotarget. 2017;8(59):99757–99771. | ||
Wang J, Song W, Shen W, et al. MicroRNA-200a suppresses cell invasion and migration by directly targeting GAB1 in hepatocellular carcinoma. Oncol Res. 2017;25(1):1–10. | ||
Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798–808. | ||
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. | ||
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | ||
Jia Q, Zhou W, Yao W, et al. Downregulation of YAP-dependent NUPR1 promotes tumor-repopulating cell growth in soft matrices. Oncogenesis. 2016;5:e220. | ||
Kang W, Huang T, Zhou Y, et al. miR-375 is involved in Hippo pathway by targeting YAP1/TEAD4-CTGF axis in gastric carcinogenesis. Cell Death Dis. 2018;9(2):92. | ||
Zhang Y, Xia H, Ge X, et al. CD44 acts through RhoA to regulate YAP signaling. Cell Signal. 2014;26(11):2504–2513. | ||
Patel SH, Camargo FD, Yimlamai D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology. 2017;152(3):533–545. | ||
Valero V, Pawlik TM, Anders RA. Emerging role of Hpo signaling and YAP in hepatocellular carcinoma. J Hepatocell Carcinoma. 2015;2:69–78. | ||
Stanger BZ. Quit your YAPing: a new target for cancer therapy. Genes Dev. 2012;26(12):1263–1267. | ||
Huggett MT, Jermyn M, Gillams A, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110(7):1698–1704. | ||
Shan L, Jiang H, Ma L, Yu Y. Yes-associated protein: a novel molecular target for the diagnosis, treatment and prognosis of hepatocellular carcinoma. Oncol Lett. 2017;14(3):3291–3296. | ||
Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. 2016;29:26–33. | ||
Yuan Y, Li D, Li H, Wang L, Tian G, Dong Y. YAP overexpression promotes the epithelial-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol Med Rep. 2016;13(1):237–242. | ||
Lu J, Yang Y, Guo G, et al. IKBKE regulates cell proliferation and epithelial-mesenchymal transition of human malignant glioma via the Hippo pathway. Oncotarget. 2017;8(30):49502–49514. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.