")
Back to Journals » International Journal of Nanomedicine » Volume 11
Downregulation of B-cell lymphoma/leukemia-2 by overexpressed microRNA 34a enhanced titanium dioxide nanoparticle-induced autophagy in BEAS-2B cells
Authors Bai W, Chen Y, Sun P, Gao A
Received 5 November 2015
Accepted for publication 7 March 2016
Published 6 May 2016 Volume 2016:11 Pages 1959—1971
DOI https://doi.org/10.2147/IJN.S99945
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Wenlin Bai,1,2,* Yujiao Chen,1,2,* Pengling Sun,1,2 Ai Gao1,2
1Department of Occupational Health and Environmental Health, School of Public Health, 2Beijing Key Laboratory of Environmental Toxicology, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Titanium dioxide (TiO2) nanoparticles (TNPs) are manufactured worldwide for a wide range of applications and the toxic effect of TNPs on biological systems is gaining attention. Autophagy is recognized as an emerging toxicity mechanism triggered by nanomaterials. MicroRNA 34a (miR34a) acts as a tumor suppressor gene by targeting many oncogenes, but how it affects autophagy induced by TNPs is not completely understood. Here, we observed the activation of TNP-induced autophagy through monodansylcadaverine staining and LC3-I/LC3-II conversion. Meanwhile, the transmission electron microscope ultrastructural analysis showed typical morphological characteristics in autophagy process. We detected the expression of miR34a and B-cell lymphoma/leukemia-2 (Bcl-2). In addition, the underlying mechanism of TNP-induced autophagy was performed using overexpression of miR34a by lentivirus vector transfection. Results showed that TNPs induced autophagy generation evidently. Typical morphological changes in the process of autophagy were observed by the transmission electron microscope ultrastructural analysis and LC3-I/LC3-II conversion increased significantly in TNP-treated cells. Meanwhile, TNPs induced the downregulation of miR34a and increased the expression of Bcl-2. Furthermore, overexpressed miR34a decreased the expression of Bcl-2 both in messenger RNA and protein level, following which the level of autophagy and cell death rate increased after the transfected cells were incubated with TNPs for 24 hours. These findings provide the first evidence that overexpressed miR34a enhanced TNP-induced autophagy and cell death through targeted downregulation of Bcl-2 in BEAS-2B cells.
Keywords: titanium dioxide nanoparticles, autophagy, miR34a, Bcl-2, lentivirus, cell death, cytotoxicity
Introduction
Traditionally, nanomaterials have been considered as objects with at least one of their three dimensions in the range of 1–100 nm, which possess dramatically different physicochemical properties compared to fine particles of the same composition. Titanium dioxide (TiO2) nanoparticles (TNPs), a kind of nanomaterials, are extensively used in domestic and cosmetic products, medical devices, additives in pharmaceuticals, food colorants, and sunscreen owing to their typical characteristics of surface adsorption, photocatalysis, and ultraviolet absorption.1–5 Among the three well known crystallographic structures (anatase, rutile, and brookite) of titanium dioxide, only anatase and rutile are applied commercially and commonly.6 Meanwhile, TiO2 anatase has been suggested to exert a greater toxic effect than TiO2 rutile.7,8 It is possible for industrial or commercial TNPs to spread into the air of indoor or outdoor atmosphere during the process of production, use, distribution, and recycle.9 Thus, there exists a considerable risk for nano-TiO2 to potentially enter into human bodies via many routes, such as inhalation (respiratory tract), ingestion (gastrointestinal tract), dermal penetration (skin), and injection (blood circulation).10 As the most common route of TNPs to enter human body, the respiratory tract has become the primary target organ system. Regarding the toxicity of TNPs, lungs seem to be the main target organ for toxicity studies.3 The number of studies on pulmonary toxicity also outweighs studies of other exposure routes, emphasizing its importance especially with reference to environmental and occupational exposures.11 TiO2 has been classified as a Group 2B carcinogen, which is possibly carcinogenic to humans, by the International Agency for Research on Cancer after lung tumors developed in rats exposed to high concentrations of TiO2 for 2 years.12,13 The published studies showed that TNPs induced oxidative DNA damage, lipid peroxidation, and micronuclei formation and increased hydrogen peroxide and nitric oxide production.7–8,14 Yet, the underlying mechanisms of TNPs toxicity have not been clarified. Recently, autophagy was considered as an emerging toxicity mechanism occurring in several nanomaterials, such as nanosized fullerene, rare earth oxides, copper oxide, and silica.15–18
Autophagy, also called macroautophagy, is a highly regulated intracellular self-catabolic degradation process for the lysosomal degradation and recycling of organelles as well as unfolded and aggregated proteins so as to maintain cellular homeostasis. During autophagy, parts of the cytoplasm are sequestered into characteristic double-membrane vesicles, autophagosomes, which subsequently fuse with late endosomes or lysosomes, forming the autolysosome.19,20 Autophagy, therefore, serves as a natural and essential defense mechanism against inflammatory, infectious, neurodegenerative, and neoplastic disorders, and deregulation of this pathway has been implicated in the pathogenesis of numerous human diseases.21–24 Activation of autophagy was considered as an attempt for a cell to maintain cellular homeostasis by sequestering and degrading exogenous materials entered into the cytoplasm. However, the toxicological consequence of autophagy dysfunction is regarded as a potential mechanism of cell death.18 A few studies showed the biological response of the TNPs associated with autophagy;24–26 however, they provided limited evidence to consider the underlying mechanisms.
Autophagy is regulated by a complex network that consists of different signaling pathways and autophagy-related genes.27 Dysregulation of autophagy has been implicated in numerous human diseases.23,28 A recent study has confirmed that a lot of microRNAs (miRNAs) are involved in the regulation of autophagic process.29 miRNAs are a class of endogenously expressed, ~22-nucleotide noncoding RNAs that affect many cellular processes30 and human diseases, including cancer,31,32 by posttranscriptionally regulating gene expression.33 They guide the binding of the RNA-induced silencing complex to partial complementarity regions within 3′ untranslated regions of target messenger RNA (mRNA) molecules, resulting in mRNA degradation and/or translational inhibition.34 MicroRNA 34a (miR34a), a tumor suppressor, belongs to one of several evolutionarily conserved families of miR34 and was originally identified as a TP53-targeted miRNA.35 miR34a was reported in the deregulation of multiple types of cancer through the suppression of many oncogene targets related to autophagy, proliferation, apoptosis, and invasion, such as B-cell lymphoma/leukemia-2 (Bcl-2) and cyclinD1.35–38 Furthermore, it has been reported that miR-34 decreased the expression of Bcl-2, leading to an increase in apoptosis.39 As one of the key regulators of apoptosis and autophagy, Bcl-2 protein possesses four conserved Bcl-2 homology domains (BH1–4) and can inhibit Beclin1-dependent autophagy by binding to Beclin1 under starvation conditions.40 Yet, it is not clear whether miR34a is capable of strengthening TNP-related autophagy through targeted regulating expression of Bcl-2.
In this study, we examined the impact of miR34a on Bcl-2 mRNA and protein expression and its molecular actions in the regulation of TNP-induced autophagy in vitro with human bronchial epithelial normal BEAS-2B cells. Prior to conducting toxicity experiments, we detected the characterization of TNPs, which is essential for nanotoxicity studies, by transmission electron microscope (TEM) and dynamic light scattering measurements. To investigate the autophagy triggered by TNPs, we performed a sequence of assessments, including ultrastructural analysis, monodansylcadaverine (MDC) staining, and LC3-I/LC3-II conversion after BEAS-2B cell exposure to TNPs for 24 hours. We detected the expression of miR34a and Bcl-2 with or without overexpression of miR34a to analyze the underlying mechanisms of the TNP-induced autophagy.
Materials and methods
Characterization of the TNPs
Commercial grade anatase TNPs were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) with a primary particle size of less than 25 nm. After examining the particle size and distribution of the TNPs by TEM (JEOL, Tokyo, Japan) and ImageJ software, we measured the hydrodynamic sizes and zeta potential of TNPs (25 μg/mL) in ultrapure water as stock media and serum-free Dulbecco’s Modified Eagle’s Medium (DMEM) as culture media by Zetasizer (Malvern Nano-ZS90, Malvern Instruments, Malvern, UK). In addition, the TNP suspensions were dispersed using a sonicator for 10 minutes (300 W) (Bioruptor UDC-200; Diagenode s.a., Seraing, Belgium).
Cell culture and the TNP exposure
BEAS-2B cells were purchased from Cell Resource Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, People’s Republic of China). The cells were cultured at 37°C in a humidified incubator with 5% CO2, and maintained in DMEM (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum (MP Biomedicals, Santa Ana, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. For tests, 3×105 cells were seeded into 6-well plates to attach for 24 hours. After being washed with phosphate-buffered saline (PBS) twice, the cells were exposed to suspensions of TNPs in DMEM (6.25, 12.5, and 25 μg/mL) for 24 hours. The equivalent volume of DMEM without TNPs was used as a control group.
Cellular internalization of the TNPs and ultrastructural analysis
BEAS-2B cells were cultured in a cell culture dish (100 mm in diameter) in DMEM. After attachment for 24 hours, the cells were incubated with TNPs dispersed in DMEM (25 μg/mL) for another 24 hours. Then, we employed TEM to detect whether generation of autophagy was triggered by TNPs. After being washed twice with PBS and trypsinized, the BEAS-2B cells were then centrifuged for 10 minutes at 1,500 r/minutes. Then, the cell pellets were washed by 0.1 M phosphate buffer for three times and postfixed for 3 hours with 1% osmic acid. After that, we performed a series of processes (50%, 70%, 80%, 90%, and 100% alcohol, and 100% acetone) to dehydrate for the cell samples. After being embedded in epoxy resin and stained by lead citrate and uranyl acetate, the cell samples were imaged using a TEM (JEM2100; JEOL, Tokyo, Japan).
MTT assay
The cytotoxicity induced by TNPs was detected by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. About 5×103 BEAS-2B cells with or without infection were seeded into a 96-well plate for attachment, then the cells were incubated with different concentrations of TNPs (6.25, 12.5, and 25 μg/mL) at 37°C for 24 hours. Then, 10 μL MTT was added at 5 mg/mL for another 4-hour incubation. Then, 150 μL dimethylsulfoxide was added to each well and mixed thoroughly for 10 minutes. A microplate reader (Thermo Multiscan MK3; Thermo Fisher Scientific, Waltham, MA, USA) was used to measure the optical density at 492 nm.
Autophagy measurement
MDC, which is usually used to stain autophagosomes, is a specific marker of autophagic vacuoles. In this study, MDC was employed to detect the generation and level of autophagy in BEAS-2B cells exposed to the different concentrations of TNPs. Briefly, ~3×105 cells were seeded into a 6-well plate for attachment for 24 hours, then cells incubated with TNPs (6.25, 12.5, and 25 μg/mL) for 24 hours were stained with 0.05 mM MDC (Sigma-Aldrich Co.) at 37°C for 30 minutes in dark. The observation of MDC-stained autophagic vacuoles was conducted by a laser scanning confocal microscopy (Leica TCS SP5, Leica Microsystems, Wetzlar, Germany).
Immunoblot assay
After different treatments of the cells and extraction of lysate proteins, the protein concentration was measured using the bicinchoninic acid protein assay (Thermo Fisher Scientific). The equal amounts of proteins (25 μg) were loaded onto 15% SDS-PAGE and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, Billerica, MA, USA). After membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.05% Tween-20 for 1 hour, immunoblotting was conducted by incubating with LC3 and β-actin rabbit antibodies (1:1,000) (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C. The membranes were then washed with tris-buffered saline containing 0.05% Tween-20 and incubated with a horseradish peroxidase-conjugated antirabbit IgG secondary antibody (Cell Signaling Technology) for 1 hour at room temperature. After being washed three times with tris-buffered saline containing 0.05% Tween-20, the proteins bound with the antibody were detected using the ECL chemiluminescence reagent (Thermo Fisher Scientific). We used the Image Lab™ Software (Bio-Rad Laboratories Inc., Hercules, CA, USA) to perform the densitometric analysis of the Western blot results.
Lentivirus infection
Lentivirus vectors were constructed in Genechem (Shanghai, People’s Republic of China). These lentivirus vectors contain a target gene (miR34a) or empty lentiviral vectors and a green fluorescent protein marker. To generate stable clones, 1×105 BEAS-2B cells were seeded into a 12-well plate for 24 hours. Afterwards, 5×106 transducing units of lentivirus with 500 μL of enhanced infection solution and polybrene (5 μg/mL) were mixed to infect BEAS-2B cells at 70% confluence according to the manufacturer’s instructions (Genechem).
RNA isolation and quantificational real-time polymerase chain reaction
Total RNA was extracted with TRIzol Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Reverse transcribed and mature miRNAs were quantified using Hairpin-it™ miRNA RT-PCR Quantitation Kit (GenePharma Co., Ltd., Shanghai, People’s Republic of China). miRNA expression level was normalized using U6 snRNA as an internal control. Quantificational real-time polymerase chain reaction for Bcl-2 with specific primers from Sangon Biotech (Shanghai, People’s Republic of China) was performed using RevertAid First Strand Kit (Thermo Fisher Scientific) and Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific) according to the manufacturer’s protocol. β-Actin was used as an endogenous control to normalize mRNA levels. The sequences of primers were as follows: Bcl-2: forward: 5′GGATTGTGGCCTTCTTTGAG3′; reverse: 5′TACCCAGCCTCCGTTATCCT3′; β-actin: forward: 5′TGAGACCTTCAACACCCCAG3′; reverse: 5′GCCATCTCTTGCTCGAAGTC3′. Real-time reverse transcription-polymerase chain reaction was performed on Bio-Rad (CFX96™ optics module). Each sample was replicated three times. Data were analyzed by comparing cycle threshold values.
Statistical analysis
Results were expressed as mean ± standard deviation of three independent experiments. Least significant difference test was used to compare the mean of two samples while one way analysis of variance was used for group mean. In all cases, P<0.05 was considered to be statistically significant. This study was approved by the Ethical Committee of Capital Medical University (Beijing, People’s Republic of China).
Results
Characterization of the TNPs
In the current study, we first characterized the commercially purchased TNPs by TEM. A near-spherical particle shape is shown in Figure 1A, and the mean pristine particle diameter of 22.07±8.93 nm was obtained after we measured 500 random particles using ImageJ (Figure 1B). Hydrodynamic measurements of both TNPs (25 μg/mL) in ultrapure water and serum-free DMEM at different time points by dynamic light scattering are shown in Table 1. TNPs in DMEM exhibited a larger hydrodynamic size than in ultrapure water, which suggested that TNPs formed aggregates that were not dispersed thoroughly by ultrasonication in both dispersion mediums due to the Van der Waals force and hydrophobic interaction with surrounding media. This is consistent with reports of poor dispersion in other serum-free media,41 presumably due to decreased protein corona levels that act as surfactants to reduce particle aggregation. Zeta potentials can provide quantitative information on the stability of the particles, which confirm that the particles are more likely to remain dispersed if absolute values of zeta potentials are higher than 30 mV.42 As shown in Table 1, zeta potential values in ultrapure water and DMEM varied between -27.5 and -18.3 mV, which represented the actual condition that the BEAS-2B cells were exposed in media.
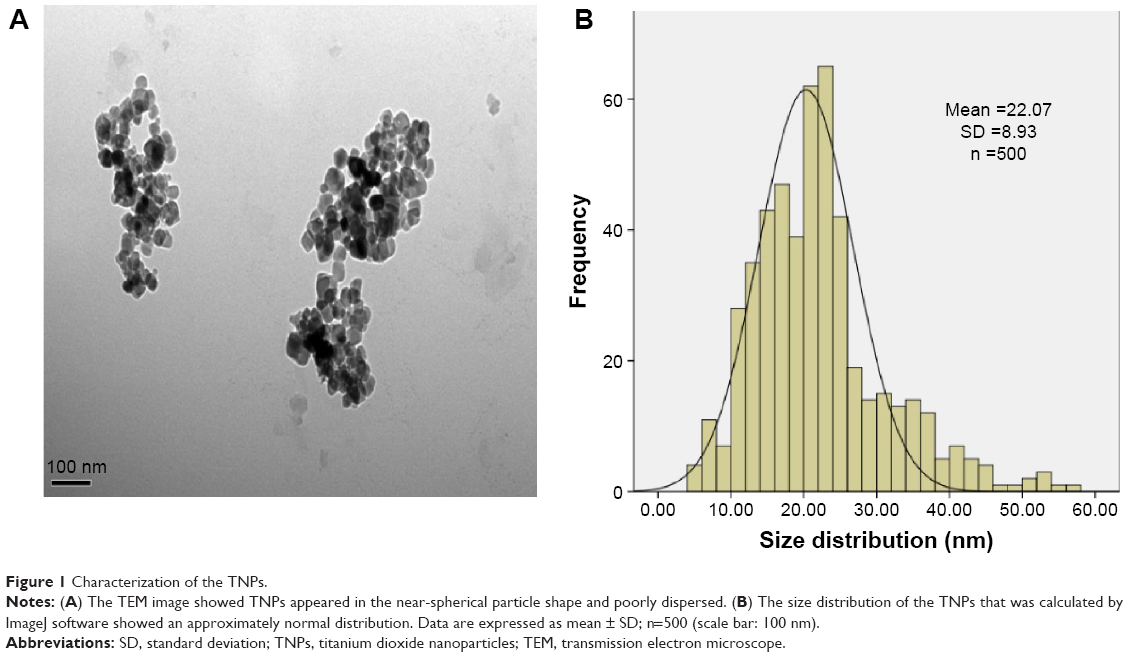 | Figure 1 Characterization of the TNPs. |
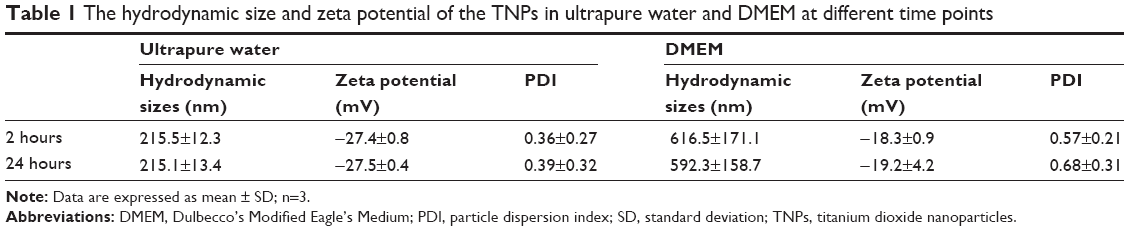 | Table 1 The hydrodynamic size and zeta potential of the TNPs in ultrapure water and DMEM at different time points |
The TNPs internalization
Taken the importance of TNPs intracellular localization in the toxicity evaluation, the cellular uptake of TNPs by BEAS-2B cells was examined by TEM. Compared with Figure 2A, a number of TNPs were dispersed as free or membrane-bound aggregates in cytoplasm (Figure 2B). The TNPs located in organelles, including mitochondria (Figure 2B) and lysosomes (Figure 2B), induced destruction of organelle structures, such as mitochondrial swelling, cristae rupturing, and disappearance (Figure 2B).
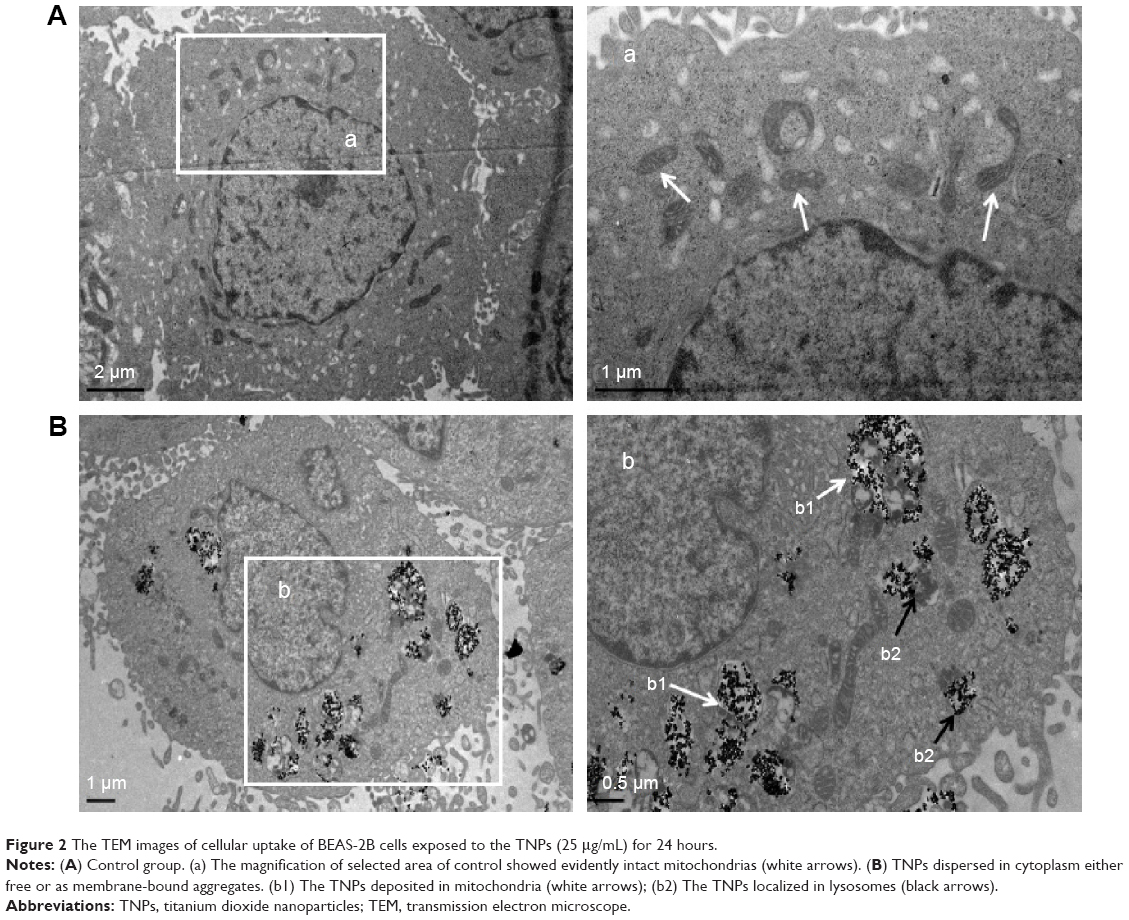 | Figure 2 The TEM images of cellular uptake of BEAS-2B cells exposed to the TNPs (25 μg/mL) for 24 hours. |
Cytotoxicity of BEAS-2B cells induced by TNPs
After exposing to TNPs for 24 hours, cell viability was determined to evaluate the possible toxicity of TNPs on BEAS-2B cells. As indicated in Figure 3, viability of BEAS-2B cells induced by TNPs was significantly lower than that of control. Our results suggested that TNP induced cytotoxicity in a dose-dependent manner.
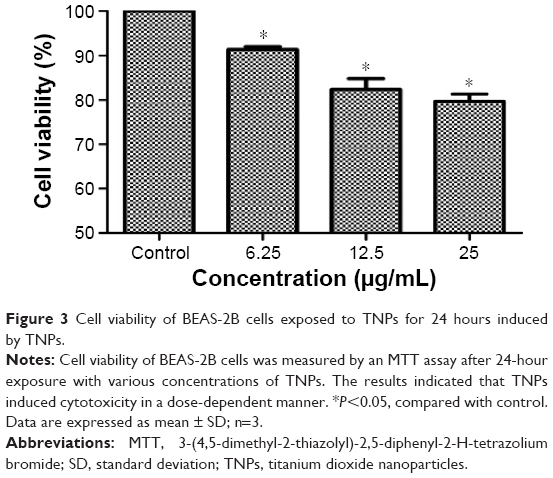 | Figure 3 Cell viability of BEAS-2B cells exposed to TNPs for 24 hours induced by TNPs. |
MDC staining of autophagic vacuoles
MDC was employed to measure autophagy activation of BEAS-2B cells after 24-hour treatment with TNPs. Identification of MDC staining was performed under a confocal microscope, as shown in Figure 4A and B; both the fluorescence intensity and the numbers of MDC-labeled vacuoles were increased in a dose-dependent manner. In addition, the vacuoles preferentially emerged in the cytosol and accumulated in the perinuclear regions. In contrast, a low level of fluorescence signal was detected in the control group.
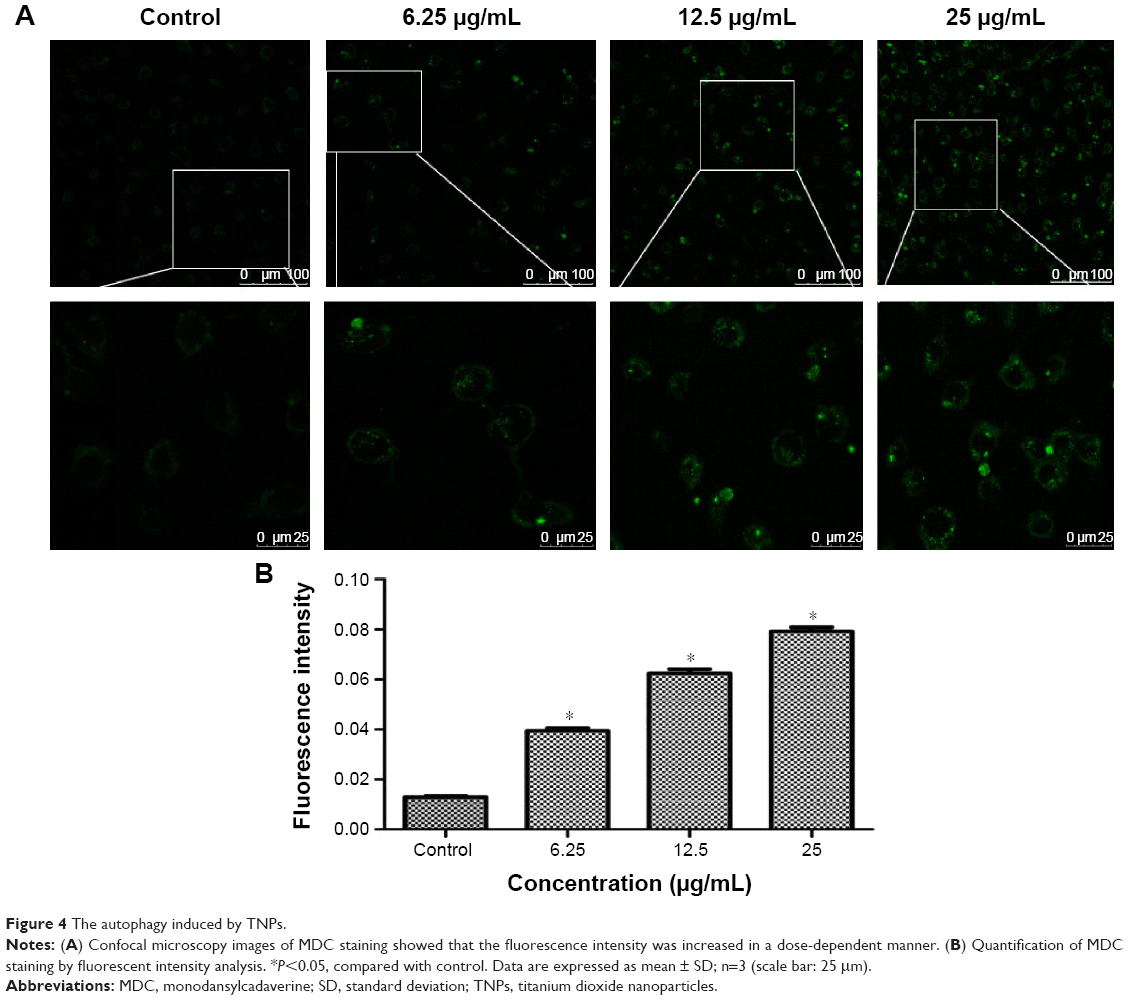 | Figure 4 The autophagy induced by TNPs. |
The observation of autophagic ultrastructural features
As a golden standard of autophagy measurement, TEM was used to show the autophagic ultrastructural features, thus to confirm the activation of TNP-induced autophagy. In Figure 5, cells treated with TNPs evidently manifested the accumulation of autophagic vacuoles (Figure 5B–F) in comparison with control cells (Figure 5A). As shown in Figure 5B, a double-membranous phagophore swallows parts of the cytoplasm; Figure 5C shows an autophagosome combined with cytoplasmic contents after being swallowed by a phagophore; the autophagosomes have merged with lysosomes (Figure 5D). Figure 5E and F show the autolysosomes with cytoplasmic contents inside while the contained cytoplasmic materials are partially degraded. These TEM images proved the presence of different stages of autophagic vacuoles and strongly suggest the accumulation of autophagic vacuoles in cells exposed to TNPs.
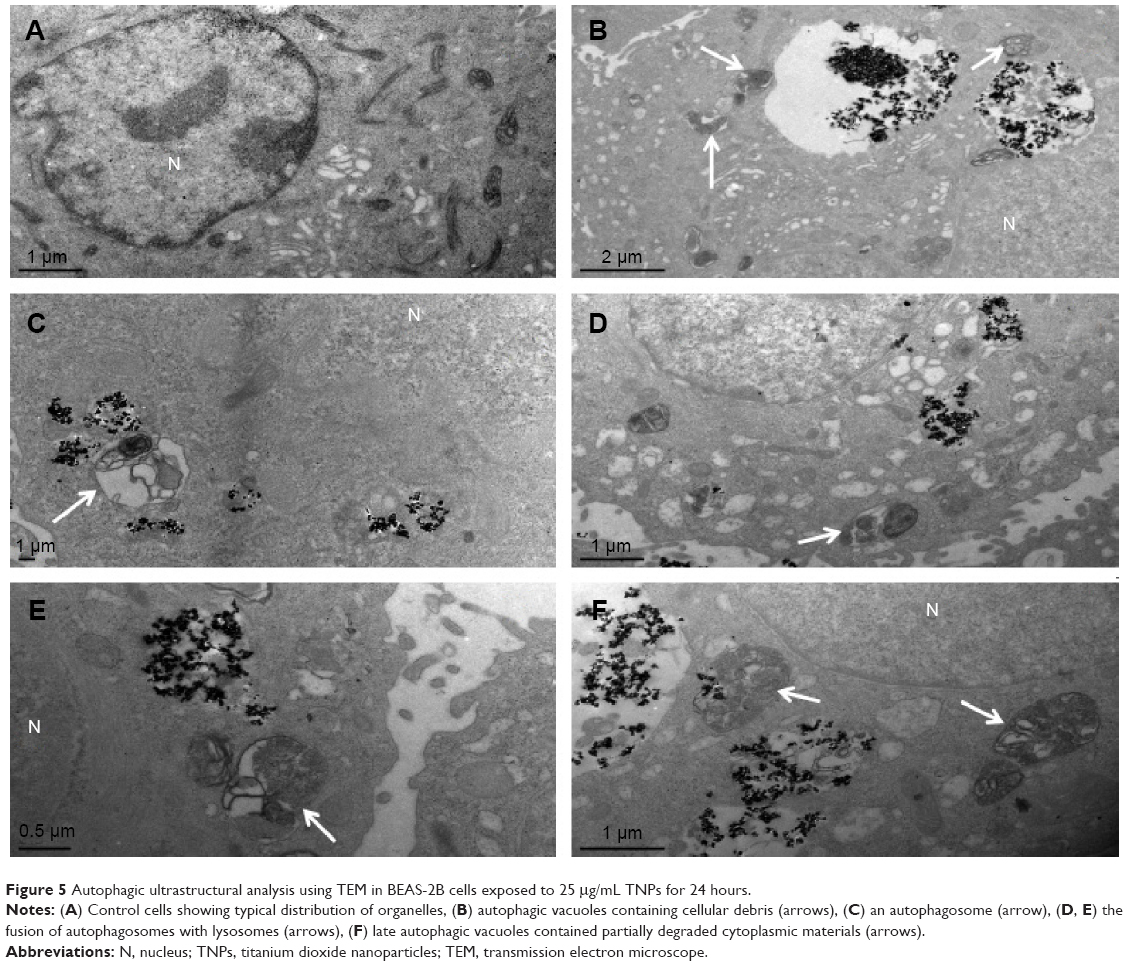 | Figure 5 Autophagic ultrastructural analysis using TEM in BEAS-2B cells exposed to 25 μg/mL TNPs for 24 hours. |
The LC3-I/LC3-II conversions
As the standard molecular marker of autophagy, LC3 protein is widely used in evaluating autophagy and intently associated with autophagosomes. During autophagy, LC3 protein usually performs a conversion from cytosolic LC3-I into its enzymatic counterpart LC3-II, which is bound to the membrane of autophagosomes.43 Autophagy triggered by TNPs was confirmed by assessing the expression levels of LC3-I and LC3-II. Our results demonstrated the increasing expression of LC3-II with dosage elevations of TNPs (Figure 6A). Further analysis showed that the ratio of LC3-II/LC3-I was increased significantly, which suggested that the autophagy in TNP-treated BEAS-2B cells increased in a dose-dependent manner (Figure 6B).
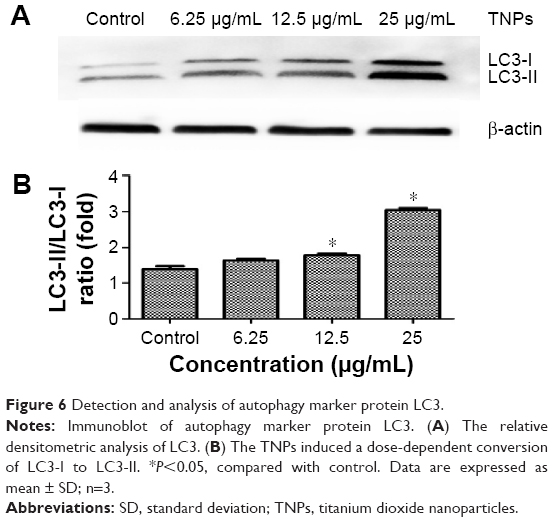 | Figure 6 Detection and analysis of autophagy marker protein LC3. |
Different expression of miR34a and Bcl-2 induced by TNPs
After exposure to various concentrations of TNPs (0, 6.25, 12.5, and 25 μg/mL) for 24 hours, the total RNAs and protein of the cells were extracted. Expression of miR34a and target gene Bcl-2 mRNA was detected by real-time PCR. Then, the protein level of Bcl-2 was measured by Western blot. Results indicated that TNPs decreased the expression of miR34a (Figure 7A) while the Bcl-2 was upregulated significantly in a dose-dependent manner both in mRNA (Figure 7B) and protein (Figure 7C and D) level in cells treated by TNPs (P<0.05).
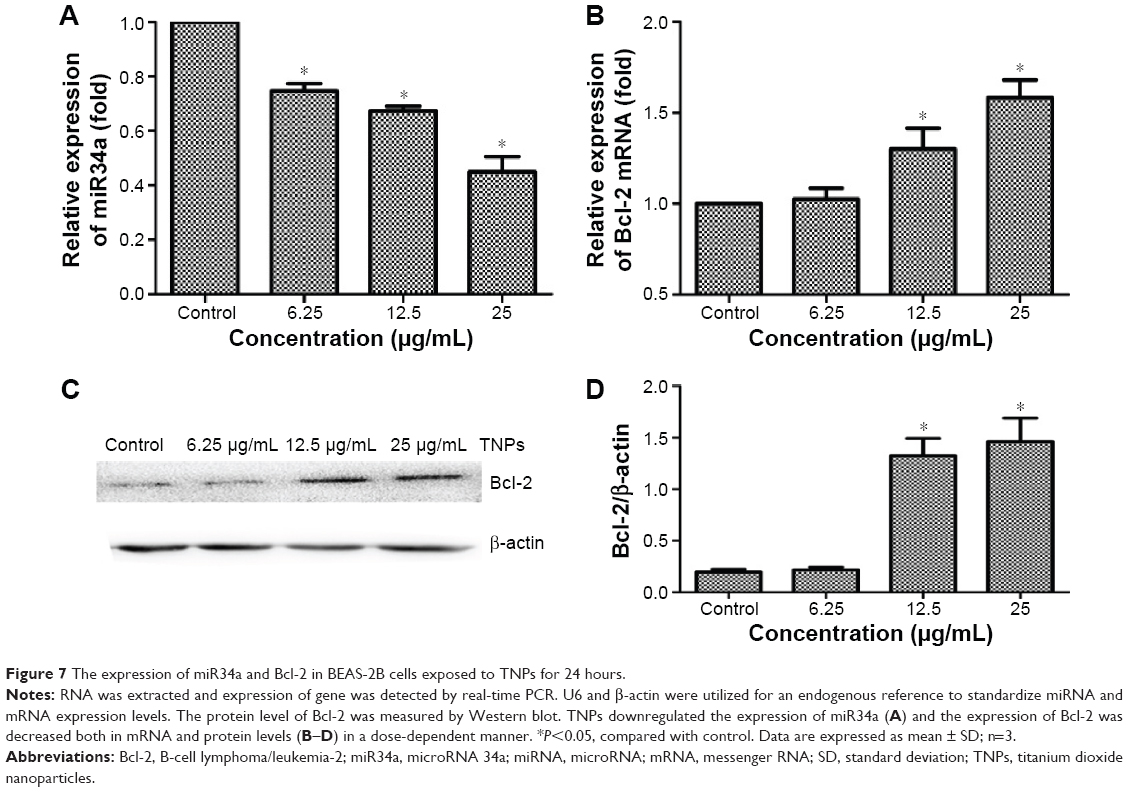 | Figure 7 The expression of miR34a and Bcl-2 in BEAS-2B cells exposed to TNPs for 24 hours. |
Overexpression of miR34a was achieved by lentivirus vector transfection
BEAS-2B cells were transfected by lentivirus vectors with miR34a or empty lentiviral vectors for 72 hours. As shown in Figure 8A, the ratio of green fluorescent protein expression was up to 80%. Further, transfection efficiency was verified by detecting the expression of miR34a. From the result in Figure 8B, we confirmed that miR34a was overexpressed successfully.
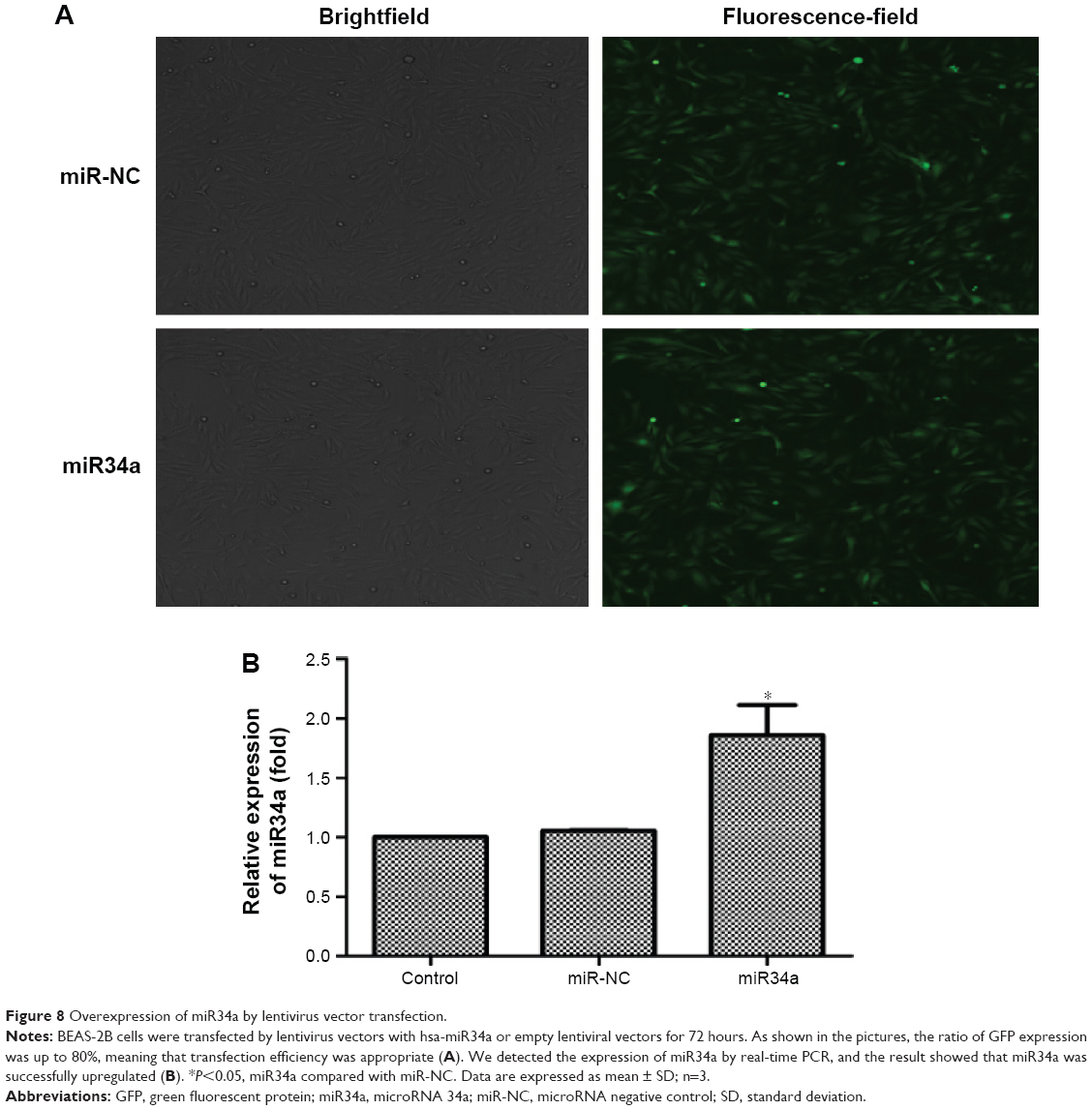 | Figure 8 Overexpression of miR34a by lentivirus vector transfection. |
TNP-induced miR34a and Bcl-2 alteration were regulated by overexpressed miR34a
Subsequently, we treated the transfected BEAS-2B cells with 25 μg/mL TNPs for 24 hours. Results (Figure 9A and B) showed that overexpression of miR34a attenuated the decrease of miR34a and increase of Bcl-2 mRNA induced by TNPs. We observed that (Figure 9C and D) TNPs induced an increase of Bcl-2 protein level (P<0.05), while overexpressed miR34a altered the TNP-induced expression of Bcl-2 protein in the same way as with Bcl-2 mRNA (P<0.05).
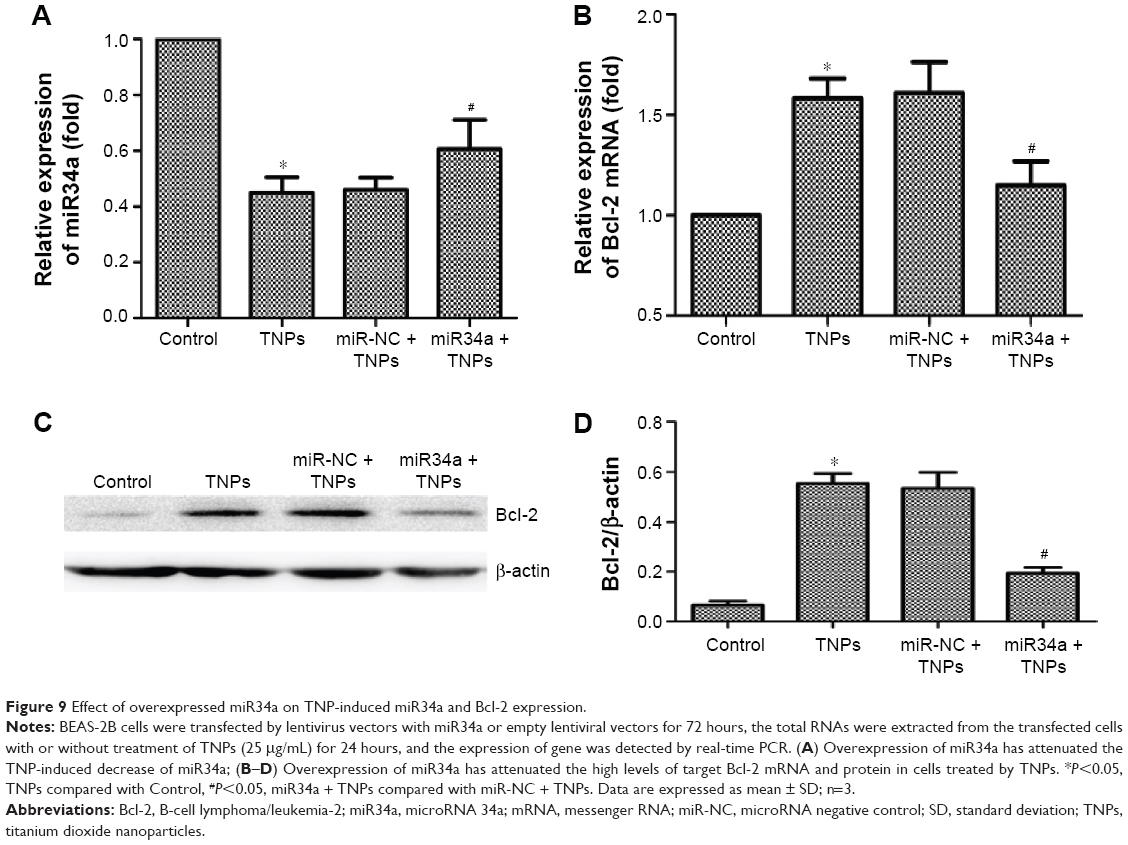 | Figure 9 Effect of overexpressed miR34a on TNP-induced miR34a and Bcl-2 expression. |
Overexpressed miR34a enhanced TNP-induced autophagy and cell death
We detected the expression of LC3-I and LC3-II to assess the effect of miR34a on TNP-induced autophagy. Results showed that the ratio of LC3-II/LC3-I in the group of overexpressed miR34a with TNPs was increased following the lower level of Bcl-2 protein compared with the cells with empty lentiviral vectors that were treated with TNPs (Figure 10A and B). Meanwhile, we found that overexpression of miR34a induced a lower level of cell viability in TNPs treated cells (Figure 11). This suggested that overexpression of miR34a increased the level of TNP-induced autophagy and cell death through targetedly decreasing the expression of Bcl-2.
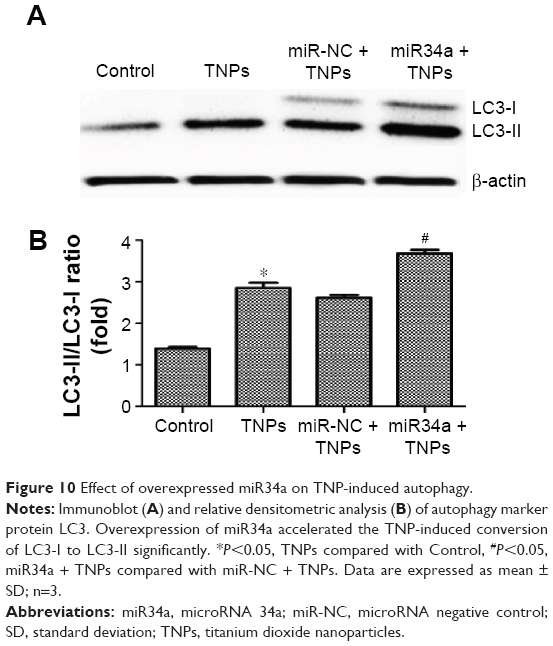 | Figure 10 Effect of overexpressed miR34a on TNP-induced autophagy. |
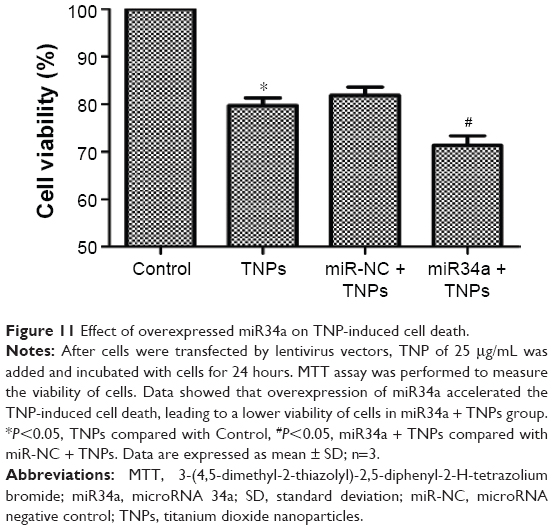 | Figure 11 Effect of overexpressed miR34a on TNP-induced cell death. |
Discussion
Autophagy can potentially be an emerging toxicity mechanism of cells for the degradation and clearance of insoluble nanoparticles.15 Remarkably, autophagy has been discovered in cells exposed to TNPs.20,21,24–26 However, the underlying mechanistic insights into autophagy induced by TNPs are still poorly investigated. Recently, there is growing interest in the topic of how epigenetic processes modulate cellular behavior in response to environmental chemicals. This study aimed to observe the effect of autophagy induced by TNPs and the regulation of miR34a in this process. Our findings provided the evidence for the first time that overexpressed miR34a enhanced TNP-induced autophagy and cell death through targetedly downregulating Bcl-2 in BEAS-2B cells.
Currently, the cellular uptake of nanomaterials is necessary for understanding their biological behavior in cells. Our results showed that TNPs were internalized and mainly dispersed in cytoplasmic or peri-region of the nucleus either as free or membrane-bound aggregates. The internalized TNPs could attack organelles, such as mitochondria and lysosomes, directly. It was reported that the cytotoxicity of nanoparticles was linked closely to the mitochondria and lysosomal damage in both normal and cancer cells.17,43 To gain closer mechanistic insight into TNP-induced biological effects, we measured the cell viability. Our data revealed that TNPs induced decrease of cell viability in a dose-dependent manner. This may be correlated with the enlarged amount of TNPs uptaken by BEAS-2B cells, since higher concentrations of nanoparticles localizing inside cells could initiate more cytotoxicity.43
Autophagy is a catabolic process involving the sequestration and degradation of cell’s own components, such as waste or unfolded and aggregated proteins and organelles, through the lysosomal machinery for maintaining cellular homeostasis.44 Autophagic recycling, therefore, yields simple metabolites for use in cell growth or bioenergetics, and can destroy damaged structures to prevent inflammatory, infectious, neurodegenerative, and neoplastic disorders, and deregulation of this pathway has been implicated in the pathogenesis of numerous human diseases.22,33,45 The process of autophagy in BEAS-2B cells exposed to TNPs was also presented in the TEM images, which was similar to the TNP-induced autophagy noted in keratinocytes.26 Besides, to further monitor autophagy triggered by TNPs in BEAS-2B cells, MDC staining and LC3-I/LC3-II conversion were employed according to the guidelines.46 MDC staining revealed that TNPs induced autophagy activation in a dose-dependent manner. When autophagy is activated, LC3 is converted from cytosolic LC3-I into enzymatic LC3-II. Consequently, the ratio of LC3-II to LC3-I is a standard marker for the detection of autophagy.47 In our research, it was observed that the ratio of LC3-II/LC3-I increased dramatically, which was in line with the results of the MDC staining and the ultrastructural analysis, suggesting that TNPs were the autophagy activator in BEAS-2B cells.
Autophagy is controlled by an ever-growing number of regulators, signalers, and miRNAs. miRNAs are a new class of small noncoding RNAs that have been highly conserved during evolution and recently estimated to regulate gene expression at posttranscriptional and translational levels. Importantly, miRNAs play a critical role in a broad range of biological processes, including proliferation, differentiation, apoptosis, and stress response, linking them to numerous human diseases, including cancer.48 In particular, miRNAs were well characterized to impact the core autophagy pathway, including autophagy induction, vesicle nucleation, vesicle elongation, retrieval, and fusion between autophagosomes and lysosomes.33 Indeed, several miRNAs were significantly altered by TNPs exposure, suggesting that those miRNAs may be involved in the regulation of mRNA translation.48 miR34a, a highly conserved miRNA, has recently emerged as a key tumor suppressor in multiple malignancies through the suppression of multiple targets.35
In our study, TNPs downregulated the miR34a while autophagy and expression of Bcl-2 increased. Autophagy activated by TNPs could be considered as a stressful process of degradation and clearance of the insoluble TNPs for cells, following which upregulation of Bcl-2 by miR34a was observed to limit overactivation of autophagy and prevent further cell death. It was reported that upregulation of miR34a could decrease the expression of a series of targets, such as Bcl-2, cyclin D1, cyclin-dependent kinase 6, Forkhead box protein P1, Notch 1, and Sirtuin 1.38 As a member of the antiapoptotic family, Bcl-2 interacts with Beclin 1 (H3 domain), which is well documented to arrest autophagy after freeing from Bcl-2 inhibition.49 Therefore, miR34a probably regulates autophagy through Bcl-2/Beclin-1-PI3KIII pathway, via targeting Bcl-2.21,50–53 As indicated in our study, Bcl-2 was upregulated both in mRNA and protein levels in line with the increase of autophagy induced by TNPs. It seemed counter to the statement that Bcl-2 can inhibit Beclin-1-dependent autophagy.54,55 However, it is reported that Bcl-2 was upregulated accompanied with the autophagy induced by serum starvation and rapamycin in neuroblastoma SH-SY5Y cells.56 Also, studies have shown that suppression of Bcl-2 further increased autophagic activity and cell death.57
Further, we performed overexpression of miR34a by lentivirus vector transfection and analyzed the function of miR34a and Bcl-2 in regulating autophagy. Results showed that overexpression of miR34a attenuated the increase of Bcl-2 mRNA and protein induced by TNPs. And the ratio of LC3-II to LC3-I increased significantly compared with the TNP-treated cells containing empty lentiviral vectors. Usually, autophagy was regarded as a way for a cell to maintain the homeostasis by sequestering and degrading the exogenous materials. However, cell death is regarded as a potential mechanism of the toxicological consequence of autophagy dysfunction.18 Subsequently, we observed that overexpression of miR34a induced a lower level of cell viability, suggesting an increase of cell death rate in TNP-treated cells. This implied that downregulation of Bcl-2 expression by overexpressed miR34a enhanced the TNP-induced autophagy and then resulted in a higher level of cell death.
Together, we suggest that autophagy may play an important role in determining the survival of cells when cells were treated by TNPs. Besides, miR34a may vitally participate in this process due to its ability to regulate Bcl-2 expression. However, there are more underlying mechanisms that are still needed to be clarified in TNP-induced autophagy.
Acknowledgments
This work has been supported by grants from the Scientific Research Common Program of Beijing Municipal Commission of Education (KM201410025008), the National Natural Science Foundation of China (81472957, 81172639), the Beijing Natural Science Foundation (7142020), and the Importation and Development of High-Caliber Talents Project of Beijing Municipal Institutions (CIT&TCD201404187).
Disclosure
The authors report no conflicts of interest in this work.
References
Hougaard KS, Jackson P, Jensen KA, et al. Effects of prenatal exposure to surface-coated nanosized titanium dioxide (UV-Titan). A study in mice. Part Fibre Toxicol. 2010;7:16. | ||
Magaye R, Zhao JS, Bowman L, Ding M. Genotoxicity and carcinogenicity of cobalt-, nickel- and copper-based nanoparticles (Review). Exp Ther Med. 2012;4(4):551–561. | ||
Menard A, Drobne D, Jemec A. Ecotoxicity of nanosized TiO2. Review of in vivo data. Environ Pollut. 2011;159(3):677–684. | ||
Robertson TA, Sanchez WY, Roberts MS. Are commercially available nanoparticles safe when applied to the skin? J Biomed Nanotechnol. 2010;6(5):452–468. | ||
Weir A, Westerhoff P, Fabricius L, Hristovski K, von Goetz N. Titanium dioxide nanoparticles in food and personal care products. Environ Sci Technol. 2012;46(4):2242–2250. | ||
Tsyganov I, Maitz MF, Wieser E, Prokert F, Richter E, Rogozin A. Structure and properties of titanium oxide layers prepared by metal plasma immersion ion implantation and deposition. Surf Coat Technol. 2003;174:591–596. | ||
Petkovic J, Zegura B, Stevanovic M, et al. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology. 2011;5(3):341–353. | ||
Xue CB, Wu JH, Lan FL, et al. Nano titanium dioxide induces the generation of ROS and potential damage in HaCaT cells under UVA irradiation. J Nanosci Nanotechnol. 2010;10(12):8500–8507. | ||
Zhang X, Li W, Yang Z. Toxicology of nanosized titanium dioxide: an update. Arch Toxicol. 2015;89(12):2207–2217. | ||
Shi H, Magaye R, Castranova V, Zhao J. Titanium dioxide nanoparticles: a review of current toxicological data. Part Fibre Toxicol. 2013; 10:15. | ||
Murashov V. Occupational exposure to nanomedical applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(2):203–213. | ||
Lee KP, Trochimowicz HJ, Reinhardt CF. Pulmonary response of rats exposed to titanium dioxide (TiO2) by inhalation for two years. Toxicol Appl Pharmacol. 1985;79(2):179–192. | ||
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Cobalt in hard metals and cobalt sulfate, gallium arsenide, indium phosphide and vanadium pentoxide. IARC Monogr Eval Carcinog Risks Hum. 2006;86:1–294. | ||
Tedja R, Marquis C, Lim M, Amal R. Biological impacts of TiO2 on human lung cell lines A549 and H1299: particle size distribution effects. J Nanopart Res. 2011;13(9):3801–3813. | ||
Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. | ||
Yu L, Lu Y, Man N, Yu SH, Wen LP. Rare earth oxide nanocrystals induce autophagy in HeLa cells. Small. 2009;5(24):2784–2787. | ||
Yu YB, Duan JC, Yu Y, et al. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater. 2014;270:176–186. | ||
Zabirnyk O, Yezhelyev M, Seleverstov O. Nanoparticles as a novel class of autophagy activators. Autophagy. 2007;3(3):278–281. | ||
Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. 2012;4(6):a008813. | ||
Kroemer G, Galluzzi L, Vandenabeele P, et al; Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. | ||
Chio CC, Lin JW, Cheng HA, et al. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch Toxicol. 2013;87(3):459–468. | ||
Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012;4(10): a008839. | ||
Yang ZF, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2): 124–131. | ||
Yu KN, Chang SH, Park SJ, et al. Titanium dioxide nanoparticles induce endoplasmic reticulum stress-mediated autophagic cell death via mitochondria-associated endoplasmic reticulum membrane disruption in normal lung cells. PLoS One. 2015;10(6):e0131208. | ||
Yu KN, Sung JH, Lee S, et al. Inhalation of titanium dioxide induces endoplasmic reticulum stress-mediated autophagy and inflammation in mice. Food Chem Toxicol. 2015:85;106–113. | ||
Zhao Y, Howe JLC, Yu Z, et al. Exposure to titanium dioxide nanoparticles induces autophagy in primary human keratinocytes. Small. 2013;9(3):387–392. | ||
Zhao GX, Pan H, Ouyang DY, He XH. The critical molecular interconnections in regulating apoptosis and autophagy. Ann Med. 2015;47(4):305–315. | ||
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. | ||
Jing Z, Han WD, Sui XB, Xie JS, Pan HM. Interaction of autophagy with microRNAs and their potential therapeutic implications in human cancers. Cancer Lett. 2015;356(2):332–338. | ||
Lee SK, Calin GA. Non-coding RNAs and cancer: new paradigms in oncology. Discov Med. 2011;58:245–254. | ||
Cho WCS. MicroRNAs in cancer – from research to therapy. Biochim Biophys Acta. 2010;1805(2):209–217. | ||
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH. MicroRNA regulation of core apoptosis pathways in cancer. Eur J Cancer. 2011;47(2):163–174. | ||
Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis. 2012;33(11):2018–2025. | ||
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. | ||
Li XJ, Ren ZJ, Tang JH. MicroRNA-34a: a potential therapeutic target in human cancer. Cell Death Dis. 2014;5:e1327. | ||
Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73(2):473–477. | ||
Liu K, Huang J, Xie M, et al. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy. 2014;10(3):442–452. | ||
Xiao Z, Li CH, Chan SL, et al. A small-molecule modulator of the tumor-suppressor miR34a inhibits the growth of hepatocellular carcinoma. Cancer Res. 2014;74(21):6236–6247. | ||
Tanaka N, Toyooka S, Soh J, et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol Rep. 2013;29(6):2169–2174. | ||
Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ. 2006;13(8):1339–1350. | ||
Ji Z, Jin X, George S, et al. Dispersion and stability optimization of TiO2 nanoparticles in cell culture media. Environ Sci Technol. 2010;44(19):7309–7314. | ||
Jiang JK, Oberdorster G, Biswas P. Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies. J Nanopart Res. 2009;11(1):77–89. | ||
Lu X, Qian JC, Zhou HJ, et al. In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine. 2011;6:1889–1901. | ||
Duan JC, Yu YB, Li Y, et al. Toxic effect of silica nanoparticles on endothelial cells through DNA damage response via Chk1-dependent G2/M checkpoint. PLoS One. 2013;8(4):e62087. | ||
Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease REPLY. N Engl J Med. 2013;368(19):1845–1846. | ||
Klionsky DJ. Coming soon to a journal near you – The updated guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2014;10(10):1691. | ||
Sun L, Li Y, Liu XM, et al. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicology In Vitro. 2011;25(8):1619–1629. | ||
Halappanavar S, Jackson P, Williams A, et al. Pulmonary response to surface-coated nanotitanium dioxide particles includes induction of acute phase response genes, inflammatory cascades, and changes in microRNAs: a toxicogenomic study. Environ Mol Mutagen. 2011;52(6):425–439. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Li LS, Yuan LJ, Luo JM, Gao J, Guo JL, Xie XM. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin Exp Med. 2013;13(2):109–117. | ||
Tian LL, Zhang JR, Ge JC, et al. MicroRNA-205 suppresses proliferation and promotes apoptosis in laryngeal squamous cell carcinoma. Med Oncol. 2014;31(1):785. | ||
Wu ZW, Lu HF, Sheng JF, Li LJ. Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS Lett. 2012;586(16):2459–2467. | ||
Yan DS, Dong XD, Chen XY, et al. Role of microRNA-182 in posterior uveal melanoma: regulation of tumor development through MITF, BCL2 and cyclin D2. PLoS One. 2012;7(7):e40967. | ||
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. | ||
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. | ||
Xu HD, Wu D, Gu JH, et al. The pro-survival role of autophagy depends on Bcl-2 under nutrition stress conditions. PLoS One. 2013;8(5):e63232. | ||
Zhang XD, Wang Y, Wu JC, et al. Down-regulation of Bcl-2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J Neurosci Res. 2009;87(16):3600–3610. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.