")
Back to Journals » Cancer Management and Research » Volume 6
DNA repair in cancer: emerging targets for personalized therapy
Authors Abbotts R, Thompson N, Madhusudan S
Received 7 December 2013
Accepted for publication 5 January 2014
Published 19 February 2014 Volume 2014:6 Pages 77—92
DOI https://doi.org/10.2147/CMAR.S50497
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Rachel Abbotts, Nicola Thompson, Srinivasan Madhusudan
University of Nottingham, Academic Unit of Oncology, Division of Oncology, School of Medicine, Nottingham University Hospitals, City Hospital Campus, Nottingham, UK
Abstract: Genomic deoxyribonucleic acid (DNA) is under constant threat from endogenous and exogenous DNA damaging agents. Mammalian cells have evolved highly conserved DNA repair machinery to process DNA damage and maintain genomic integrity. Impaired DNA repair is a major driver for carcinogenesis and could promote aggressive cancer biology. Interestingly, in established tumors, DNA repair activity is required to counteract oxidative DNA damage that is prevalent in the tumor microenvironment. Emerging clinical data provide compelling evidence that overexpression of DNA repair factors may have prognostic and predictive significance in patients. More recently, DNA repair inhibition has emerged as a promising target for anticancer therapy. Synthetic lethality exploits intergene relationships where the loss of function of either of two related genes is nonlethal, but loss of both causes cell death. Exploiting this approach by targeting DNA repair has emerged as a promising strategy for personalized cancer therapy. In the current review, we focus on recent advances with a particular focus on synthetic lethality targeting in cancer.
Keywords: biomarker, drug target, synthetic lethality
Introduction
Genomic deoxyribonucleic acid (DNA) is at risk of damage from endogenous metabolic byproducts, spontaneous base modifications, and exogenous sources such as ultraviolet (UV) light, ionizing radiation, and chemical agents. Unrepaired DNA damage is a major source of potentially mutagenic lesions that drive carcinogenesis. To promote genomic stability, mammalian cells have evolved highly conserved DNA damage sensor mechanisms that can initiate: 1) induction of apoptosis to eliminate heavily damaged cells; 2) transcriptional response, which causes changes in the transcriptional profile that may promote cell survival; 3) DNA damage tolerance; 4) activation of DNA damage checkpoints and modulation of cell-cycle progression to allow time for DNA repair; and/or 5) initiation of DNA repair to restore genomic stability.
DNA repair pathways are so essential that germline mutations within DNA repair genes are associated with cancer predisposition syndromes such as hereditary non-polyposis carcinoma coli (HNPCC) or breast cancer susceptibility protein (BRCA)-deficient breast and ovarian cancer syndromes.1,2 In addition, polymorphic variants that confer suboptimal DNA repair capacity could also influence cancer susceptibility and prognosis.3,4 Furthermore, the anticancer activity of chemotherapy (such as alkylating agents and platinum compounds) and radiotherapy is to a large extent directly related to their ability to induce DNA damage. The DNA repair capacity of cancer cells to recognize and repair chemo/radiotherapy-induced damage is therefore also an important mechanism for therapeutic resistance that negatively impacts upon clinical outcomes.5 Pharmacological inhibition of DNA repair may increase cytotoxicity of anticancer agents, reverse treatment resistance and improve therapeutic efficacy.6 Recent evidence indicates that inhibitors of DNA repair also offer the opportunity to target genetic differences that exist between normal and tumor tissue.7,8
In the current review, we provide an overview of major DNA repair pathways in mammalian cells. We will then focus on the emerging DNA repair drug targets for personalized cancer therapy.
DNA repair pathways
DNA repair pathways operate in mammalian cells to maintain genomic integrity (Figure 1). Loss of efficiency of one or more DNA repair mechanisms, whether by germline inheritance or sporadic mutation, accelerates the rate of accumulation of additional mutations by 100–1,000 times, with selective pressure favoring those mutations that drive carcinogenesis – the “mutator phenotype”.9,10
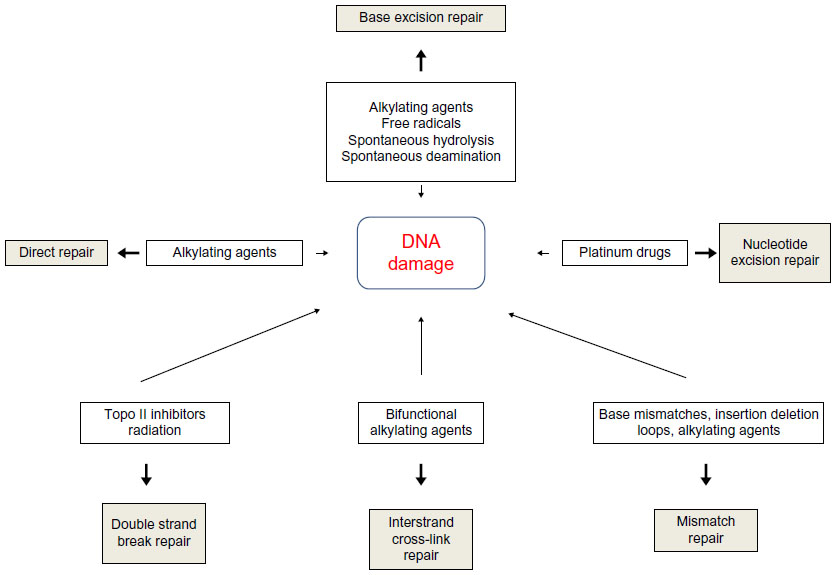 | Figure 1 Types of DNA damage and DNA repair pathways. |
Direct repair
A number of mechanisms exist to directly reverse certain DNA-damage lesions in single-step processes. Direct reversal of the oxidative lesion O6-methylguanine is carried out by the suicide enzyme methylguanine methyltransferase (MGMT) via an active site Cys145 that acts as a methyl recipient, followed by rapid ubiquitin-induced degradation. MGMT expression is one of several factors governing response to alkylating chemotherapy agents.11,12 The 2-oxoglutarate/iron-dependent dioxygenases, alkylated DNA repair protein alkB homolog 2 (ABH2) and alkylated DNA repair protein alkB homolog 3 (ABH3) also repair various alkylation adducts, including 1-methyladenine and 1-ethyladenine, by oxidative dealkylation. ABH2 preferentially acts on double-stranded DNA, possibly in the vicinity of replication forks, whereas ABH3 is involved in single-stranded DNA and ribonucleic acid (RNA) repair.11 ABH2 and ABH3 knockout mice are viable with no cancer phenotype, although ABH2-deficient mice do spontaneously accumulate 1-methyladenine adducts and are hypersensitive to exogenous alkylating agents.13 UV-induced damage such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) are repaired via photolyases: 50–55 kDa flavoproteins that contain chromophoric groups (flavin adenine dinucleotides) that become activated when illuminated with visible or near-UV light, allowing transfer of an electron to the lesion to destabilize the interpyrimidine bonds.14
Base excision repair
Base excision repair (BER) is responsible for detection and repair of damage caused by a number of mechanisms, including alkylation, oxidation, ring saturation, single-strand breaks (SSBs), and base deamination. Although complex, with at least two subpathways (short patch and long patch), BER generally proceeds via: 1) recognition and removal of a damaged base by a DNA glycosylase to form an apurinic/apyrimidinic (AP) site intermediate, 2) cleavage of the phosphodiester backbone 5′ to the AP site by AP endonuclease 1 (APE1), 3) removal of the 5′ sugar fragment, 4) incorporation of the correct base by a DNA polymerase, and 5) sealing of the strand break by a DNA ligase (Figure 1).14–21 Given the wide range of substrate lesions and potential mutagenic sequelae of failed repair, several BER gene mutations have been linked to human disease, including autosomal recessive familial adenomatous polyposis (MYH mutation), primary immunodeficiency disorders (uracil DNA glycosylase mutation), and neurological disorders (mutations in auxiliary genes such as aprataxin, tyrosyl-DNA phosphodiesterase 1, or polynucleotide kinase 3′-phosphatase). Furthermore, large numbers of single-nucleotide polymorphisms in BER genes have been identified, with variable effect on repair capacity and pathological consequences (reviewed in Wilson et al).22
SSB repair
SSB repair (SSBR) is most accurately considered a BER-related pathway, given the similarity of substrates and shared protein members. SSBR repairs single-strand discontinuities arising from a variety of sources, including reactive oxygen species (ROS), base deamination, and BER intermediates. It also repairs breaks introduced by DNA topoisomerase 1 (topo I) activity, which transiently introduces a DNA nick to relax DNA during transcription and replication, but which can fail to reseal the nick if in close proximity to polymerases or other DNA lesions.23,24 SSBR requires effective surveillance and damage detection, for which PARP1 (poly[ADP-ribose] polymerase 1) is believed to play an essential role. On detecting an SSB, PARP1 rapidly becomes bound and poly(ADP-ribosyl)ated, protecting the nick ends from undesirable recombination and allowing the recruitment of the molecular scaffold protein X-ray repair cross-complementing protein 1 (XRCC1) for ongoing repair. As with BER, end processing follows damage recognition and may be undertaken by a large range of proteins (depending upon the termini damage present), each of which requires interaction with XRCC1 for efficient activity. ROS-related damage often results in 3′-phosphate and 3′-phosphoglycolate modifications, which are processed by polynucleotide kinase (PNK) 3′-phosphatase (PNKP) and APE1 respectively. Topo I-associated SSBs require processing by TDP1 (tyrosyl-DNA phosphodiesterase 1), whereas 5′-adenosine monophosphate-SSBs resulting from abortive DNA ligase activity at existing SSBs are processed by aprataxin. Repair can then proceed via short- or long-patch gap filling and end ligation as in the classical BER pathway.25 SSBR may also play a role in replication-associated damage repair.25,26 When the replication machinery encounters an unrepaired SSB, fork collapse occurs, with the creation of a one-ended double-strand break (DSB) on one chromatid, and an SSB on the other. The DSB is processed by components of the homologous recombination (HR) pathway to allow RAD51-mediated template switching and reformation of the replication fork. Without repair, the associated SSB would be converted to a further DSB on replication fork restart, and hence would represent an irrevocably unrepairable lesion. SSBR end-processing and long-patch BER are probably involved in replication-coupled SSBR, as highlighted by the transcriptional activation of the critical SSBR enzyme XRCC1 by replication-associated transcription factors, such as forkhead box protein M1 (FOX M1) and E2F-1.27,28
Nucleotide excision repair
Nucleotide excision repair (NER) recognizes and repairs base lesions associated with distortion of the DNA helical structure, including UV-induced photoproducts not eliminated by direct repair, and an array of bulky adducts induced by various exogenous chemical agents. Two subpathways of NER exist: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). TC-NER removes lesions from the transcribed DNA strand of transcriptionally active genes when encountered by RNA polymerase II, restoring transcriptional activity and preventing apoptosis. GG-NER performs this process with poor efficiency, instead removing lesions on non-transcribed strands and transcriptionally inert genes to avoid replication fork stalling and chromosomal breakages.29 In GG-NER, damage recognition is sensed by various proteins, including the xeroderma pigmentosum (XP), complementation group C (XPC)-RAD23B complex (helix distortions), UV-damaged DNA-binding protein 1 (DDB1), and UV-damaged DNA-binding protein 2 (DDB2) (UV damage), and XPA (unknown substrate).30 In TC-NER, recognition is mediated by stalling of RNA polymerase II at a damaged site. Recognition factor binding in both pathways is associated with localized distortion to allow repair factor access to the damaged site. Transcription factor IIH (TFIIH), a nine-subunit complex including the DNA helicases XP complementation group B (XPB) and XP complementation group D (XPD), is recruited to unwind the DNA local to the damaged site. Dual incision around the lesion is performed by structure-specific endonucleases XP complementation group G (XPG) (3′ incision) and the excision repair cross-complementing group 1 (ERCC1)-XP complementation group F (XPF) complex (5′ incision), resulting in cleavage of a 24–29 nucleotide fragment. In common with BER and mismatch repair (MMR), proliferating cell nuclear antigen (PCNA) is then recruited to coordinate DNA polymerase repair synthesis and DNA ligase nick joining.29 Given the critical role NER plays in repairing UV-induced damage, it is unsurprising that mutation within NER genes can lead to UV hypersensitivity. XP is an autosomal recessive syndrome that manifests as photosensitivity, neurological abnormalities, and predisposition to skin and other cancers. XP is characterized by marked clinical and genetic heterogeneity, with causative mutations falling into one of seven complementation groups that span many NER factors. Different mutations in the same NER genes can give rise to alternative phenotypes that do not include cancer predisposition, namely Cockayne syndrome (variable UV sensitivity, premature aging, and physical and mental retardation) and trichothiodystrophy (TTD; variable UV-sensitivity, premature aging, ichthyosis, brittle hair, and short stature).31 This heterogeneity may result from the bifunctionality of NER factors. For example, the XPD complementation group is associated with several disease-specific mutations that may cause XP, combined Cockayne syndrome and XP, or TTD. When the causative mutation is associated with XP, the NER function of TFIIH is deficient, correlating with a phenotype of severe UV-sensitivity and cancer predisposition. TFIIH also has a role in transcriptional initiation of RNA polymerase II, in which the helicase subunits (XPB and XPD) unwind the DNA at the promoter region to allow transcription complex access. XPD mutations affecting this function may have normal NER (non-UV-sensitive TTD), or defective CPD but intact 6-4PP repair (UV-sensitive TTD), accounting for the reduced malignancy risk.
MMR
MMR recognizes and repairs errors introduced during replication. DNA polymerases possess 3′-5′ exonuclease activity to excise incorrectly paired bases in newly synthesized DNA. Failure of this proofreading process leads to mispair persistence, forming a substrate for MMR. MMR also recognizes and repairs insertion/deletion loops (IDLs), particularly within microsatellite DNA – hence, “microsatellite instability” is recognized as a hallmark of MMR failure.32,33 If microsatellite instability manifests within tumor suppressor genes, it can produce frame-shift mutations that contribute to carcinogenesis – a common feature of certain cancers, including colorectal, endometrial, ovarian, and gastric cancer (reviewed in Li).34 MMR error recognition is initiated by heterodimers of MSH (Escherichia coli MutS homolog) ATPases. MutSα comprises MSH2 and MSH6, and recognizes base mismatches and small 1-2 nucleotide IDLs. MutSβ, comprising MSH2 and MSH3, binds to larger IDLs of up to 16 nucleotides. Loss of MSH2 results in the absence of both MMR subpathways, and hence cancer predisposition in preclinical models35 and in the clinical setting (as HNPCC).36 Once bound to the DNA substrate, the MutS heterodimer recruits MutLα, a heterodimer comprised of MutL homolog 1 (MLH1) and PMS2, homologs of the E. coli ATPase MutL. MLH1 is critical to MMR, with deletion or promoter methylation producing a comparable phenotype to MSH2 mutation.37,38 The MutS/MutL complex functions as a sliding clamp, undergoing an ATP-dependent conformational change that results in release from the mismatch site followed by translocation in both directions from the mismatch. Translocation continues until a strand break is encountered, such as the 3′ terminus of the leading strand, or the 5′ or 3′ termini of Okazaki fragments (200–1,000 nucleotide lagging strand fragments generated and later ligated during replication). Exonuclease-1 (EXO1) is loaded at the strand break and degrades the strand back towards the mismatch site, allowing repair using the parent strand template by the high fidelity DNA polymerase δ (Polδ). DNA ligase I seals the gap.33,34
Non-homologous end joining
Along with HR repair, non-homologous end-joining is one of the two major pathways that exist for repair of DSBs. Non-homologous end joining (NHEJ) essentially involves processing of the terminal nucleotides to allow end ligation, in a manner that restores molecular integrity but may not maintain sequence fidelity. Damage recognition in NHEJ is performed by the Ku70/Ku80 heterodimer, which binds to the DSB ends with high affinity, possibly tethering the broken ends together. Ku binding recruits and activates the DNA-dependent protein kinase DNA-PKcs, forming the DNA-PK complex that phosphorylates other repair proteins including XRCC4-like factor (XLF, also known as Cernunnos), Werner syndrome helicase (WRN), DNA ligase IV (LIG4), and XRCC4. Additionally, DNA-PK is able to autophosphorylate, allowing NHEJ regulation. DNA end-processing prepares damaged terminal nucleotides for ligation. Dependent on the nature of the damage, this may require variable combinations of repair factors. Damaged DNA overhangs can be removed by nucleases such as Artemis, while other factors such as WRN, PNK, and aprataxin and PNK-like factor also have roles. Nucleotide incorporation by DNA polymerases λ and μ replaces nucleotides cleaved during end-processing in a non-template dependent manner, and represents a significant source of error in the pathway. Once processed, ends are ligated by the LIG4/XRCC4/XLF complex.39–41
NHEJ is able to resolve a large variety of DNA DSB damage, and is thought to be the major pathway for DSB repair in eukaryotes, occurring throughout the cell cycle. Accurate repair in NHEJ is likely if the DSB possesses fully complementary single-stranded ends. In the absence of complementary ends, repair is dependent on annealing of short microhomologous sequences (four or fewer nucleotides) within the overhanging ends, which leads to the introduction of potentially mutagenic deletions or insertions.42 NHEJ also has a critical role in V(D)J recombination to recombine the variable region in B-cell and T-cell receptors – in this context, low fidelity is beneficial to maximize diversity.43
Microhomology-mediated end joining
The observation that NHEJ can proceed in the absence of key factors, including DNA-PKcs and Ku, without significant input from the HR pathway, has led to the theory that at least one backup NHEJ pathway exists.44 It appears that this pathway functions when classical NHEJ fails, either due to enzymatic deficiency or failure to interact at certain DNA lesions. This alternate mechanism, known as microhomology-mediated end joining (MMEJ), is reliant on the annealing of microhomologous regions of 5–25 base pairs. To uncover these microhomologies, binding of the NHEJ heterodimer Ku and the HR factor RAD51 must be inhibited, possibly by members of the PARP family, to allow 5′-3′ nucleolytic resection by the MRN complex (MRE11-RAD50-NBS1). During this process, replication protein A (RPA) binds to the single-stranded DNA ends to prevent self-complementization. Once microhomologous regions are uncovered, the DNA overhangs anneal. End processing proceeds as required, via XRF-ERCC1 flap cleavage and polymerase-mediated gap-filling. Ligation appears to involve DNA ligase I (LIG1) and III (LIG3).42,45 Unlike NHEJ, MMEJ is always associated with sequence loss and is therefore always mutagenic. Indeed, microhomologies can frequently be demonstrated at chromosome breakpoints in human cancer cells.45
Single-strand annealing
Like MMEJ, single-strand annealing also involves 3′ end resection to uncover homologous regions that can anneal directly under RAD52 control. The size of homologous repeat sequences utilized in single-strand annealing (such as ~300 bp Alu repeats) requires more extensive end resection creating a large flap. Flap removal is catalyzed by RAD1/RAD10 nucleases, under SAW1/SLX4 (single-strand annealing weakened 1/structure-specific endonuclease subunit 4) guidance, resulting in a large deletion event. Significant sequence diversity within repeat elements probably suppresses this pathway to a relatively minor role in DSB repair.40,46–60
HR
HR utilizes a homologous DNA sequence as a template for DNA synthesis and gap filling to ensure error-free repair.40,46–60 For this reason, HR is the predominant mechanism for DSB repair during cellular replication. Cell cycle control is exerted by a dependence on cyclin-dependent kinase activity, which is upregulated in S and G2 phases, although high levels in M phase are associated with HR suppression due to concurrent BRCA2 phosphorylation. HR is also suppressed during G1, when use of the homologous chromosome as a template would result in loss of heterozygosity. Pathway choice for DSB repair is probably also guided by the repair substrate. Two-ended DSBs formed by fracture of a duplex molecule can generally be accurately repaired by simple end ligation via the NHEJ pathway, which in mammalian cells is the predominant mechanism of repair of this type of damage.61 Conversely, one-ended DSBs occurring when a replication fork encounters an SSB or distorting base lesion require template-guided repair to prevent inappropriate annealing leading to large-scale rearrangements or insertions/deletions.
In general, HR requires: 1) damage recognition, 2) end resection mediated by the MRN complex, 3) RAD51-dependent homology-directed strand invasion and repair synthesis, 4) dissociation from the template strand, and 5) end ligation. The classical model is synthesis-dependent strand annealing, which occurs at two-ended DSBs. Following damage recognition, 3′ strand resection coordinated by the MRN complex occurs on both fractured strands. MRN interacts with CtIP (also known as RBBP8 [retinoblastoma binding protein 8]) to promote end resection and generate 3′ single-strand DNA overhangs. These are bound by the protective RPA to prevent self-annealing. RAD51 binds the DNA ends, in combination with associated proteins including BRCA2, RAD52, RAD54, RAD54B, and the RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3). The resultant nucleoprotein filament invades the sister chromatid or homologous chromosome to search for homologous regions to form heteroduplex DNA. Polymerase η catalyzes 3′ extension of the invading strand using the sister strand as a template. The non-template DNA strand is displaced into a D-loop, with a Holliday junction forming at the crossover between the hetero- and homoduplex DNA. This junction can slide along the DNA in either direction (“branch migration”), facilitated by numerous proteins (WRN, BLM, p53, RAD54, BLAP75, MSH2/MSH6) via mechanisms that are yet to be elucidated. Branch migration results in strand dissociation upon reaching the terminus of the invading strand. Once released, the newly synthesized strand anneals beyond the original breakpoint, where it is bound by RPA and RAD52 to coordinate recruitment of factors involved in end processing (eg, XPF/ERCC1 for flap removal), gap filling (by DNA polymerases) and end ligation (by DNA ligases). The repaired strand can then be used as a template for DNA synthesis on the non-invading strand. As a result, sequence information is copied from the template region into the breakpoint (“gene conversion”), potentially resulting in loss of heterozygosity. An alternative model theorizes the formation of a double Holliday junction through simultaneous strand invasion by both DSB 3′ ends, possibly as a mechanism for DSB repair during meiosis. Strand dissociation in this model requires cleavage at the Holliday junction. Depending on the orientation of cleavage, this might result in a crossover event, which could account for the large-scale sequence exchanges that can be demonstrated in meiotic cells.
It is now thought that the primary repair substrates for HR are one-ended DSBs formed by replication fork collapse at the site of an unrepaired SSB or base lesion. Repair of such lesions occurs by the break-induced replication pathway, which initially proceeds in a similar manner to two-ended DSB repair, with 3′ resection creating an overhang which invades the sister chromatid and anneals to a homologous region in a RAD51-mediated mechanism. Cleavage of the single Holliday junction restores the replication fork, allowing replication to continue. Dependent on the orientation of Holliday junction cleavage, this mechanism can result in the leading strand template becoming ligated to the newly synthesized lagging strand, resulting in a sister chromatid exchange.
Evidence to support replication fork collapse as the primary substrate for HR is based upon comparison of the recombination products formed following repair of induced SSBs and spontaneous recombination events. Restriction endonuclease-induced two-ended DSBs result in short tract gene conversion events, consistent with the synthesis-dependent strand annealing model of HR described above. In contrast, camptothecin exposure induces one-ended DSBs by stabilizing DNA–topo-I interactions to prevent re-ligation of topo I-induced SSBs, thus leading to replication fork collapse. Repair of camptothecin-induced damage results in sister chromatid exchanges and long tract gene conversions. Similarly, impaired repair of SSBs in XRCC1- or PARP1-deficient cells, or following PARP inhibitor exposure, increases one-ended DSB formation, associated with increased formation of γH2AX and RAD51 foci – markers of HR activity.8,62 Spontaneous recombination outcomes are more similar in spectrum to failed SSBR or camptothecin-related repair products than to those formed during the repair of endonuclease-induced two-ended DSBs, suggesting that replication fork collapse forms the primary repair substrate for HR.62
Crosslink repair
A number of DNA repair mechanisms play a role in the repair of interstrand crosslinks (ICLs), a highly toxic form of damage that can stall and collapse replication forks, potentially leading to DNA rearrangement, mutation, or cell death. ICLs are cytotoxic at densities as low as 40 per cell, because they cause DNA distortion and prevent strand dissociation, impacting upon DNA synthesis and replication.63
Fanconi anemia (FA) is an autosomal recessive condition associated with predisposition to acute myelogenous leukemia and other malignancies, progressive bone marrow failure, short stature, and developmental delay. Fourteen complementation groups have been identified, with evidence suggesting that an FA core complex containing Fanconi anemia, complementation group A (FANCA), FANCB, FANCC, FANCE, FANCF, FANCG, and FANCL localizes to DNA damage and activates FANCD2, which in turn co-localizes with BRCA1. Recent studies also implicate XPF mutations in FA.64,65 Given that FA is associated with hypersensitivity to crosslinking agents, it is believed that the genetic basis may be a defect in ICL repair, although much of the pathway remains to be elucidated. The covalent link of the ICL causes localized DNA distortion and prevents replication-mediated DNA unwinding, leading to replication fork stalling. This is recognized by FANCM and associated proteins, which recruits the FA core complex and other repair proteins, including FANCD2-FANCI. The FA core complex possesses ubiquitin ligase activity, which monoubiquitinates FANCD2-FANCI, allowing interaction with Fanconi-associated nuclease 1 (FAN1) and DNA polymerase ν (Polν, POLN). FAN1 has 5′-3′ exonuclease and 5′-flap endonuclease activity, causing DNA cleavage (known as “unhooking”) alongside the ICL, converting the stalled replication fork into a one-ended DSB on the sister chromatid. A number of other nucleases may mediate DNA cleavage 3′ to the ICL, including MUS81-EME1, XPF-ERCC1, and SLX1-SLX4, thus excising the damaged region. PCNA is recruited, coordinating a switch to translesion synthesis (TLS) involving REV1 and DNA polymerase ζ to extend the nascent strand beyond the ICL site.66 Subsequently, the 3′ strand of the sister chromatid DSB is reintegrated into the homologous duplex, forming a double Holliday junction, and replication is restarted via HR in a RAD51-dependent manner.67–70
DNA damage tolerance mechanisms
Unrepaired lesions (particularly bulky adducts, intercalations, crosslinks, and helix distortions) can block progression of the replication fork. DNA damage tolerance mechanisms allow the replication machinery to bypass these lesions prior to repair. This allows replication to proceed without affecting the viability of dividing cells, but can increase the risk of propagating mutations to the daughter population.71
The DNA polymerase-mediated TLS pathway may proceed by one of two models. In polymerase-switching TLS, fork progression is stalled when the replicative helicase encounters a DNA lesion. This triggers the recruitment of specialized TLS polymerases that insert one or more nucleotides opposite the damaged base before the replicative polymerase is “switched” back into the replication machinery. The alternative “gap-filling” TLS model involves reinitiation of replication downstream from the damage lesion, resulting in a single-strand gap that is filled by one of the TLS polymerases. Polymerases that have been implicated in TLS include Polν, Polθ (A family polymerases), Polζ (B family polymerase), Polβ, Polλ, Polμ, and terminal deoxynucleotidyl transferase (X family polymerases), Polη, Polκ, Polι, and Rev1 (Y family polymerases). Polymerase choice may be damage-specific: for example, Polη may bypass UV-induced CPDs, whereas Rev1 may function in AP site bypass.72 TLS fidelity is polymerase-specific, highly variable, and may be an important source of genomic instability and susceptibility to cancer and other diseases.73
The HR pathway may also play a role in bypassing unrepaired lesions during replication, via a subpathway known as template switching. In this mechanism, the stalled daughter strand invades the sister chromatid in an HR-mediated mechanism to utilize the complementary parent strand as a template for synthesis, creating a Holliday junction that is resolved prior to resumption of replication. An alternative HR-related model is that of fork regression. The presence of a single-strand base lesion blocks replication on one parent strand, whilst replication persists on the complementary strand beyond the point of damage. Regression of the replication fork allows transient annealing of the daughter strands into a Holliday intermediate (“chicken foot” structure), providing an alternative template for synthesis across the damaged region.74,75
Global DNA damage response
The cellular DNA damage response involves activation of cell cycle checkpoints to induce a cell cycle arrest while repair mechanisms, transcriptional modulation, and/or apoptotic pathways are activated. DNA damage is detected by sensor proteins, which may overlap with specific repair pathway damage sensors. Checkpoint-specific sensors include: ataxia telangiectasia mutated (ATM), which primarily detects DSBs; ataxia telangiectasia and Rad3 related (ATR), which detects UV-induced and other damage; and RAD17-replication factor C (RFC complex) in conjunction with the RAD9-RAD1-HUS1 (9-1-1) complex, which can detect multiple damage types. Damage sensors interact with a wide range of mediator proteins, including BRCA1, MDC1 (mediator of DNA damage checkpoint 1), 53BP1 (p53 binding protein 1), and Claspin, which are required for downstream activation of Chk1 (activated downstream from ATR signaling) and Chk2 (activated downstream from ATM signaling). Chk1 and -2 kinases phosphorylate the phosphotyrosine phosphatases Cdc25A, -B, and -C, leading to their inactivation. As a result, the Cdc phosphatases are unable to dephosphorylate the cyclin-dependent kinases that promote cell cycle transition: Cdk2, which promotes the G1/S transition, and Cdc2 phosphotyrosine, which promotes the G2/M transition.
Three main checkpoints exist: G1/S, intra-S phase, and G2/M. The G1/S checkpoint is activated by damage that prevents initiation of replication, via the ATM-Chk2-Cdc25A or ATR-Chk1-Cdc25A pathways, and is maintained by Chk1 or Chk2-mediated phosphorylation of p53, which leads to p21-mediated inactivation of Cdk2. The intra-S phase checkpoint is activated by replication fork stalling, and is probably initiated both by the specialized checkpoint sensors (via inactivation of the S phase promoters cyclin E/Cdk2) and by various repair proteins such as the MRN complex (MRE11/RAD50/NBS1) and BRCA1. Activation by the latter group of sensors is thought to also activate a second pathway via phosphorylation of SMC1 (structure and maintenance of chromosomes 1) and SMC3 (structure and maintenance of chromosomes 3), which promotes recombination repair to recover stalled or collapsed replication forks. The G2/M checkpoint, which prevents initiation of mitosis in the presence of DNA damage, operates via the ATM and ATR-mediated to regulate G2/M transition via inactivation of Cdc25C/Cyclin B.14 Maintenance of the G2/M arrest requires transcriptional repression of Cdc2 and Cyclin B expression, mediated via activation of p53 and p21.
Activation of p53 plays an important role in apoptotic signaling. It has been implicated in activation of the intrinsic apoptosis pathway by shifting the balance of the multifunctional B-cell lymphoma-2 (Bcl-2) family away from Bcl-2 survival signaling towards induction of proapoptotic factors such as Bax and PUMA (p53 upregulated modulator of apoptosis), which contribute to caspase cascade activation.76 Furthermore, p53 has been linked to activation of Fas and DR5/KILLER death receptors, which activate the extrinsic apoptosis pathway that also contributes to caspase-mediated cell death.77
Clinical implications of DNA repair in cancer
Alterations in expression of DNA repair may influence cancer biology and influence aggressive phenotypes. Germ-line polymorphism of the POLB gene (rs3136797) encoding a Polβ variant with a low catalytic activity has been recently shown to induce cellular transformation and may be associated with increased cancer susceptibility.78 About 30% of human tumors appear to express Polβ variant proteins (such as K289M or I260M) which can induce cellular transformation in vitro, associated with an aggressive mutator phenotype.79
Overexpression of DNA repair factors may promote cell survival in established tumors. For example, ROS generated during increased metabolic activity in cancer cells generate DNA damaging lesions such as AP sites, oxidative base damage, DNA SSBs, and DNA DSBs. If unrepaired, such DNA lesions could be deleterious to the cancer cell. Moreover, hypoxic and acidic tumor microenvironments can promote further oxidative stress in cancer cells. Although ROS scavenging systems (such as glutathione and thioredoxin, superoxide dismutases, catalases, and peroxidases) do operate in cancer cells, capacity is limited, eventually leading to ROS-induced DNA damage. Therefore cancer cells also utilize the DNA repair machinery to process DNA damaging lesions and maintain cellular survival. Clinical evidence supports the hypothesis that overexpression of DNA repair factors may have prognostic and predictive significance in patients (reviewed in Abbotts and Madhusudan).21
Taken together, this evidence suggests that targeting DNA repair is a valid anticancer strategy. A detailed discussion is beyond the scope of this article, as several recent comprehensive reviews are available.80–83 Here, we focus on the current status of PARP inhibitors in cancer therapy.
The most advanced class of DNA repair inhibitors to date are PARP inhibitors, which disrupt the BER-related SSBR pathway. PARP1 senses and binds to DNA strand breaks, catalyzing (auto-) poly(ADP-ribosyl)ation of target proteins to induce localized chromatin relaxation and assembly of an XRCC1-LIG3-PNKP repair complex. A number of potential PARP inhibitors have been identified, usually with nonspecificity within the PARP family due to high sequence homology at the active site. In vitro and in xenograft models, PARP inhibitors have been demonstrated to potentiate the action of a wide variety of damaging agents, including platinums, the alkylating agents temozolomide and cyclophosphamide, the nucleoside analogue gemcitabine, the topoisomerase inhibitor irinotecan, and ionizing radiation.84,85 Several PARP inhibitors have entered the clinical setting in Phase I–III studies in combination with various chemotherapeutic agents, although results have been mixed (reviewed recently by Davar et al).86 For example, the Pfizer compound rucaparib (AG-014699; Pfizer, Inc., New York, NY, USA) has been evaluated in Phase I and II in combination with temozolomide in malignant melanoma, demonstrating successful PARP inhibition at a tissue level and probable anticancer activity, but significant myelosuppression causing dose-limiting toxicity.87 Similar toxicity has been noted with olaparib (AZD2281; AstraZeneca, London, UK) in combination with paclitaxel, carboplatin, or cisplatin and gemcitabine in Phase III trials in gastric cancer.88 Myelosuppression or other dose-limiting toxicities have not been noted with iniparib (BSI-201; Sanofi, Paris, France), which has been evaluated at Phase II in metastatic triple-negative breast cancer in combination with gemcitabine and carboplatin. A significantly improved median overall survival was demonstrated compared with gemcitabine and carboplatin alone, without increased toxicity. However, a Phase III trial failed to meet co-primary endpoints of overall and progression-free survival,89 and after further disappointing results in a Phase III non-small-cell lung cancer trial, iniparib has been suspended from further development.90 It should be noted that doubts have been raised about iniparib’s ability to inhibit PARP activity. Although initially believed to noncompetitively inhibit PARP1 by association with the DNA binding domain, more recent studies have failed to demonstrate target inhibition.91 A good safety profile was also observed with veliparib (ABT-888; Abbott Laboratories, Abbott Park, IL, USA) in combination with temozolomide. This was associated with positive early results in metastatic colorectal and BRCA-deficient breast cancers, although in advanced melanoma the combination was associated with poor response and no progression-free or overall survival improvement. Many additional Phase I and II trials are currently underway, in combination with a variety of agents, including carboplatin, 5-fluorouracil and oxaliplatin, cisplatin and paclitaxel, topotecan, gemcitabine, and radiotherapy (reviewed in Davar et al).86 Other PARP inhibitors, including orally bioavailable agents, are currently also under Phase I investigation.86,92
The data presented above suggest that the clinical utility of PARP inhibitors in combination with chemotherapy may be limited in tumors in view of narrow therapeutic index. However, evolving preclinical and clinical evidence provides compelling data that DNA repair inhibitor use could be targeted more effectively by utilizing a synthetic lethality strategy.
Synthetic lethality
Synthetic lethality exploits inter-gene relationships where the loss of function of either of two related genes is nonlethal, but loss of both causes cell death. This offers the potential to specifically target cancer cells through inhibition of a gene known to be in a synthetic lethal relationship with a mutated tumor suppressor gene.93
PARP1 inhibition in BRCA deficiency
The best-characterized synthetic lethality relationship is between BRCA mutation and PARP1 inhibition.94–96 PARP1 plays a role in the BER-related pathway of SSBR. Inhibition of SSBR is associated with accumulation of DSBs, which can be exploited in a subset of cancers possessing defects in DSB repair. BRCA1 and -2 have long been identified as tumor suppressors, being mutated in an inherited cancer predisposition that increases susceptibility to breast and ovarian tumors.97 Both BRCA gene products have a role in the HR DNA repair pathway.98 In BRCA-deficient cells, loss of effective HR leads to DSB persistence and cell death (Figure 2). As heterozygosity at a BRCA allele is associated with effective HR, DSB accumulation induced by PARP inhibition specifically occurs only in tumor cells with acquired BRCA−/− homozygosity.7,8 Furthermore, loss of BRCA2 function has been linked to hyperactivation of PARP1 (an observation replicated in loss of other HR factors), enhancing the cytotoxic effect.99
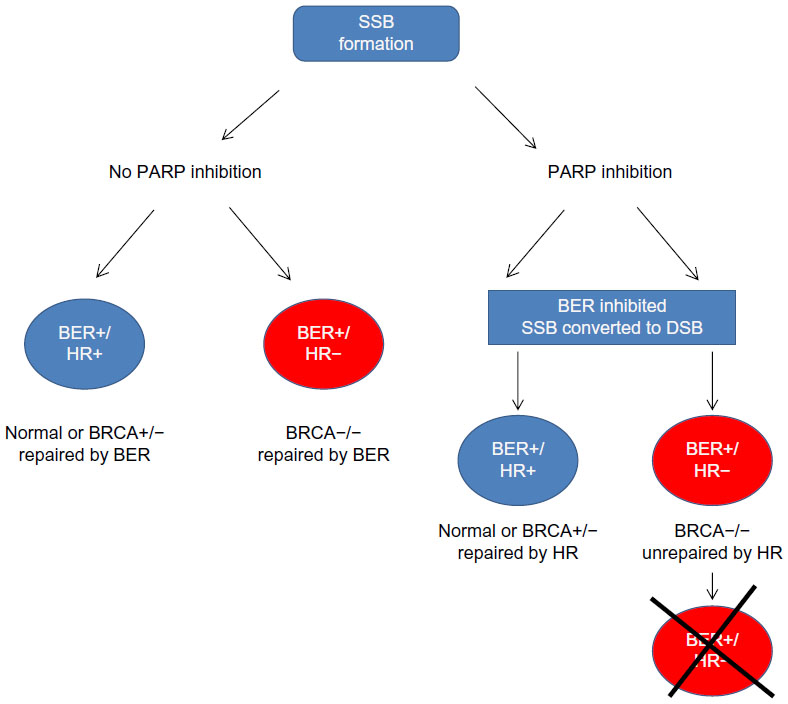 | Figure 2 Synthetic lethality in BRCA−/− cells upon PARP inhibition. PARP inhibition leads to SSBs. During replication SSBs get converted to DSBs. BRCA−/− cells are deficient in HR and hence unable to repair DSBs. DSB accumulation leads to cell death. In cells where PARP is proficient, SSBs are repaired by BER irrespective of BRCA status. There is no DSB generation, and cells continue to survive. |
PARP inhibition has been hypothesized to cause persistence of endogenously generated SSBs, inducing collapse of replication forks and formation of lethal DSBs.100 However, the exact mechanism of PARP inhibition has not been fully elucidated. Because of its role in the related SSBR pathway, PARP1 has occasionally and erroneously been described as essential for BER, although knockdown models have demonstrated this to be inaccurate. Unlike many BER factors, PARP1 is not essential for viability, nor is it required for repair of BER substrates such as alkylation damage – in actuality, PARP1 has been demonstrated to reduce BER kinetics.101 Interestingly, the mode of PARP1 inactivation may impact the biological consequences. For example, small interfering RNA (siRNA)-mediated PARP1 knockdown does not induce significant cytotoxicity in BRCA-deficient cells,7 while small molecule inhibition induces SSB accumulation after alkylating agent treatment and is well documented to be synthetically lethal in BRCA-deficient cells.101 This is probably because the BER intermediate single-strand nick is a substrate for transient PARP1 binding, hence accounting for slowed BER kinetics in the presence of PARP1. In this model, PARP inhibitor binding may trap PARP1 onto an SSB, whether formed spontaneously, exogenously, or by BER.102 As a result, downstream repair (whether by SSBR or BER) is prevented, leading to toxic DSBs during replication.103 Phase I and II trials of PARP inhibitors have demonstrated favorable efficacy and limited toxicity in BRCA-related breast and ovarian cancers.104 An initial Phase I study of olaparib in a cohort enriched for BRCA1/2 mutation carriers demonstrated evidence of in vivo anti-PARP activity (using PARP activity measurement in peripheral blood mononuclear cells and the surrogate marker of γH2AX induction, which accumulates at DSBs) and evidence of response in 40% BRCA carriers.96 This led to Phase II trials in breast or ovarian cancer associated with BRCA1/2 mutation, demonstrating further favorable data suggesting antitumor efficacy in this cohort.105,106 Phase III trials in BRCA-mutated ovarian cancer are currently planned.107,108 Likewise, Phase II investigation of rucaparib in BRCA1/2-mutated breast or ovarian cancer demonstrates PARP activity inhibition and evidence of tumor response.109 The oral PARP1/2 inhibitor niraparib (MK4827; Merck & Co, Inc., Whitehouse Station, NJ, USA) has also been evaluated at Phase I to possess an acceptable safety profile and probable antitumor activity.110 “BRCAness” refers to a subset of breast cancers, including “triple negative” (estrogen-, progesterone-, and HER2 [human epidermal growth factor receptor 2]-negative) and “basal phenotype” cancers, that possess molecular and histopathological similarity to BRCA-deficient tumors, which have been successfully targeted in vitro by PARP inhibition.111,112 Similarly, high-grade serous/undifferentiated ovarian cancers (HGSOC) are commonly associated with somatic or epigenetic loss of BRCA1/2.113 Phase II investigations of olaparib in these cohorts initially suggested that there may be a role for PARP inhibition in HGSOC,114 although Phase III development was terminated after it was deemed unlikely that reported progression-free survival would translate to an overall benefit.115 It should be noted that BRCA deficiency does not translate to PARP inhibitor response in all patients, and resistance may be a significant problem in future development. Two groups independently described the deletion of a previously identified BRCA2 mutation in PARP inhibitor-resistant cancer cells that led to restoration of the open reading frame, and hence HR proficiency.116,117 Furthermore, in BRCA1-deleted cells, loss of expression of the HR protein 53BP1 appears to partially restore HR competency and abrogate the ATM-dependent checkpoint response, limiting the resultant cell cycle arrest triggered by DSB accumulation after DNA damaging agent exposure.118
Alternative synthetic lethality partners for BER
The discovery of the synthetic lethality relationship between PARP1 and BRCA suggests that other tumor-specific defects in DSB repair factors may be therapeutically targeted by PARP inhibition. Germline mutations in the HR protein RAD51D have been identified as conferring susceptibility to ovarian cancer and may offer a target for PARP inhibitors in a small subset of women.119 Recent evidence suggests single agent cytotoxicity of PARP inhibitors in cells with reduced expression of ATM, the checkpoint activator that is activated by DSBs.120,121 Similar results have been observed in cells deficient in expression of the HR protein MRE11,122 and following in vitro downregulation of Artemis or LIG4, both of which function within the NHEJ pathway.123,124 Other potential synthetic lethality partners in PARP inhibition identified on a high-throughput siRNA screen include the DSB-induced checkpoint activator ATR, and a variety of factors that have been associated with bypass of stalled replication or transcription forks, including PCNA, DDB1, and XAB2 (XPA-binding protein 2).125 Conversely, SSBR factors other than PARP1 are potential synthetic lethality partners in DSB repair loss, as observed by the cytotoxicity induced by inhibitors of ATM or DNA-PKcs following knockdown of the BER protein XRCC1.126 Given its critical role in BER, targeting APE1 also presents a promising alternative. Sultana et al121 recently demonstrated that novel small molecule APE1 inhibitors are able to induce AP site accumulation, DSBs, cell cycle arrest, and cytotoxicity in BRCA2- or ATM-deficient cells. The synthetic lethality relationship between HR and APE1 was confirmed by cytotoxicity observed following ATM inhibitor exposure in APE1−/− cells.121
In addition to targeting tumors harboring germline defects in HR, Seo et al highlighted a possible treatment strategy in sporadic tumors by demonstrating that inhibition of APE1 DNA repair function induces targeted cytotoxicity in cell lines cultured in acidic environments.127 Tumor microenvironments are commonly acidic and have been associated with upregulation of BER proteins, including APE1. Conversely, other DNA repair mechanisms, including HR, are often downregulated under such conditions.128,129 Identification of tumors with BER upregulation and HR depletion may therefore offer an opportunity to exploit synthetic lethality through APE1 inhibition.
Recent evidence suggests that relationships between BER and non-HR DNA repair pathways may hold potential for synthetic lethality. For example, 8-oxoguanine base lesions, which are induced by metabolic ROS and can cause mutagenic GC→TA transversions if unrepaired, may be processed by both BER and MMR. Mutations in the MMR genes MLH1 or MSH2 are implicated in HNPCC and some sporadic colorectal cancers. SiRNA inhibition of the BER constituent DNA polymerases β/γ have been demonstrated to be selectively lethal in MLH1/MSH2 mutant cell lines, suggesting a synthetic lethality relationship.130 It remains to be established whether additional factors such as APE1 may have a role in this capacity.
Phosphatase and tensin homolog mutation as a synthetic lethality target
Phosphatase and tensin homolog (PTEN) is a negative regulator of the anti-apoptotic PI3K/Akt pathway. In addition to its inositol phosphatase function, PTEN has recently been implicated in the maintenance of genomic integrity.131–136 On the basis of evidence supporting an HR defect associated with PTEN mutation, Mendes-Pereira et al38 tested for synthetic lethality in HCT116 colorectal tumor cells transfected with a PTEN-mutant cDNA clone. Homozygosity for PTEN mutation was associated with a 20-fold increase in sensitivity to PARP inhibitors, which was replicated in a panel of commonly cultured tumor cell lines and in mouse xenografts. Ectopic expression of wild type PTEN into a PTEN-deficient prostate cancer cell line abrogated this effect, as did induced expression of a PTEN phosphatase domain mutant, suggesting that PTEN’s influence over HR lies out with its phosphatase function. Ectopic expression of RAD51 in a PTEN-deficient cell line was also able to overcome PARP inhibitor sensitivity, supporting the proposed link between PTEN mutation and reduced RAD51 expression. Similar results were demonstrated in endometrioid endometrial carcinoma, in which PTEN is mutated in up to 80% of patients.137 In primary PTEN−/− mouse astrocytes, reduced transcription of the RAD51 paralogs was associated with sensitivity to PARP inhibition,138 while PTEN disruption in colorectal cancer cells resulted in reduced MRE11 accumulation at DSBs that is also associated with PARP inhibitor sensitivity.139 In lung cancer cells, PTEN deficiency potentiated the synergistic effect of olaparib and cisplatin combination treatment,140 while rucaparib sensitized PTEN-deficient prostate cancer cells to ionizing radiation,141 with both reports highlighting delayed DSB repair kinetics as a likely mechanism.
In the clinical setting, there is anecdotal evidence of successful targeting of PTEN deficiency with PARP inhibitor. Forster et al142 have presented a case study of usage of the PARP inhibitor olaparib in a patient with platinum-responsive metastatic endometrioid endometrial adenocarcinoma. Treatment was initiated following development of brain metastases, on the basis of previous sensitivity with platinum agents, which are highly effective in HR deficiency. A partial response on magnetic resonance imaging was noted at 10 weeks, followed by a progression-free survival of 8 months. Tumor biopsy demonstrated PTEN mutation with wild type BRCA1 and -2 status.142
However, there is not a consensus regarding the synthetic lethality relationship between PTEN deficiency and PARP inhibition. In studies on PTEN-null prostate139 and lung143 cancer cells, no enhanced DNA damaging agent or PARP inhibitor sensitivity was observed. Furthermore, in a Phase I trial in BRCA mutation carriers and sporadic cancer, PTEN status did not correlate with antitumor activity of niraparib.144
Although PARP inhibitor-induced synthetic lethality in PTEN loss has been most widely studied, a recent report from Mereniuk et al145 provides evidence that other proteins within the SSBR pathways may also be valid targets. A forward transfection screen of nearly 7,000 siRNAs was performed using A549 lung cancer cells stably depleted of the BER end-processing enzyme PNKP. This screen identified PTEN as a potential synthetic lethal partner, a result then validated by: 1) repeat siRNA downregulation of PTEN in PNKP-null MCF7 (Michigan Cancer Foundation-7) breast cancer cells; and 2) PNKP inhibitor exposure in PTEN-null HCT116 (colorectal) and PC3 (prostate) cancer cells. Furthermore, PNKP inhibitor treatment in PTEN-deficient cells is associated with accumulation of DSBs, increased apoptosis, and reduced clonogenic survival in a manner analogous to published reports of PARP inhibition in BRCA mutation. The authors hypothesize that PTEN loss is associated with strand-break accumulation that may require PNKP-mediated end processing for repair. Although a mechanism for strand breakage is not presented, sensitivity to PNKP inhibitor could not be abrogated by ectopic RAD51 expression in PTEN-null PC3 cells, in keeping with previous reports that synthetic lethality in this cell line is not mediated via RAD51 loss.
Developing biomarkers for synthetic lethality response
BRCA mutation, although an excellent marker of HR deficiency, comprises a small subset of breast and ovarian patients. Identification of other synthetic lethality relationships is ongoing, as described above. A number of individual DNA repair proteins may therefore be informative regarding HR deficiency and synthetic lethality response (reviewed in Martin et al).130 An alternative approach to identify patients who may benefit from PARP (or BER) inhibition is to use gene expression profiles that predict responsiveness. While data regarding PARP inhibitor response is currently limited, there is a large body of evidence related to anthracycline or platinum sensitivity. As both classes are associated with DSB induction (via intercalation/topoisomerase inhibition and crosslink formation, respectively), it may be predicted that response to such agents would also equate to PARP inhibitor response. By correlating DNA repair gene expression microarray data with anthracycline sensitivity in triple-negative breast cancer146 and platinum sensitivity in epithelial ovarian cancer,147 two groups have been able to develop “BRCAness gene signatures” which reproducibly predict treatment response. In the ovarian cancer study, this was further analyzed in BRCA2-mutated pancreatic cancer cell clones to predict for RAD51 foci formation as a marker of HR, and for sensitivity to PARP inhibition. Independently, RAD51 foci formation in tumor samples has been developed as a functional assay for HR status.148 Primary cultures of epithelial ovarian cancer cells from ascitic fluid can be assessed for γH2AX and RAD51 foci, which correlate with in vitro response to the PARP inhibitor.
Assays have also been developed for monitoring effective PARP inhibition on treatment. In multiple clinical trials, PARP activity in peripheral mononuclear blood cells (measured as cellular levels of poly[ADP]-ribose polymers detected by immunofluorescence or enzyme-linked immunosorbent assay) has been used as a marker of effective inhibition.149,150 Surrogate markers, such as comet assay assessment of DNA damage level, or DSB estimation by RAD51 or γH2AX foci after treatment, have also been used.151 However, it is important to note that Phase II studies of olaparib have indicated that the maximum tolerated dose, determined by conventional dose escalation, may induce a better clinical response than the lowest effective PARP inhibitory dose.105,106
Conclusion
DNA repair mechanisms play an essential role in promoting genomic stability. Defective DNA repair may predispose to cancer. On the other hand, impaired DNA repair capacity in cancer cells may influence a favorable response to chemotherapy and radiotherapy. Recent evidence demonstrates that overexpression of DNA repair factors has prognostic and predictive significance in cancer patients. More recently, DNA repair has emerged as a new area for anticancer drug discovery. Use of DNA repair inhibitors in combination with chemotherapy or radiotherapy can increase cancer cell killing, although combination strategies can lead to profound normal tissue toxicity. The strategy of synthetic lethality to exploit interrelationships between DNA repair pathways appears to bypass many problems associated with combination strategies. The recent success of PARP inhibitors in BRCA-deficient breast and ovarian cancer clearly suggests that additional factors within DNA repair are likely to be promising synthetic lethality targets in the future and have the potential to transform the therapeutic landscape in cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. | |
Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5(3):250–259. | |
Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11(12):1513–1530. | |
Frosina G. Commentary: DNA base excision repair defects in human pathologies. Free Radic Res. 2004;38(10):1037–1054. | |
Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205(2):275–292. | |
Madhusudan S, Hickson ID. DNA repair inhibition: a selective tumour targeting strategy. Trends Mol Med. 2005;11(11):503–511. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | |
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | |
Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34(9):2311–2321. | |
Lambros MB, Natrajan R, Geyer FC, et al. PPM1D gene amplification and overexpression in breast cancer: a qRT-PCR and chromogenic in situ hybridization study. Mod Pathol. 2010;23(10):1334–1345. | |
Eker AP, Quayle C, Chaves I, van der Horst GT. DNA repair in mammalian cells: direct DNA damage reversal: elegant solutions for nasty problems. Cell Mol Life Sci. 2009;66(6):968–980. | |
Lord CJ, Ashworth A. Biology-driven cancer drug development: back to the future. BMC Biol. 2010;8:38. | |
Natrajan R, Weigelt B, Mackay A, et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Res Treat. 2010;121(3):575–589. | |
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. | |
Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85(11):1053–1071. | |
Nilsen H, Krokan HE. Base excision repair in a network of defence and tolerance. Carcinogenesis. 2001;22(7):987–998. | |
Izumi T, Wiederhold LR, Roy G, et al. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193(1–2):43–65. | |
Dianov G, Allinson SL, Budworth H, Sleeth KM. Mammalian base excision repair. In: Caldecott KW, editor. Eukaryotic DNA Damage Surveillance and Repair. Kluwer Academic/Plenium Publishers; 2003:1–22. | |
Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. | |
Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66(6):981–993. | |
Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36(5):425–435. | |
Wilson DM 3rd, Kim D, Berquist BR, Sigurdson AJ. Variation in base excision repair capacity. Mutat Res. 2011;711(1–2):100–112. | |
Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3(6):430–440. | |
Pommier Y, Redon C, Rao VA, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res. 2003;532(1–2):173–203. | |
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619–631. | |
Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35(5):1569–1577. | |
Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol. 2007;27(3):1007–1016. | |
Chen D, Yu Z, Zhu Z, Lopez CD. E2F1 regulates the base excision repair gene XRCC1 and promotes DNA repair. J Biol Chem. 2008;283(22):15381–15389. | |
Sugasawa K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res. 2010;685(1–2):29–37. | |
Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18(1):64–72. | |
de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21(3):453–460. | |
Iorns E, Lord CJ, Grigoriadis A, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS One. 2009;4(4):e5120. | |
Sourisseau T, Maniotis D, McCarthy A, et al. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. Embo Mol Med. 2010;2(4):130–142. | |
Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. | |
Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat Rev Clin Oncol. 2010;7(4):197–208. | |
Martin SA, Lord CJ, Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clin Cancer Res. 2010;16(21):5107–5113. | |
Martin SA, Hewish M, Lord CJ, Ashworth A. Genomic instability and the selection of treatments for cancer. J Pathol. 2010;220(2):281–289. | |
Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. Embo Mol Med. 2009;1(6–7):315–322. | |
Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–650. | |
Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584(17):3703–3708. | |
Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res. 2011;711(1–2):61–72. | |
McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24(11):529–538. | |
de Villartay JP. V(D)J recombination deficiencies. Adv Exp Med Biol. 2009;650:46–58. | |
Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131(2):223–225. | |
Katsura Y, Sasaki S, Sato M, et al. Involvement of Ku80 in microhomology-mediated end joining for DNA double-strand breaks in vivo. DNA Repair (Amst). 2007;6(5):639–648. | |
Schipler A, Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013;41(16):7589–7605. | |
Kavanagh JN, Redmond KM, Schettino G, Prise KM. DNA double strand break repair: a radiation perspective. Antioxid Redox Signal. 2013;18(18):2458–2472. | |
Shi L, Oberdoerffer P. Chromatin dynamics in DNA double-strand break repair. Biochim Biophys Acta. 2012;1819(7):811–819. | |
Murray JM, Stiff T, Jeggo PA. DNA double-strand break repair within heterochromatic regions. Biochem Soc Trans. 2012;40(1):173–178. | |
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. | |
Kobayashi J, Iwabuchi K, Miyagawa K, et al. Current topics in DNA double-strand break repair. J Radiat Res. 2008;49(2):93–103. | |
Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst). 2007;6(7):923–935. | |
Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. | |
Cahill D, Connor B, Carney JP. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci. 2006;11:1958–1976. | |
van den Bosch M, Lohman PH, Pastink A. DNA double-strand break repair by homologous recombination. Biol Chem. 2002;383(6):873–892. | |
Karran P. DNA double strand break repair in mammalian cells. Curr Opin Genet Dev. 2000;10(2):144–150. | |
Featherstone C, Jackson SP. DNA double-strand break repair. Curr Biol. 1999;9(20):R759–R761. | |
Hanada K, Ikeda H. [Double strand break repair via DNA end-joining]. Tanpakushitsu Kakusan Koso. 1999;44(Suppl 12):1838–1844. Japanese. | |
Kanaar R, Hoeijmakers JH, van Gent DC. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8(12):483–489. | |
Tsukamoto Y, Ikeda H. Double-strand break repair mediated by DNA end-joining. Genes Cells. 1998;3(3):135–144. | |
Sargent RG, Brenneman MA, Wilson JH. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol Cell Biol. 1997;17(1):267–277. | |
Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25(16):7158–7169. | |
Lawley PD, Phillips DH. DNA adducts from chemotherapeutic agents. Mutat Res. 1996;355(1–2):13–40. | |
Bogliolo M, Schuster B, Stoepker C, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–806. | |
Kumaresan KR, Sridharan DM, McMahon LW, Lambert MW. Deficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cells. Biochemistry. 2007;46(50):14359–14368. | |
Ho TV, Scharer OD. Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environ Mol Mutagen. 2010;51(6):552–566. | |
McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2(8):483–490. | |
Hinz JM. Role of homologous recombination in DNA interstrand crosslink repair. Environ Mol Mutagen. 2010;51(6):582–603. | |
Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11(7):467–480. | |
Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333(6038):84–87. | |
Nicolay NH, Helleday T, Sharma RA. Biological relevance of DNA polymerase beta and translesion synthesis polymerases to cancer and its treatment. Curr Mol Pharmacol. 2012;5(1):54–67. | |
Yamanaka K, Lloyd RS. Functions of translesion DNA polymerases: implications for cancer risk and opportunities as therapeutic targets. In: Madhusudan S, Wilson DM 3rd, editors. DNA Repair and Cancer: from Bench to Clinic. Boca Raton, USA: CRC Press; 2013. | |
McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18(1):148–161. | |
Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101(3):417–425. | |
Long DT, Kreuzer KN. Fork regression is an active helicase-driven pathway in bacteriophage T4. Embo Rep. 2009;10(4):394–399. | |
Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci. 2003;116(Pt 20):4077–4085. | |
Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicol. 2002;181–182:475–481. | |
Yamtich J, Nemec AA, Keh A, Sweasy JB. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012;8(11):e1003052. | |
Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3(8):998–1001. | |
Bapat A, Fishel M, Kelley MR. Going ape as an approach to cancer therapeutics. Antioxid Redox Signal. 2009;11(3):651–668. | |
Basu B, Yap TA, Molife LR, de Bono JS. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr Opin Oncol. 2012;24(3):316–324. | |
Megnin-Chanet F, Bollet MA, Hall J. Targeting poly(ADP-ribose) polymerase activity for cancer therapy. Cell Mol Life Sci. 2010;67(21):3649–3662. | |
Mizuarai S, Kotani H. Synthetic lethal interactions for the development of cancer therapeutics: biological and methodological advancements. Hum Genet. 2010;128(6):567–575. | |
Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–2737. | |
Ossovskaya V, Li L, Bradley C, Sherman B. Abstract 2311: activity of BSI-201, a potent poly(ADP-ribose) polymerase (PARP1) inhibitor, alone and in combination with topotecan in human ovarian xenografts. 99th AACR Annual Meeting; 2008; San Diego, CA. | |
Davar D, Beumer JH, Hamieh L, Tawbi H. Role of PARP inhibitors in cancer biology and therapy. Curr Med Chem. 2012;19(23):3907–3921. | |
Plummer R, Lorigan P, Evans J, et al. First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM). In: ASCO Annual Meeting. Atlanta, GA; J Clin Oncol. 2006;24:Abstract 8013. | |
AstraZeneca. Efficacy and safety study of olaparib in combination with paclitaxel to treat advanced gastric cancer. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01924533. NLM identifier: NCT01924533. Accessed August 26, 2013. | |
Sanofi-aventis. Sanofi-aventis reports top-line results from phase III study with BSI-201 in metastatic triple-negative breast cancer [press release]. Paris, France: Sanofi-aventis; January 27, 2011. Available from: http://en.sanofi.com/Images/13666_20110127_BSI_en.pdf. Accessed August 26, 2013. | |
Sanofi-aventis. Sanofi provides update on phase 3 studies of two investigational compounds [press release]. Paris, France: Sanofi-aventis; June 3, 2013. Available from: http://en.sanofi.com/Images/33127_20130603_rdupdate_en.pdf. Accessed August 26, 2013. | |
Liu X, Shi Y, Maag DX, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18(2):510–523. | |
Glendenning J, Tutt A. PARP inhibitors – current status and the walk towards early breast cancer. Breast. 2011;20 Suppl 3:S12–S19. | |
Rehman FL, Lord CJ, Ashworth A. Synthetic lethal approaches to breast cancer therapy. Nat Rev Clin Oncol. 2010;7(12):718–724. | |
Tuma RS. Combining carefully selected drug, patient genetics may lead to total tumor death. J Natl Cancer Inst. 2007;99(20):1505–1506, 1509. | |
Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Phamacol. 2008;8(4):363–369. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | |
Miki Y, Swensen J, Shattuckeidens D, et al. A strong candidate for the breast and ovarian-cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. | |
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182. | |
Gottipati P, Vischioni B, Schultz N, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70(13):5389–5398. | |
Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283(5747):593–596. | |
Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39(8):3166–3175. | |
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. | |
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–393. | |
Balmana J, Domchek SM, Tutt A, Garber JE. Stumbling blocks on the path to personalized medicine in breast cancer: the case of PARP inhibitors for BRCA1/2-associated cancers. Cancer Discov. 2011;1(1):29–34. | |
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. | |
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | |
AstraZeneca. Olaparib monotherapy in patients with BRCA mutated ovarian cancer following first line platinum based chemotherapy. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01844986. NLM identifier: NCT01844986. Accessed June 14, 2013. | |
AstraZeneca. Olaparib treatment in BRCA mutated ovarian cancer patients after complete or partial response to platinum chemotherapy. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01874353. NLM identifier: NCT01874353. Accessed August 26, 2013. | |
Drew Y, Ledermann JA, Jones A, et al. Abstract 3104: Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2–mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. ASCO Annual Meeting. Chicago, IL: J Clin Oncol. 2011;29:3104. | |
Schelman WR, Sandhu SK, Moreno Garcia V, et al. First-in-human trial of a poly(ADP)-ribose polymerase (PARP) inhibitor MK-4827 in advanced cancer patients with antitumor activity in BRCA-deficient tumors and sporadic ovarian cancers (SOC). ASCO Annual Meeting. Chicago, IL: J Clin Oncol. 2011;29:Abstract 3102. | |
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | |
Giorgetti G, Galizia E, Bianchi F, et al. Brcaness phenotype and methylation of BRCA1 promoter in sporadic breast cancers. Ann Oncol. 2007;18:52–52. | |
Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–3576. | |
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | |
AstraZeneca. AstraZeneca updates on olaparib and TC-5214 development programmes [press release]. London, UK: AstraZeneca; December 20, 2011. Available from: http://www.astrazeneca.com/Media/Press-releases/Article/20111220-az-updates-olaparib-TC5214-development. Accessed August 29, 2012. | |
Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. | |
Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. | |
Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. | |
Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879–882. | |
Williamson CT, Kubota E, Hamill JD, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. Embo Mol Med. 2012;4(6):515–527. | |
Sultana R, McNeill DR, Abbotts R, et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer. 2012;131(10):2433–2444. | |
Vilar E, Bartnik CM, Stenzel SL, et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011;71(7):2632–2642. | |
Kurosawa A, Saito S, So S, et al. DNA ligase IV and artemis act cooperatively to suppress homologous recombination in human cells: implications for DNA double-strand break repair. PLoS One. 2013;8(8):e72253. | |
Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9(6):1775–1787. | |
Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst). 2008;7(12):2010–2019. | |
Sultana R, Abdel-Fatah T, Abbotts R, et al. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 2013;73(5):1621–1634. | |
Seo Y, Kinsella TJ. Essential role of DNA base excision repair on survival in an acidic tumor microenvironment. Cancer Res. 2009;69(18):7285–7293. | |
Bindra RS, Gibson SL, Meng A, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65(24):11597–11604. | |
Bindra RS, Schaffer PJ, Meng A, et al. Alterations in DNA repair gene expression under hypoxia: elucidating the mechanisms of hypoxia-induced genetic instability. Ann N Y Acad Sci. 2005;1059:184–195. | |
Martin SA, McCabe N, Mullarkey M, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–248. | |
Gupta A, Yang Q, Pandita RK, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8(14):2198–2210. | |
Puc J, Keniry M, Li HS, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7(2):193–204. | |
Shen WH, Balajee AS, Wang JL, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. | |
Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2013; 319(2):125–129. | |
Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–5453. | |
Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121(Pt 3):249–253. | |
Dedes KJ, Wetterskog D, Mendes-Pereira AM, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75. | |
McEllin B, Camacho CV, Mukherjee B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–5464. | |
Fraser M, Zhao H, Luoto KR, et al. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012;18(4):1015–1027. | |
Minami D, Takigawa N, Takeda H, et al. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res. 2013;11(2):140–148. | |
Chatterjee P, Choudhary GS, Sharma A, et al. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS One. 2013;8(4):e60408. | |
Forster MD, Dedes KJ, Sandhu S, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011;8(5):302–306. | |
Pappas G, Zumstein LA, Munshi A, Hobbs M, Meyn RE. Adenoviral-mediated PTEN expression radiosensitizes non-small cell lung cancer cells by suppressing DNA repair capacity. Cancer Gene Ther. 2007;14(6):543–549. | |
Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–892. | |
Mereniuk TR, El Gendy MA, Mendes-Pereira AM, et al. Synthetic lethal targeting of PTEN-deficient cancer cells using selective disruption of polynucleotide kinase/phosphatase. Mol Cancer Ther. 2013;12(10):2135–2144. | |
Rodriguez AA, Makris A, Wu MF, et al. DNA repair signature is associated with anthracycline response in triple negative breast cancer patients. Breast Cancer Res Treat. 2010;123(1):189–196. | |
Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. | |
Mukhopadhyay A, Elattar A, Cerbinskaite A, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res. 2010;16(8):2344–2351. | |
Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–6591. | |
Lubbers LS, Rowe BA, Hodge LM, et al. PISA, a novel pharmacodynamic assay for assessing poly(ADP-ribose) polymerase (PARP) activity in situ. J Pharmacol Toxicol Methods. 2010;61(3):319–328. | |
Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–7923. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.