")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
DNA Methylation Profiling in a Cigarette Smoke-Exposed Mouse Model of Airway Inflammation
Authors Li P , Peng J, Chen G, Chen F, Shen Y, Liu L, Chen L
Received 7 April 2022
Accepted for publication 12 September 2022
Published 1 October 2022 Volume 2022:17 Pages 2443—2450
DOI https://doi.org/10.2147/COPD.S369702
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Russell
Ping Li,1,* Junjie Peng,1,* Guangxi Chen,1,2,* Fangying Chen,1,3 Yongchun Shen,1 Lin Liu,4 Lei Chen1
1Laboratory of Pulmonary Diseases and Department of Respiratory and Critical Care Medicine, West China Hospital, West China School of Medicine, Sichuan University, Chengdu, People’s Republic of China; 2Department of Sleep Medicine, Jiujiang First People’s Hospital, Jiujiang, People’s Republic of China; 3Department of Tuberculosis, the Third People’s Hospital of Tibet Autonomous Region, Lhasa, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, 363 Hospital, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Chen, Department of Respiratory and Critical Care Medicine, West China Hospital, West China School of Medicine, Sichuan University, Chengdu, People’s Republic of China, Email [email protected] Lin Liu, Department of Respiratory and Critical Care Medicine, 363 Hospital, Chengdu, People’s Republic of China, Email [email protected]
Purpose: DNA methylation, a major epigenetic modification, has been documented to play an important role in chronic obstructive pulmonary disease (COPD). In this study, we aimed to profile the DNA methylation patterns in a mouse model of airway inflammation induced by cigarette smoke (CS), a foremost risk factor of COPD.
Material and Methods: To establish a model of airway inflammation, wild-type mice were exposed to mainstream CS or room air for 2 hours twice daily, 6 days per week for consecutive 4 weeks. Lung tissues of the mice were collected for genome-wide DNA methylation analysis by liquid hybridization capture-based bisulfite sequencing, which were used for intersection analysis with gene expression by cDNA microarray to identify candidate methylated genes. Then, functional enrichment analyses with protein–protein interaction (PPI) network regarding these genes were conducted to explore the potential mechanisms.
Results: After 4-week CS exposure, the level of DNA methylation accompanied by a subacute airway inflammation was markedly enhanced, and 2002 differentially methylated genes (DMGs) were annotated, including 565 DMGs contained methylations in gene promoters, which were used for intersection with the differentially expressed genes. Then, 135 candidate methylated genes were further selected by the intersection, among which 58 genes with functional methylated modification were finally identified. Further analyses revealed candidate methylated genes were significantly enriched in a complicated network of signals and processes, including interleukins, toll-like receptors, T-cells differentiation, oxidative stress, mast cells activation, stem cells proliferation, etc., as well as the 58 functional methylated genes were partially located at key positions in PPI network, especially CXCL1, DDX58 and JAK3.
Conclusion: This study suggests CS exposure significantly enhances DNA methylated level, and the potential functional methylated genes are closely related to complicated inflammatory-immune responses, which may provide some new experimental evidence in understanding the epigenetic mechanisms of CS-induced airway inflammation in COPD.
Keywords: chronic obstructive pulmonary disease, airway inflammation, cigarette smoke, DNA methylation, liquid hybridization capture-based bisulfite sequencing
Introduction
Chronic obstructive pulmonary disease (COPD) is a common, heterogeneous chronic respiratory disease characterized by persistent respiratory symptoms and airflow limitation.1 Cigarette smoke (CS) has been regarded as a major environmental risk factor for COPD. CS-induced airway inflammation significantly contributes to COPD pathogenesis,1 but its molecular mechanisms are not fully understood.
DNA methylation, as a crucial epigenetic modification, plays a key role in regulating tissue-specific gene expression and complex traits.2 In recent years, some studies have documented that abnormal DNA methylation may lead to alternative expression of specific genes, especially the pro- and anti-inflammatory genes,3 which is closely associated with COPD susceptibility, lung function decline and exacerbation.4,5 Moreover, it is increasingly recognized that DNA methylation could be an important link between environmental and genetic factors.6,7 Emerging evidences have demonstrated environmental stimuli such as CS, could alter gene methylation patterns,8 and CS-induced DNA methylated modification was reported in several studies.9,10 However, the DNA methylation patterns in CS-induced airway inflammation are not well-profiled.
Therefore, we established a CS-exposed mouse model of airway inflammation to profile the genome-wide DNA methylation by liquid hybridization capture-based bisulfite sequencing (LHC-BS) and uncover the underlying mechanisms.
Materials and Methods
Animal Model
In our former article, a CS-exposed mouse model of airway inflammation has been established.11 Briefly, wild-type (WT) C57BL/6 mice (7–9 weeks old, 20–22 g weight) were divided into two experimental groups: WT (control) group and CS group, n=10 mice per group. All mice were specific pathogen-free and kept on a 12-h light/12-h dark cycle, at a room temperature of 22±2℃, with free access to food and water. The mice in CS group were exposed to Marlboro cigarettes (Marlboro®, Philips Morris, United States, with 1.0 mg nicotine and 11 mg tar per cigarette) with mainstream CS for 2 hours twice daily, 6 days per week for consecutive 4 weeks using a Baumgartner-Jaeger CSM2082i automated cigarette smoking machine (CH Technologies, West-Wood, NJ, USA), which output a smoke concentration of 320–340 mg TPM/m3. The control mice were exposed to filtered air according to the same schedule. Thereafter, all mice were anesthetized intraperitoneally with pentobarbital sodium and sacrificed by femoral artery transection. Hematoxylin-eosin (HE) stain in mouse lung tissues was used to display the CS-induced airway inflammation. The study protocol was in accordance with the European Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (1986) and approved by the Panel on Laboratory Animal Care of West China School of Medicine of Sichuan University (approval number: 2020394A).
LHC-BS
Lung tissues from the mice (n=3 per group) were randomly selected for genomic DNA extraction by DNeasy Blood Tissue Kit (Qiagen, Germany) and subsequently used for library construction. In brief, 1μg genomic DNA was sonicated to random fragments with approximately 200–300bp. After purification, the DNA library was prepared using SureSelectXT Mouse methyl-seq Library Prep Kit (Agilent Technologies, USA). Afterward, DNA was bisulfite-treated using EZ DNA Methylation-GoldTM Kit (Zymo Research, Cat.D5006) according to the supplier’s instruction. Finally, purified libraries were quantified by the Bioanalyzer analysis system (Agilent) and real-time PCR assay. The libraries were then sequenced on Illumina Novaseq PE150, the detailed process was described previously.12
DNA Methylation Data Analyses
The DNA methylation data were analyzed as described in detail previously.13 Briefly, after removing low-quality reads, then the cleans LHC-BC reads were aligned to the reference genome by Bismark software v0.19.0 with bowtie2 (version 2.3.4.2).14 The DNA methylation rate of cytosine was assessed by the number of supporting methylated readings divided by the total number of readings covering the cytosine. Methylation levels were analyzed using the R package, methylKit (version v1.6.1)15 and eDMR16 for differentially methylated regions (DMRs) of interest (eg, promoters, CpG islands), which may reveal more biological relevance. DMR was defined as adjusted P<0.05 and the absolute differential methylation levels (absolute meth.diff >5%). Finally, the related differentially methylated genes (DMGs) were located and annotated in the DMRs by the ChIPseeker software.
cDNA Microarray
The data of gene expression by cDNA microarray have been informed in our former study.11 Briefly, according to the manufacturer protocols, the total RNA from mouse lung tissue was extracted using the miRNeasy kit (Qiagen). After purification, the total RNA was generated to cDNA, which was biotin-labeled library to be hybridized on GeneChip Mouse Gene 2.0 STArray (Affymetrix, USA) covering more than 39,000 transcripts. Next, Affymetrix GeneChip Scanner 3000 7G (Affymetrix) was used for signal detection. The microarray results were assessed by Expression Console software (Affymetrix) and Transcriptome Analysis Console software (Affymetrix). Then, the expression of each gene was estimated by the lognormal-normal model. Only differentially expressed genes (DEGs) at least 1.2-fold upregulated or downregulated were used for the further study.
Intersection Analyses
To select the candidate genes with potential functional methylated modification, CS-induced (CS vs WT) DMGs located in promoters and CS-induced DEGs were intersected. The functional methylated genes were finally identified according to the reversed effects of methylation in promoters on gene transcription.
Functional Enrichment Analyses
To identify biological function of the candidate genes, especially those with functional methylated modification, also called functional methylated genes, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses, using ClueGO (Version 2.5.8)17 which integrates GO terms as well as KEGG pathways and creates a functionally organized GO/pathway term network within Cytoscape (Version 3.9).17 The GO and KEGG enrichment analyses were used with a kappa score of 0.5, showing ontologies with P values <0.05. Other settings were all default.
Protein–Protein Interaction (PPI) Network
To further identify the interactions among the candidate genes, especially the functional methylated genes, PPI network analysis was performed using STRING18 and displayed using CytoScape (Version 3.9).19 To calculate the degree of connectivity among the candidate genes in PPI network, the plug-in “Cytohubba” of Cytoscape20 was conducted to score each node gene, using 10 different algorithms, and the top 10 node genes by each algorithm were obtained, respectively (Table S1).
Statistical Analysis
Data for the methylation level (%) were presented as median (interquartile range, IQR). All the statistical analyses in this study were performed with R software packages (R Foundation for Statistical Computing, Vienna, Austria). P<0.05 was considered statistically significant.
Results
DNA Methylation Data
Based on the LHC-BS method, we generated 15 Gbp raw sequence data on average for each sample (Table S2). Over 79% of mapped reads covered ~77% of the target regions, with an average of 57× sequencing depth per CpG and 13% duplication rata. For the downstream analysis, the duplicated sequence reads were filtered.
CS Exposure Induced Airway Inflammation and DNA Functional Methylated Modifications
The 4-week consecutive CS exposure significantly induced a subacute airway inflammation (Figure 1A) and meanwhile increased the DNA methylation level (Figure 1B). Then, 2002 DMGs were annotated with 1009 hypomethylated, 853 hypermethylated and 140 both hypo- and hypermethylated (Figure 1C). It is well-known that DNA methylation occurs almost in CpG islands that are primarily located in promoters, which is closely correlated with gene expression regulation according to a counter-regulation rule.21 Subsequently, among these 2002 DMGs, 565 DMGs (363 hypomethylated, 196 hypermethylated, 6 both hypo- and hypermethylated, Figure 1D) contained methylations in gene promoters, which were intersected with the DEGs. Finally, 135 candidate methylated genes were selected after the intersection analysis (Figure 1E), and 58 genes from the 135 candidate genes, with functional methylated modification in promoters were identified.
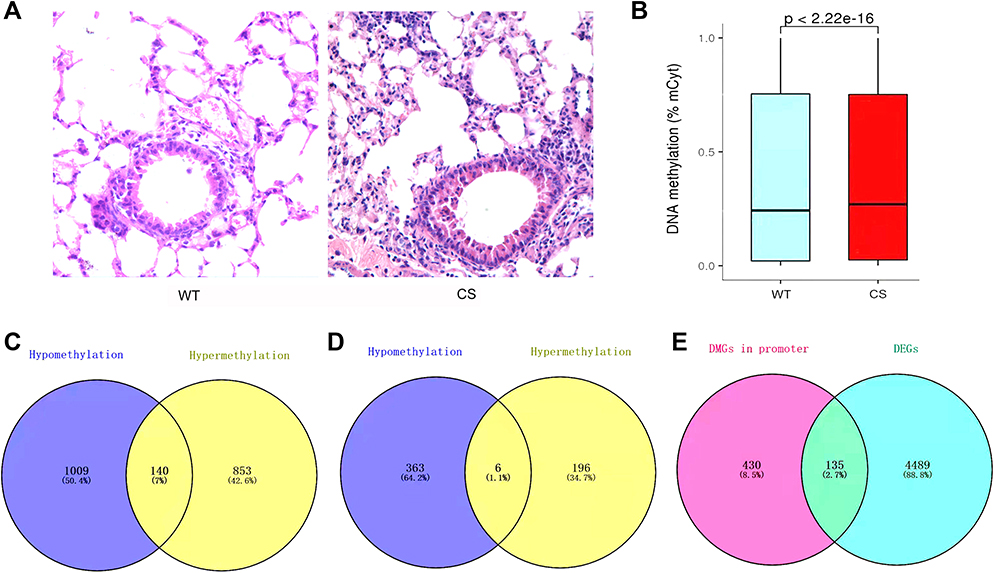 |
Figure 1 (A) The representative images of hematoxylin-eosin (HE) stain in mouse lung tissues (×400). (B) The methylation level (%) in wild-type (WT) and cigarette smoke (CS) groups. The medians with interquartile ranges (IQRs) for the WT and CS groups were 0.243 (0.733) and 0.27 (0.726) respectively. (C) Venn diagram of DMGs between WT and CS groups. (D) Venn diagram of the differentially methylated genes (DMGs) in promoter. (E) Venn diagram of intersection between DMGs in promoter and differentially expressed genes (DEGs). |
The Functional Methylated Genes Were Correlated with CS-Induced Inflammatory-Immune Responses
Functional enrichment analyses indicated that the 135 candidate genes were significantly correlated with immune-inflammatory responses via a complicated network of signals and processes (Table S3), including interleukins, toll-like receptors, T-cells differentiation, oxidative stress, mast cells activation, stem cells proliferation, etc (Figure 2), and the candidate genes, especially the 58 functional methylated genes were in part located at key positions in PPI network (Figure 3), and further calculation by 10 different algorithms indicated the top 10 candidate genes, respectively. After taking intersection of the top 10 candidate genes in each algorithm, CXCL1, DDX58 and JAK3 were identified as the hub genes with functional methylated modification (Table S1).
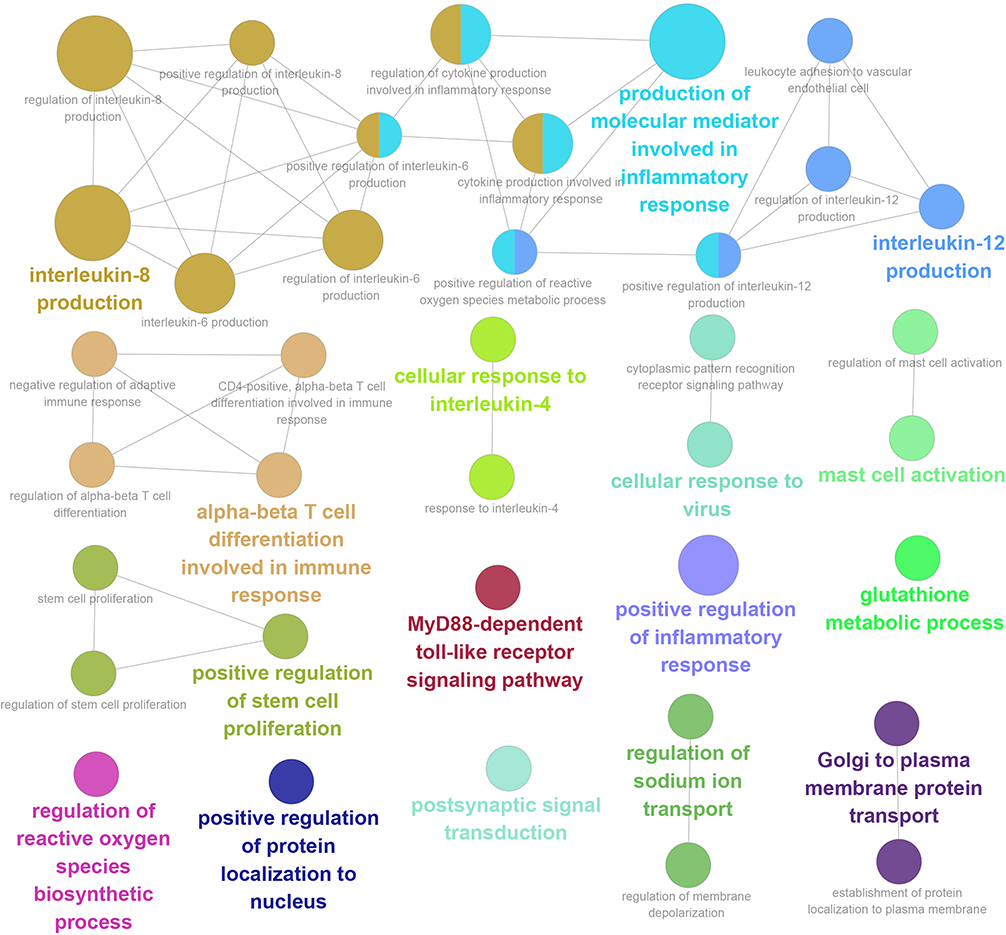 |
Figure 2 Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) in the candidate genes. Each node represented a signal/process, and the same signals/processes were showed with the same colors. Cluster analyses were performed on these signals/processes, and the significant signals/processes were indicated with bold and bigger nodes in the same cluster. |
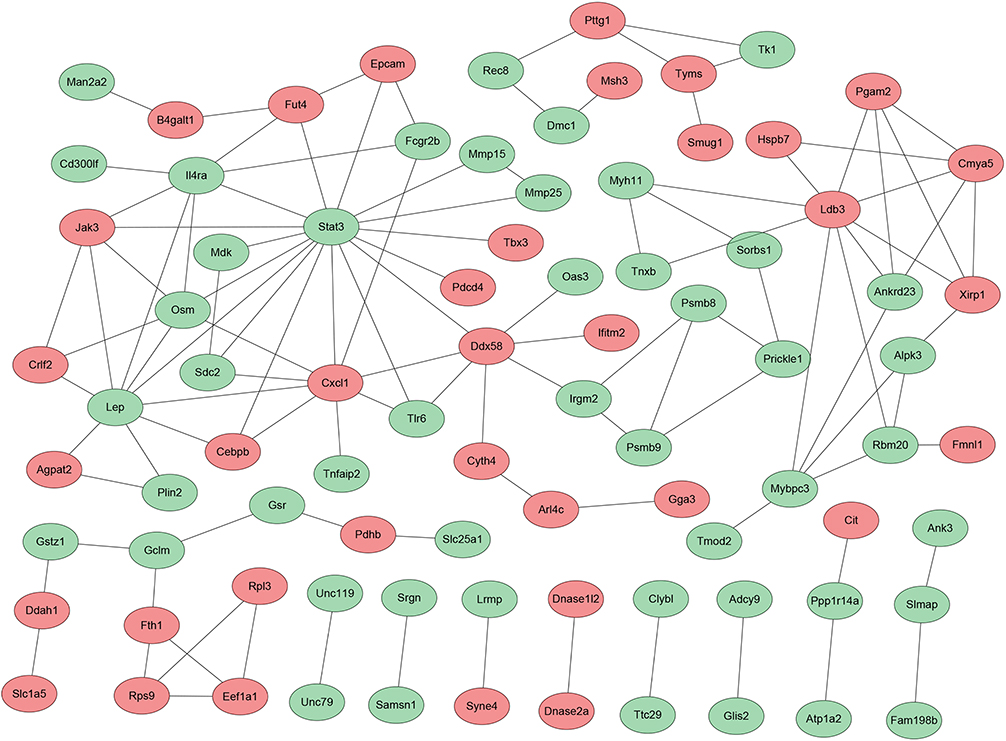 |
Figure 3 Protein–protein interaction network of the candidate genes. Each node represented a candidate gene and the undirected link between two nodes indicated an edge. Red circles pionted the functional methylated genes in promoter. |
Discussion
CS, as a foremost risk factor for COPD, has been shown to alter DNA methylation, which is associated with the initiation and progression of COPD.9,10 CS-induced DNA methylation plays a regulatory role in COPD, whereas the mechanisms remain not fully explained. In the present study, 135 overlapping candidate genes, regarding airway inflammation induced by CS exposure, were initially selected using an intersection model, and the functional DNA methylated modification in 58 genes were subsequently identified, which might significantly contribute to CS-induced airway inflammation in COPD. In addition, functional enrichment analyses suggested these candidate genes were significantly related to the immune-inflammatory responses via a complicated network of signals and processes, including interleukins, toll-like receptors, T-cells differentiation, oxidative stress, mast cells activation, stem cells proliferation, etc. All these signals and processes were reported to be contributors to inflammatory injury in COPD,10,22,23 and our data further indicated a potentially important role of methylated modifications in these signals/processes in CS-induced airway inflammation.
Noticeably, the present study novelly reported the functional methylated modifications in genes related to mast cells activation and stem cells proliferation induced by CS exposure. It has been documented that CS exposure can lead to an increase in the number of mast cells in the bronchial mucosa,24 which plays a crucial role in excessive activation of innate immune system and is associated with lung function decline and airway/vascular remodeling in COPD.25–27 On the other hand, since the failure of lung regeneration is considered as a major mechanism of inflammatory injury in COPD,28 stem cells significantly contribute to the maintenance and repair of lung tissue,29 partly owing to the anti-inflammatory effects of stem cells via an increase in M2 macrophages, resulting in inflammation resolution and repair enhancement.30 However, the mechanisms regarding mast cells and stem cells in COPD have not been well-elucidated. As suggested in this study, functional methylated genes related to mast cells activation and stem cells proliferation were identified, which would be potential targets for intervention in CS-induced airway inflammation in COPD.
In addition, based on the PPI network analysis with 10 different algorithms, the hub genes with functional methylated modification were further identified in this study, including CXCL1, DDX58 and JAK3, which might significantly contribute to CS-induced airway inflammation in COPD. CXCL1, a member of the chemokine subfamily of CXC, is markedly increased in the lungs of COPD patients, which is correlated with the degree of airflow limitation and the increased proportion of neutrophils, facilitating neutrophilic inflammation of COPD.22,31,32 In this study, functional methylated CXCL1 was enriched in positive regulation of stem cell proliferation and reactive oxygen species metabolic process. DDX58, also called RIG-I, is a pattern recognition receptor (PRR) involved in viral double-stranded (ds) RNA recognition and regulation of antiviral innate immune response,33 which senses cytoplasmic viral nucleic acids and activates a downstream signaling cascade leading to the production of type I interferons and pro-inflammatory cytokines.34,35 In addition to the cellular response to virus, our study indicated functional methylated DDX58 was closely related to the production and regulation of IL-6 and IL-8. JAK3 is a member of Janus kinase (JAK) family of tyrosine kinases involved in cytokine receptor-mediated intracellular signal transduction. It is predominantly expressed in immune cells and participates in inflammatory-immune responses through tyrosine phosphorylation of interleukin receptors, which is associated with COPD.36,37 Functional methylated JAK3 participated in IL-12 regulation and production, cellular response to IL-4 and regulation of T cell differentiation. Moreover, functional methylated DDX58 and JAK3 might have potential crosstalk with another PRR, TLR6. Importantly, these hub genes (CXCL1, DDX58 and JAK3) have been reported to be targets of DNA methylated modification in schizophrenia,38 hand, foot and mouth disease (HFMD),39 and breast cancer.40 The present results further suggested functional DNA methylation modification in these hub genes might play a crucial role in CS-induced airway inflammation in COPD.
However, two limitations in this study should be considered. First, the sample size of each group was relatively small, although the minimum requirement for biological repeat was reached. Second, the main findings were needed to be verified in COPD patients in the future.
Overall, in this CS-exposed mouse model of airway inflammation, the DNA methylated level was significantly enhanced, and the functional methylated genes were closely related to a complicated network of signals and processes associated with inflammatory-immune responses, which might provide some new experimental evidence regarding epigenetic mechanisms underlying CS-induced airway inflammation in COPD.
Abbreviations
COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; CXCL1, C-X-C Motif Chemokine Ligand 1; DDX58, DExD/H-Box Helicase 58; DEGs, differentially expressed genes; DMGs, differentially methylated genes; DMRs, differentially methylated regions; GO, Gene Ontology; HFMD, hand, foot and mouth disease; IQR, interquartile range; JAK3, Janus Kinase 3; KEGG, Kyoto Encyclopedia of Genes and Genomes; LHC-BS, Liquid hybridization capture-based bisulfite sequencing; PPI, protein–protein interaction; PRR, pattern recognition receptor; TLR6, Toll Like Receptor 6; TPM, total particulate matter.
Data Sharing Statement
The datasets generated and analyzed during the present study are available from the corresponding authors on reasonable requests.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported in part by grant 81970040 from the National Natural Science Foundation of China and grant 18PJ410 from the Health and Family Planning Commission of Sichuan Province.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. Gold executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
2. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi:10.1038/nbt.1685
3. Sundar IK, Mullapudi N, Yao H, Spivack SD, Rahman I. Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Curr Opin Pulm Med. 2011;17(4):279–285. doi:10.1097/MCP.0b013e3283477533
4. Busch R, Qiu WL, Lasky-Su J, Morrow J, Criner G, DeMeo D. Differential DNA methylation marks and gene comethylation of COPD in African-Americans with COPD exacerbations. Respir Res. 2016;17(15):143. doi:10.1186/s12931-016-0459-8
5. de Vries M, van der Plaat DA, Nedeljkovic I, et al. From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res. 2018;19(1):212. doi:10.1186/s12931-018-0904-y
6. Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics. 2016;11(10):730–739. doi:10.1080/15592294.2016.1226451
7. Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi:10.1038/nature05919
8. Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi:10.1371/journal.pone.0063812
9. Cheng L, Liu J, Li B, Liu S, Li X, Cigarette Smoke-Induced TH. Hypermethylation of the GCLC gene is associated with COPD. Chest. 2016;149(2):474–482. doi:10.1378/chest.14-2309
10. Vucic EA, Chari R, Thu KL, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50(5):912–922. doi:10.1165/rcmb.2013-0304OC
11. Chen M, Wang T, Shen Y, et al. Knockout of RAGE ameliorates mainstream cigarette smoke-induced airway inflammation in mice. Int Immunopharmacol. 2017;50:230–235. doi:10.1016/j.intimp.2017.06.018
12. Gao F, Wang J, Ji G, et al. Clustering of cancer cell lines using a promoter-targeted liquid hybridization capture-based bisulfite sequencing approach. Technol Cancer Res Treat. 2014. doi:10.7785/tcrt.2012.500416
13. Li P, Wang T, Chen M, Chen J, Shen Y, Chen L. RAGE-mediated functional DNA methylated modification contributes to cigarette smoke-induced airway inflammation in mice. Biosci Rep. 2021;41(7). doi:10.1042/bsr20210308
14. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. doi:10.1093/bioinformatics/btr167
15. Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. doi:10.1186/gb-2012-13-10-R87
16. Li S, Garrett-Bakelman FE, Akalin A, et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinform. 2013;14(Suppl5):S10. doi:10.1186/1471-2105-14-s5-s10
17. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi:10.1093/bioinformatics/btp101
18. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi:10.1093/nar/gkw937
19. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
20. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl4):S11. doi:10.1186/1752-0509-8-s4-s11
21. Bender CM, Gonzalgo ML, Gonzales FA, Nguyen CT, Robertson KD, Jones PA. Roles of cell division and gene transcription in the methylation of CpG islands. Article. Mol Cell Biol. 1999;19(10):6690–6698. doi:10.1128/MCB.19.10.6690
22. Hikichi M, Mizumura K, Maruoka S, Gon Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis. 2019;11(S17):S2129–S2140. doi:10.21037/jtd.2019.10.43
23. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
24. Ekberg-Jansson A, Amin K, Bake B, et al. Bronchial mucosal mast cells in asymptomatic smokers relation to structure, lung function and emphysema. Respir Med. 2005;99(1):75–83. doi:10.1016/j.rmed.2004.05.013
25. Beckett EL, Stevens RL, Jarnicki AG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol. 2013;131(3):752–762. doi:10.1016/j.jaci.2012.11.053
26. Andersson CK, Mori M, Bjermer L, Löfdahl CG, Erjefält JS. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(3):206–217. doi:10.1164/rccm.200906-0932OC
27. Roos AB, Mori M, Gura HK, et al. Increased IL-17RA and IL-17RC in end-stage COPD and the contribution to mast cell secretion of FGF-2 and VEGF. Respir Res. 2017;18(1):48. doi:10.1186/s12931-017-0534-9
28. Coppolino I, Ruggeri P, Nucera F, et al. Role of stem cells in the pathogenesis of chronic obstructive pulmonary disease and pulmonary emphysema. Copd. 2018;15(5):536–556. doi:10.1080/15412555.2018.1536116
29. Kajstura J, Rota M, Hall SR, et al. Evidence for human lung stem cells. N Engl J Med. 2011;364(19):1795–1806. doi:10.1056/NEJMoa1101324
30. Glassberg MK, Csete I, Simonet E, Elliot SJ. Stem cell therapy for COPD: hope and exploitation. Chest. 2021;160(4):1271–1281. doi:10.1016/j.chest.2021.04.020
31. Charo IF, Ransohoff RM. Mechanisms of disease - The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. doi:10.1056/NEJMra052723
32. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. doi:10.1172/jci36130
33. Cadena C, Ahmad S, Xavier A, et al. Ubiquitin-dependent and -independent roles of E3 ligase RIPLET in innate immunity. Cell. 2019;177(5):1187–1200.e16. doi:10.1016/j.cell.2019.03.017
34. Saito T, Hirai R, Loo YM, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104(2):582–587. doi:10.1073/pnas.0606699104
35. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi:10.1016/j.cell.2005.08.012
36. Raivola J, Haikarainen T, Abraham BG, Silvennoinen O. Janus kinases in leukemia. Cancers. 2021;13(4):800. doi:10.3390/cancers13040800
37. Korytina GF, Akhmadishina LZ, Kochetova OV, Aznabaeva YG, Zagidullin SZ, Victorova TV. Inflammatory and immune response genes polymorphisms are associated with susceptibility to chronic obstructive pulmonary disease in Tatars population from Russia. Biochem Genet. 2016;54(4):388–412. doi:10.1007/s10528-016-9726-0
38. Zhou C, Chen J, Tang X, et al. DNA methylation and gene expression of the chemokine (C-X-C motif) ligand 1 in patients with deficit and non-deficit schizophrenia. Psychiatry Res. 2018;268:82–86. doi:10.1016/j.psychres.2018.06.067
39. Li YP, Liu CR, Deng HL, et al. DNA methylation and single-nucleotide polymorphisms in DDX58 are associated with hand, foot and mouth disease caused by enterovirus 71. PLoS Negl Trop Dis. 2022;16(1):e0010090. doi:10.1371/journal.pntd.0010090
40. Liu F, Wu H. Identification of prognostic biomarkers and molecular targets among JAK family in breast cancer. J Inflamm Res. 2021;14:97–114. doi:10.2147/jir.S284889
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.