")
Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
DNA methylation: its role in transcriptional regulation and association with lung cancer
Authors Pantazi P, Acha-Sagredo A, Papaioannou A, Nikolaidis G, Liloglou T
Received 15 September 2014
Accepted for publication 14 October 2014
Published 13 January 2015 Volume 2015:5 Pages 11—30
DOI https://doi.org/10.2147/RRBC.S53690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Nikolay Dokholyan
Paschalia Pantazi,1,2 Amelia Acha-Sagredo,1,3,4 Alexandra Papaioannou,2 Georgios Nikolaidis,1,2 Triantafillos Liloglou1
1Department of Molecular and Clinical Cancer Medicine, University of Liverpool, Liverpool, UK; 2Division of Basic Science, University of Crete Medical School, Heraklion, Greece; 3Department of Stomatology II, Oral Medicine and Pathology UFI 11/25, University of the Basque Country, Leioa, Spain; 4IKERBASQUE, Basque Foundation for Science, Bilbao, Spain
Abstract: DNA methylation is a fundamental biochemical modification that in conjunction with noncoding RNAs, histone modifications, and chromatin remodeling institutes the epigenetic machinery of mammalian cells. As a result of the second decade of intense epigenetic research and its role in human disease, substantial new mechanisms have been uncovered. However, it is well acknowledged that we have just scratched the tip of the iceberg. Epigenetic deregulation appears to be one of the foundations of major human diseases, including lung cancer, which is the most frequent cause of cancer-related deaths. Currently, significant effort is made to dissect the role of epigenetic deregulation in the development of lung cancer and utilize this knowledge in diagnostic and therapeutic applications. Taking advantage of the recent technologies in genomic research, many studies have been conducted to discover and validate abnormal DNA methylation patterns that may shed light on cancer development pathways and open new areas of potential clinical exploitation. In this article, we provide basic information of the DNA methylation involved in gene regulation and review the latest literature on potential relevant translational applications in lung cancer. Particular emphasis is given to the development and validation of DNA methylation biomarkers that may assist in the clinical management of lung cancer.
Keywords: epigenetics, biomarkers, body fluids
DNA methylation in transcriptional regulation
The term “Epigenetics” is used to define the heritable changes in phenotype or gene expression caused by mechanisms not involving primary DNA sequence alterations.1 These mechanisms include DNA methylation, histone tail modifications, nucleosomal rearrangements/remodeling, and noncoding RNAs (ncRNAs). These mechanisms interact with each other and follow a precise molecular orchestration in order to maintain or change epigenetic marks throughout the genome; this crosstalk seems to be pivotal for the maintenance of cell differentiation, growth, and homeostasis.2,3 There are many enzymes mediating this crosstalk. For instance, MeCP2, a methyl-binding domain-containing protein, decks on methylated DNA and recruits co-repressor complexes such as histone methyltransferases and histone deacetylases; thus, establishing an inactive chromatin state that hinders transcription factor accessibility and leads to loss of gene expression.4,5 It has been shown that DNA methylation status can determine nucleosome occupancy and further influence gene expression.6 Recently, it has been shown that long ncRNAs also regulate the interaction between chromatin and remodeling complexes, such as PRC2,7 by bringing them to specific genomic loci and shifting their position when necessary.8
DNA methylation is the most studied epigenetic modification in humans. It designates the addition of a methyl group (CH3) at position 5′ of the amino base cytosine within CpG dinucleotides,9 leading to the formation of 5-methylcytosine. This reaction is catalyzed by DNA methyltransferases (DNMTs), which can act de novo (mainly DNMT3a and DNMT3b) or maintain the existing methylation patterns (mainly DNMT1)10–12 assisted by accessory proteins such as UHRF1. Demethylation can be passive when the maintenance mechanism fails to copy the pattern to the newly synthesized strand following DNA replication, or an active process that occurs via a 5-hydroxymethyl cytosine intermediate catalyzed by TET proteins and encompasses the action of a number of accessory enzymes.11–17 It seems that the first oxidation of 5-methylcytosine to 5-hydroxymethyl cytosine is carried out by TET113 while several ways of thereafter transforming 5-hydroxymethyl cytosine to cytosine have been suggested and involve both nuclear and base excision repair pathways.15,16
DNA methylation is implicated in various physiological processes, such as cell differentiation, X-chromosome inactivation, gene imprinting, and genomic stability. DNA methylation alterations in human disease are well established to date,18 although the list of epigenetic-driven diseases is growing fast. Global hypomethylation19 and loci-specific hypermethylation20 have been frequently reported in cancer.21 Originally, changes in DNA methylation were studied in gene promoters, where hypermethylation was associated with downregulation of tumor suppressor genes and hypomethylation triggers upregulation of oncogenes.22 More recently, taking advantage of the development of genome-wide analysis techniques, intra- and intergenic methylation changes have also been described. Alterations of DNA methylation within the gene body can affect alternative intragenic promoters and enhancers, ncRNA expression, transposable elements mobility, alternative splicing, or polyadenylation.23,24 Intergenic methylation changes can also affect enhancers or insulators; therefore, leading to gene silencing or activation, respectively,25 or alter the expression of intergenic ncRNAs.23,24 The involvement of DNA methylation in transcriptional regulation is based on the chromatin conformation changes that are associated with it. As mentioned DNA methylation occurs in combination with other epigenetic mechanisms and determines whether a particular stretch of DNA is loose and accessible to the transcriptional machinery or is tightly wrapped around nucleosomes in a dense chromatin form prohibitive of transcription factor binding.23 The most exciting point is probably that this relationship is bidirectional; it is now known that certain transcription factors can place epigenetic marks upon binding to DNA and can therefore alter DNA methylation.26
In biomarker studies, both DNA methylation and messenger RNA (mRNA) expression compete for a place in molecular diagnostics. The main disadvantage of detecting DNA methylation only is that it does not indicate the level of expression deregulation of the affected gene(s). One would argue that expression deregulation ultimate proof is only demonstrated by protein assays; however, determining mRNA expression is providing good preliminary evidence. In the case of DNA methylation, there is no such evidence and one can hypothesize the outcome on RNA.
On the other hand, DNA methylation analysis has significant analytical and biological advantages. DNA methylation changes require at least one cell cycle, which makes this modification robust following sampling; RNA expression profiles deteriorate quickly upon removal of the sample from the body. In addition, DNA is a much more stable chemical entity than RNA; therefore, DNA methylation assays do not need special sampling requirements and are applicable to archival material, where mRNA analysis can be particularly challenging.
Lung cancer biomarkers
Lung cancer is one of the most frequently diagnosed cancers in Europe27 and the US,28 accounting for 13% of all new cases of cancer in the world.29 Lung cancer is divided into two main types: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC). NSCLC represents approximately 85% of lung tumors with adenocarcinoma, squamous cell carcinoma, and large cell carcinoma representing the main histological subtypes. The precise histomolecular classification of lung tumors plays a critical role in personalized medicine and effective treatment of patients.30 Lung cancer still remains the leading cause of cancer-related deaths worldwide.29 This is mainly due to very late diagnosis,31 and there are worldwide efforts from the lung cancer research community to introduce national screening strategies.32 There is a consensus that implementation of molecular markers can assist clinical management of lung cancer. Multiple studies focused on the development of biomarkers for early detection,33–36 metastatic risk assessment,37,38 and therapeutic stratification of patients.39–41
According to the Biomarkers Definitions Working Group, the term “biomarker” is used to describe a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention. Biomarker qualification involves four major steps: 1) discovery phase, 2) experimental validation, 3) preclinical validation, and 4) clinical validation.31,42 Numerous studies describe new biomarkers and their potential use in the clinic;43,44 however, very few of them manage to reach clinical validation.35 The main obstacles are the poor reproducibility of the studies, sample heterogeneity, methodological biases, quality control implementation, and significant interlaboratory variability in experimental performance and data analysis.45 To date, biomarkers for lung cancer are mainly used to define and monitor therapy (eg, EGFR, KRAS, or ALK mutations).46 DNA-based biomarkers present practical advantages,47 and the established evidence regarding the involvement of epigenetic deregulation in human cancer, including lung,48 sets research of DNA methylation biomarkers at very high priority in lung cancer diagnostics.49
DNA methylation detection techniques and methodologies
The need for detection of abnormally methylated DNA in tissue samples and body fluids inevitably leads to discussing methodology issues and the appropriateness of different approaches. A large number of DNA methylation detection methods and modifications exist to date, each having certain advantages and disadvantages.
There are three major approaches regarding the means of discrimination between 5′-methyl cytosine and cytosine:
- Chromatin immunoprecipitation,50 which utilizes anti-5′-methyl cytosine34 or methyl-binding domain proteins.50
- Methylation-specific restriction enzyme51-based methods, which take advantage of the differential recognition of methylated DNA sequences over their unmethylated counterparts.52
- Bisulfite-based methods, which utilize the fact that bisulfite deaminates cytosine to uracil but does not affect 5′-methyl cytosine, effectively transforming this epigenetic modification into a sequence difference.53
Regarding throughput, genome-wide or gene/locus-specific approaches are employed according to the needs. Microarrays and deep sequencing are mainly employed in the first type while dideoxy sequencing,54 pyrosequencing,55 and MALDI-TOF56 are normally utilized for medium-throughput, locus-specific applications. Historically, a few other techniques have been used, such as single-strand conformation polymorphism analysis57 and high-resolution melting.58 A very frequent method also used for locus-specific applications is methylation-specific polymerase chain reaction (MSP)53,59 and its quantitative real-time derivative qMSP60 or MethyLight.61
Various particularities can be recognized in detecting DNA methylation in body fluids. These samples are known to carry a tiny load of abnormal nucleic acids in the presence of high amounts of “contaminating” normal DNA. Exception to this is the detection of DNA methylation in the peripheral leukocytes in leukemias or for monitoring therapeutic schemes with epigenetic drugs62 where the target DNA is ample.
The reliable detection of nucleic acids associated with minimal residual disease in samples such as plasma/serum, urine, sputum, bronchial washings (BWs), saliva, is demanding and devious. The most widely used approach has been bisulfite conversion of DNA followed by MSP or qMSP. The concept here is that methylation-specific primers bearing cytosines (forward) and guanines (reverse) preferably at the 3′ end of both primers will generate amplicons only from the methylated DNA copies of the target sequence under optimal conditions. The vast experience of the research community in polymerase chain reaction amplification techniques allows, in combination with the availability of new engineered hot-start Taq (Thermus Aquaticus) polymerases and reliable thermocycling hardware, to reach high levels of fidelity. Endpoint MSP is a method that revolutionized epigenetic research.53 Its main disadvantage is the lack of quantitation efficiency as well as problems in sensitivity and specificity. The latter can be overcome by using methylation enrichment pyrosequencing, which employs pyrosequencing to confirm the status of an MSP amplicon.63 The real-time version of MSP, referred to as qMSP60 or MethyLight,61 overcomes many of the endpoint assay problems. It is highly sensitive (especially when using fluorescent probes) in visualizing minute amounts that would never been seen on gels. The use of fluorescent probes adds one more level of sequence specificity and allows for multicolor detection of internal controls to normalize for DNA input. What is currently missing from qMSP/MethyLight is an internal control of bisulfite conversion.
As mentioned, an important problem in body fluids is the low availability of DNA. To make the problem more challenging, bisulfite conversion reduces DNA quality while the subsequent cleanup limits significantly recovery of DNA quantities. It is widely accepted today in the epigenetic biomarker research community that it is unlikely to find the perfect single marker. Most studies point to the discovery of panels of biomarkers, and thus multiple assays. Therefore, the tiny amount of DNA recovered has to be split in different reactions. One way to overcome this problem is to multiplex four targets per reaction using probes with different fluorescent dyes.64 Of course the level of optimization required to prove equal amplification factors over five concentration logs between the different amplicons is significant. The abundance of each target is different; thus, in the absence of such calibration, the high-abundance target (10−1–10−2 per genome) will probably consume resources impairing amplification of the rare (≤10−3) copies. A different approach is the post-bisulfite whole-genome (bisulfitome) amplification,65 which demonstrates promising findings, but further research is required to prove the potential extent of its use.
A last important point, frequently bypassed, regarding methodology is the amount of DNA added in a qMSP reaction. qMSP users claim detection of targets in dilution with normal DNA as low as 10−4. In this case, one needs to ensure 104 genome equivalents (32 ng) are added in the reaction to have one abnormal copy keeping in mind that MSP targets only one bisulfite converted strand. This is also an important issue for consideration in whole bisulfite amplification.
Overall, massive progress has been demonstrated in the last decade in methodological approaches for DNA methylation detection in body fluids. Many academic groups have involved themselves in this research, and the contribution of the biotechnical industry must also be acknowledged. Although there are still issues regarding the quality control of the used techniques, there should be no doubt that these will be overcome and a clinically useful method will be very soon available.
DNA methylation aberrations in lung cancer
The high frequency of DNA methylation aberrations in lung cancer has been shown in a large number of studies. Development of epigenetic biomarkers, which could reach clinically validated applications, would assist in personalized management of patients with lung cancer. Identification of markers for early detection (diagnosis), risk assessment (prognosis), and therapeutic stratification of patients is of utmost importance and has been the subject of numerous studies during the last decade.66 Investigating the tumorous, dysplastic, or normal lung tissue is a more direct way to search for biomarkers when compared with surrogate tissues. The relevant literature is vast, and older studies have been previously reviewed.49 In the present review, we are summarizing the most recent (post-2012) information and attempt to evaluate the progress of DNA methylation biomarkers in the clinical management of lung cancer. We searched for reports in PubMed using combinations of keywords such as “lung cancer”, “DNA methylation”, “biomarker”, “diagnosis”, “early detection”, “prognosis”, “body fluids”, “plasma”, “serum”, “sputum”, “bronchial lavage”, and “bronchial washings”.
The findings of recent DNA methylation studies in lung cancer tissue are summarized in Table 1. This table contains 131 genes that can be categorized according to the biological processes they are involved in (Figure 1). While the vast majority of the studies analyzed in Table 1 are mainly focused on the aberrant promoter DNA methylation, studies investigating whole genome as well as gene body methylation patterns are also included. For instance, Nelson et al67 tried to identify new methylation key markers utilizing whole-genome and exonic regions of various lung cancer-related genes. Among the most thoroughly studied genes for DNA methylation in lung cancer are RASSF1, RARβ, p16 (CDKN2A), MGMT, SHOX2, APC, BRCA1, and DAPK, providing a fair list of candidates with biomarker potential. However, Do et al,68 in an attempt to confirm previous studies showing tumor-specific methylation of DNA repair genes, suggest that methylation frequency of lung cancer-associated genes like ATM, BRCA1, MLH1, and XPC is likely to be overestimated in the literature. This raises the importance of diligent validation of previous results, especially if obtained using different methodologies.
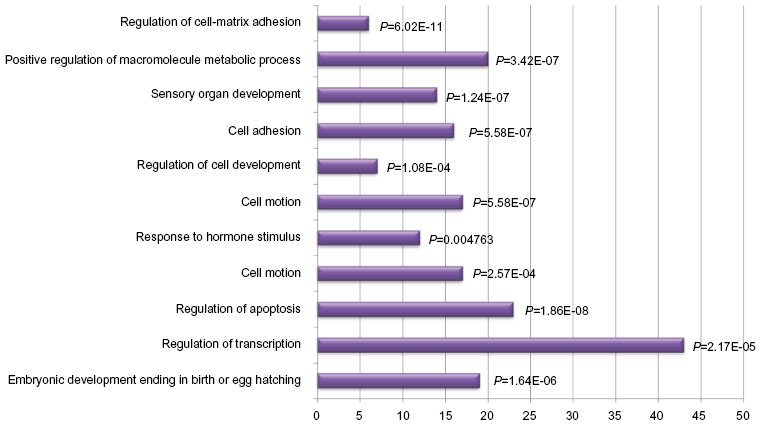 | Figure 1 Bar chart demonstrating the distribution of the most important groups describing the function of the genes with abnormal DNA methylation in lung cancer tissue. |
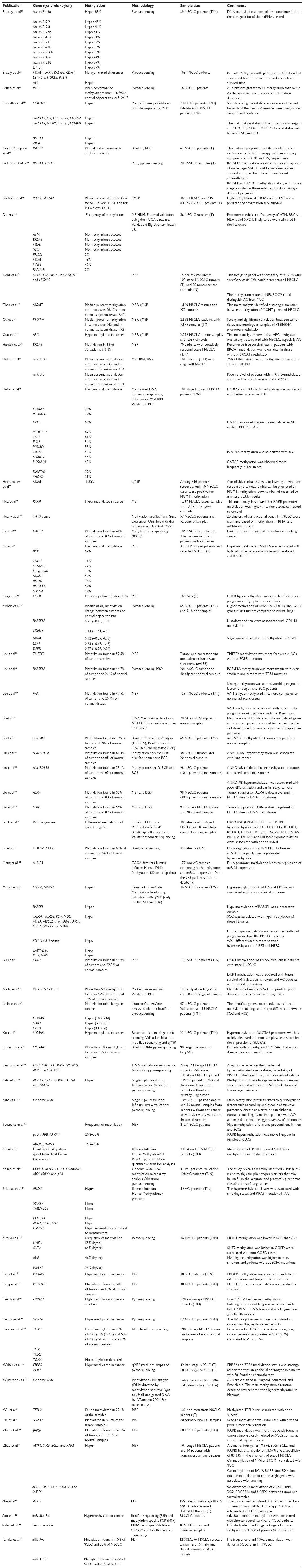 | Table 1 List of recent (post-2012) studies identifying abnormal DNA methylation patterns in human lung cancer tissue |
Diagnostic markers
As mentioned earlier, despite the advances in medicine and cancer biology, lung cancer is largely diagnosed at late stages, leading to poor prognosis and high mortality rates.31 Still the only screening method is low-dose computerized tomography scan of the lungs that is applied to high-risk individuals but includes exposure to radiation, a high rate of potential false positives, and high cost.69 Development of a biomarker-based test could boost early or more accurate diagnosis that is able to distinguish among the different types of lung cancers. Zhao et al70 proposed a panel of four genes (MYF6, SIX6, BCL2, and RARβ) that could offer a sensitivity of 93.07% and a specificity of 83.33% in the diagnosis of stage I NSCLC. A similar study shows that the methylation status of NEUROG2, NID2, RASSF1A, APC, and HOXC9 can also serve in early diagnosis of NSCLC (sensitivity 91.26%, specificity 84.62%); in particular, NEUROG2 seems to be able to discriminate between adenocarcinomas and squamous cell carcinomas.71 The DNA methylation status of miRNAs could also serve as a diagnostic marker in NSCLC. Hypermethylation in NSCLC tissue was observed for hsa-miR-34a, hsa-miR-9-2, and hsa-miR-9-333 as well as miR-193.72 Furthermore, in order to empower the precision of the diagnostic outcome of each study, several meta-analyses have been conducted.70,73–75 Taking into account the results of many individual studies, these meta-analyses confirmed that the methylation status of MGMT,70 CDKN2A,74 APC,75 and RARβ73 promoters is strongly related to lung cancer development.
Prognostic markers
Lung cancer is a highly heterogeneous malignancy. Subpopulations of patients exhibit different risk of metastatic relapse.76 Therefore, markers of prognostic value enabling prediction of the disease aggression and potential course present a long-standing clinically unmet need. The methylation status of numerous genes has been associated with patients’ survival. Recent studies have associated methylation of CDKN2A,77 BRCA1,78 HOXA-2/10,79 CHFR,80 TFPI-2,81 miR-9-3,72 and miR-34b/c82 with poor survival of patients with NSCLC and miR-886-3p83 in SCLC patients. On the other hand, hypermethylation of SHOX2, PITX2,84 DKK1,85 and CYP24A186 was related to better prognosis in NSCLC. In addition to hypermethylation, cancer-specific hypomethylated loci have been proposed as prognostic factors. More specifically, low methylation levels of SCUBE3, SYT2, KCNC3, KCNC4, GRIK3, CRB1, SOCS2, ACTA1, ZNF660, MDFI, ALDH1A3, and SRD5A2 in lung tumors was an indicator of poor survival in a recent study by Lokk et al.87 Surprisingly, although RASSF1 hypermethylation was mainly associated with poor outcome,88–90 a study by Morán et al,91 proposed its protective value.
Therapeutic stratification markers
Directly linked to prognostication is the need for biomarkers that can predict response to particular therapeutic regimens in order to facilitate personalized therapy. Currently, chemotherapy and/or radiotherapy are used based on classical histopathological criteria with very little to no input from molecular constitution of the tumor. However, as personalized medicine rapidly gains ground with the use of targeted therapies (EGFR, ALK, etc) it becomes profound that a more global genetic and epigenetic profiling of individual tumors is required. Many studies have shown that specific DNA methylation signatures can divide patients into subgroups depending on their response to specific anticancer therapy.76,92,93 Hypermethylation of IGFBP3 has been associated with lower response of NSCLC patients to cisplatin therapy,92 while methylation of miR-503 seems to also play a role in this pathway.94 Another recent study reports that unmethylated SFRP5 is a predictor of response to EGFR tyrosine kinase inhibitors therapy, independently of EGFR genotype.93 Also, a phase 2 clinical trial investigated whether MGMT promoter methylation can predict response to Temozolomide treatment in advanced aerodigestive tract (including lung cancer) and colorectal cancers. Unfortunately, the number of lung cancer patients exhibiting MGMT methylation was too small to support the power of the study, leading to uninterpretable results.95
DNA methylation detection in body fluids
The high frequency of DNA methylation detected in primary tumors led researchers to explore the feasibility of detecting these alterations in body fluids for diagnostic purposes. Body fluids represent an extremely attractive clinical material for the management of disease because they can be collected using noninvasive or minimally invasive procedures. Assays target cancer-related DNA methylation abnormalities that may be detected in circulating tumor cells or in cell-free DNA or both. However, the detection of cancer-specific DNA methylation in body fluids presents major challenges, mainly associated with low target abundance in an excess of normal DNA. Similarly to the “Lung cancer biomarkers” section, older reports have been adequately reviewed previously;49 therefore, we present the most recent updates (post-2012) in this review (Table 2).
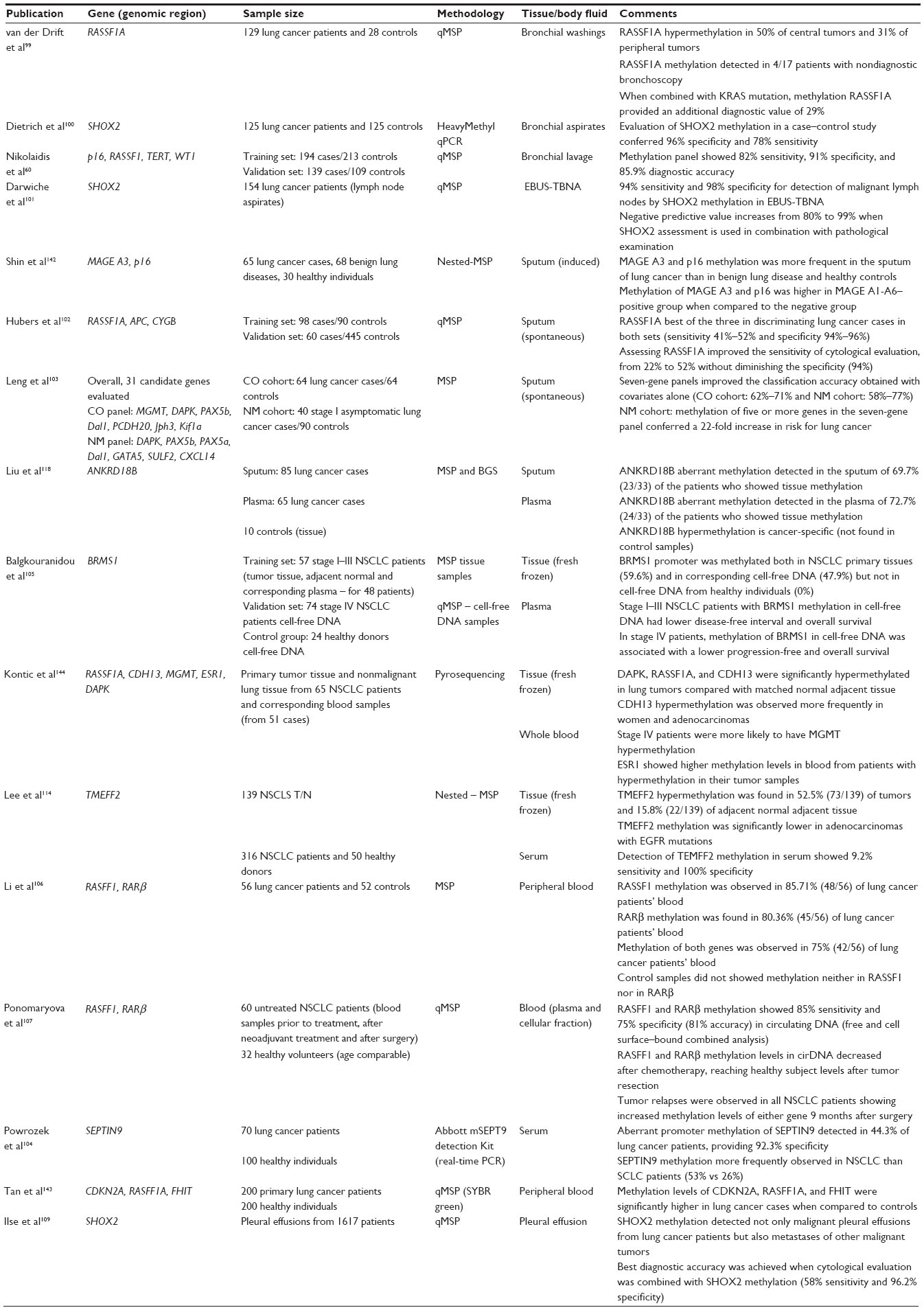 | Table 2 List of recent (post-2012) studies involving detection of DNA methylation in body fluids from patients with lung cancer |
Bronchial washings
Although bronchoscopy does not qualify as a noninvasive process, it is a standard part of the clinical workup of patients suspected for lung cancer, and currently, diagnosis on this sample type is confined to cytological examination. Unfortunately, the diagnostic efficiency of this technique is relatively poor and nearly half of the cases are missed.96,97 Therefore, it is imperative to develop molecular biomarkers capable of improving the diagnostic efficiency of lung cytology.
The feasibility detecting promoter hypermethylation in bronchial lavage was demonstrated 15 years ago,98 and this was followed by a number of relevant publications (reviewed by Liloglou et al49). More recently, the combined detection of RASSF1 methylation and KRAS mutation in BWs has been reported to aid in the detection of malignancy in false-negative or ambiguous cytological outcomes by correctly diagnosing as cancer 29% of the false-negative or doubtful cytological outcomes.99 Furthermore, a panel of DNA methylation biomarkers has been described to show a substantial gain in sensitivity of detection over standalone cytology.60 In this large retrospective case-control study, encompassing BWs from 655 individuals, a diagnostic algorithm based on the methylation profile of CDKN2A, TERT, WT1, and RASSF1 demonstrated 82% sensitivity, 92% specificity, and a diagnostic accuracy of 85.9%. Finally, DNA methylation of SHOX2 alone demonstrated a very high sensitivity in BWs (96%) but the specificity was less than optimal (78%).100 However, a significant improvement in specificity has been shown for SHOX2 methylation when endobronchial ultrasound with transbronchial needle aspiration (EBUS-TBNA) samples were used; specificity reached 99% in this study while sensitivity was improved (82%–99%).101 Moreover, the negative predictive value also increased from 80% to 99%, which from a clinical point of view, might help in avoiding further unnecessary invasive procedures.
Sputum
Sputum is another type of body fluid that has been widely tested for lung cancer biomarker qualification. Its major advantage over BWs is that it is noninvasive and can be practiced in three consecutive day collections at home and sent by post. Its major disadvantage though is the high heterogeneity of the sample in lung cell content (a large percentage of samples are inadequate and contain mainly oral epithelial cells),49 especially when it is noninduced. The methylation status of numerous genes has been recently studied (Table 2) in both spontaneous and induced sputum specimens; some of them alone102 and some of them grouped in panels.103 DNA methylation in sputum has also been shown to improve the classification accuracy. Leng et al,103 in a large case–control study using two different cohorts of patients, reported that seven-gene panels improved the prediction accuracy of standalone clinicopathological data models. More recently, RASSF1 methylation seemed to significantly aid cytological evaluation by improving sensitivity (from 22% to 52%) without diminishing specificity (94%).102
Plasma/serum
The detection of cancer-indicative biomarkers in blood has been the holy grail of cancer molecular diagnostics. Blood is not only an easily accessible and minimally invasive specimen but also highly standardized in the clinical setting across the globe. The two main problems lie with the high dilution of the potential target but also the lack of localized origin. In other words, abnormal DNA in the bloodstream could have originated anywhere in the body. Nevertheless both plasma and serum specimens from lung cancer patients have been used to detect DNA methylation aberrations in numerous genes;49 the most recent studies are listed in Table 2. Although most of these alterations seem to be highly specific, they do not reach the sensitivity levels demanded in clinical diagnostics. For instance, SEPT9 promoter hypermethylation demonstrated 44.3% sensitivity and 92.3% specificity,104 while BRMS1 exhibited 47.9% sensitivity and 100% specificity.105 So far, RASSF1A and RARβ seem to be the most promising candidates since different studies have shown that both markers combined can achieve 75%–85% sensitivity and 75%–100% specificity.106,107 Beyond diagnosis, blood has also been recently used to evaluate significance of gene promoter hypermethylation in plasma of NSCLC patients for prognosis and therapeutic monitoring.105,107 Ponomaryova et al107 report that the methylation index for RASSF1A and RARβ in plasma from lung cancer patients decreased after neoadjuvant chemotherapy and the methylation levels were comparable to those observed in healthy individuals after surgery. Furthermore, all five patients who had a relapse showed an increase in the methylation level of either gene within 9 months after surgery. More recently, the presence of promoter hypermethylation of BRMS1 in NSCLC patients’ plasma was associated with a lower overall survival for stage I–III and stage IV tumors.105
Despite the increasing number of relevant studies addressing DNA promoter methylation as a promising biomarker in body fluids, the preclinical research context and lack of clinical validation in follow-up studies in different cohorts has hampered the development of clinically useful diagnostic markers. In this regard, so far the EpiProLung BL Reflex Assay (Epigenomics, Berlin, Germany) is the only clinically validated and Conformité Européene (CE)-labeled DNA methylation biomarker for diagnosing lung cancer. The assay, which determines the methylation level of SHOX2,35 has been thoroughly validated in bronchial aspirates showing 78% sensitivity and 96% specificity.100 In addition to bronchial aspirates, SHOX2 methylation has also been analyzed in other body fluids such as plasma, pleural effusions, and transbronchial aspirates.101,108,109 Ilse et al109 noted that the detection of malignancy in pleural effusions improved when cytological examination was combined with SHOX2 assessment and that SHOX2 methylation was not only found in malignant effusions from lung cancer patients but also in effusions from malignant tumors of other origins.
Concluding summary
DNA methylation is a critical epigenetic modification affecting chromatin conformation and transcriptional activity, subsequently influencing globally the biology of the mammalian cell. Inability to maintain correct DNA methylation patterns inevitably leads to disease. The list of relevant human diseases is growing and ranges from developmental disorders to autoimmune conditions and cancer. Lung cancer is the most lethal form of human neoplasia, and epigenetic deregulation has been extensively shown in lung neoplastic tissues utilizing a wide range of methodological approaches. The feasibility of identifying abnormally methylated DNA in body fluids has been established. The focus now is moving to identifying the correct targets and improving the methodologies to reach clinical standards. Despite the significant volume of relevant literature, currently there is not enough published work to include sufficient clinical validation to allow moving into a randomized clinical trial. The latter will be the ultimate test proving the patient benefit, since early detection of lung cancer will certainly lead to a stage shift and thereafter improvement of survival of lung cancer patients.
Acknowledgments
Paschalia Pantazi and Alexandra Papaioannou are recipients of IKY Fellowships of Excellence for Post Graduate Studies in Greece – Siemens program. Amelia Acha-Sagredo is a recipient of a postdoctoral fellowship from the Basque Government. Georgios Nikolaidis is supported by the EC program “Translational Potential” (TransPOT; EC Contract 285948). Triantafillos Liloglou’s research is supported by the Roy Castle Lung Cancer Foundation, UK. This article is dedicated to our dearest friend, Lefteris Konstandinidis, who died from lung cancer on September 2, 2014.
Disclosure
The authors report no conflicts of interest in this work.
References
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. | |
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10(5):295–304. | |
Du J, Johnson LM, Groth M, et al. Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol Cell. 2014;55(3):495–504. | |
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278(6):4035–4040. | |
Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. | |
Portela A, Liz J, Nogales V, Setien F, Villanueva A, Esteller M. DNA methylation determines nucleosome occupancy in the 5′-CpG islands of tumor suppressor genes. Oncogene. 2013;32(47):5421–5428. | |
Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106(28):11667–11672. | |
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–346. | |
Daskalos A, Oleksiewicz U, Filia A, et al. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer. 2011;117(5):1027–1037. | |
Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12(2):206–222. | |
Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci. 2014;39(7):310–318. | |
Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805–811. | |
Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. | |
Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. | |
Cannon SV, Cummings A, Teebor GW. 5-Hydroxymethylcytosine DNA glycosylase activity in mammalian tissue. Biochem Biophys Res Commun. 1988;151(3):1173–1179. | |
Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33(3):344–353. | |
Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10(16):2662–2668. | |
Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19(5):698–711. | |
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92. | |
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. | |
Pfeifer GP, Rauch TA. DNA methylation patterns in lung carcinomas. Semin Cancer Biol. 2009;19(3):181–187. | |
Gokul G, Khosla S. DNA methylation and cancer. Subcell Biochem. 2013;61:597–625. | |
Kulis M, Queiros AC, Beekman R, Martin-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013;1829(11):1161–1174. | |
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. | |
Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30(2):75–84. | |
Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288(48):34287–34294. | |
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. | |
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2014. Ann Oncol. 2014;25(8):1650–1656. | |
Travis WD, Brambilla E, Riely GJ. New pathologic classification of lung cancer: relevance for clinical practice and clinical trials. J Clin Oncol. 2013;31(8):992–1001. | |
Kanne JP. Screening for lung cancer: what have we learned? Am J Roentgenol. 2014;202(3):530–535. | |
Field JK, van Klaveren R, Pedersen JH, et al; European Randomized Screening Trial Group. European randomized lung cancer screening trials: post NLST. J Surg Oncol. 2013;108(5):280–286. | |
Bediaga NG, Davies MP, Acha-Sagredo A, et al. A microRNA-based prediction algorithm for diagnosis of non-small lung cell carcinoma in minimal biopsy material. Br J Cancer. 2013;109(9):2404–2411. | |
Weber DG, Johnen G, Casjens S, et al. Evaluation of long noncoding RNA MALAT1 as a candidate blood-based biomarker for the diagnosis of non-small cell lung cancer. BMC Res Notes. 2013;6:518. | |
Schmidt B, Liebenberg V, Dietrich D, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010;10:600. | |
Lou E, Johnson M, Sima C, et al. Serum biomarkers for assessing histology and outcomes in patients with metastatic lung cancer. Cancer Biomark. 2014;14(4):207–214. | |
Nie FQ, Zhu Q, Xu TP, et al. Long non-coding RNA MVIH indicates a poor prognosis for non-small cell lung cancer and promotes cell proliferation and invasion. Tumour Biol. 2014;35(8):7587–7594. | |
Zhang H, Liu J, Yue D, Gao L, Wang D, Wang C. Clinical significance of E-cadherin, beta-catenin, vimentin and S100A4 expression in completely resected squamous cell lung carcinoma. J Clin Pathol. 2013;66(11):937–945. | |
El-Chaar NN, Piccolo SR, Boucher KM, et al. Genomic classification of the RAS network identifies a personalized treatment strategy for lung cancer. Mol Oncol. 2014;8(7):1339–1354. | |
Feng JF, Wu JZ, Hu SN, et al. Polymorphisms of the ribonucleotide reductase M1 gene and sensitivity to platin-based chemotherapy in non-small cell lung cancer. Lung Cancer. 2009;66(3):344–349. | |
Gregorc V, Novello S, Lazzari C, et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): a biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014;15(7):713–721. | |
Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med. 2012;9(5):e1001216. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Schache AG, Hall G, Woolgar JA, et al. Quantitative promoter methylation differentiates carcinoma ex pleomorphic adenoma from pleomorphic salivary adenoma. Br J Cancer. 2010;103(12):1846–1851. | |
Sandoval J, Peiro-Chova L, Pallardo FV, Garcia-Gimenez JL. Epigenetic biomarkers in laboratory diagnostics: emerging approaches and opportunities. Expert Rev Mol Diagn. 2013;13(5):457–471. | |
Korpanty GJ, Graham DM, Vincent MD, Leighl NB. Biomarkers that currently affect clinical practice in lung cancer: EGFR, ALK, MET, ROS-1, and KRAS. Front Oncol. 2014;4:204. | |
Liloglou T, Field JK. Detection of DNA methylation changes in body fluids. Adv Genet. 2010;71:177–207. | |
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. | |
Liloglou T, Bediaga NG, Brown BR, Field JK, Davies MP. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014;342(2):200–212. | |
Ballestar E, Paz MF, Valle L, et al. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003;22(23):6335–6345. | |
Hughes S, Jones JL. The use of multiple displacement amplified DNA as a control for methylation specific PCR, pyrosequencing, bisulfite sequencing and methylation-sensitive restriction enzyme PCR. BMC Mol Biol. 2007;8:91. | |
Schumacher A, Weinhausl A, Petronis A. Application of microarrays for DNA methylation profiling. Methods Mol Biol. 2008;439:109–129. | |
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–9826. | |
Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9(2):145–151. | |
Daskalos A, Nikolaidis G, Xinarianos G, et al. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer. 2009;124(1):81–87. | |
Ehrich M, Field JK, Liloglou T, et al. Cytosine methylation profiles as a molecular marker in non-small cell lung cancer. Cancer Res. 2006;66(22):10911–10918. | |
Fraga MF, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques. 2002;33(3):632,634. 636–649. | |
Kristensen LS, Hansen LL. PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin Chem. 2009;55(8):1471–1483. | |
Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes. 2008;1:54. | |
Nikolaidis G, Raji OY, Markopoulou S, et al. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer Res. 2012;72(22):5692–5701. | |
Hernandez HG, Tse MY, Pang SC, Arboleda H, Forero DA. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques. 2013;55(4):181–197. | |
Stewart DJ, Issa JP, Kurzrock R, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15(11):3881–3888. | |
Shaw RJ, Liloglou T, Rogers SN, et al. Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006;94(4):561–568. | |
Fackler MJ, Rivers A, Teo WW, et al. Hypermethylated genes as biomarkers of cancer in women with pathologic nipple discharge. Clin Cancer Res. 2009;15(11):3802–3811. | |
Vaissiere T, Cuenin C, Paliwal A, et al. Quantitative analysis of DNA methylation after whole bisulfitome amplification of a minute amount of DNA from body fluids. Epigenetics. 2009;4(4):221–230. | |
Fleischhacker M, Dietrich D, Liebenberg V, Field JK, Schmidt B. The role of DNA methylation as biomarkers in the clinical management of lung cancer. Expert Rev Respir Med. 2013;7(4):363–383. | |
Nelson HH, Marsit CJ, Christensen BC, et al. Key epigenetic changes associated with lung cancer development: results from dense methylation array profiling. Epigenetics. 2012;7(6):559–566. | |
Do H, Wong NC, Murone C, et al. A critical re-assessment of DNA repair gene promoter methylation in non-small cell lung carcinoma. Sci Rep. 2014;4:4186. | |
Tammemagi MC, Lam S. Screening for lung cancer using low dose computed tomography. BMJ. 2014;348:g2253. | |
Zhao Y, Zhou H, Ma K, et al. Abnormal methylation of seven genes and their associations with clinical characteristics in early stage non-small cell lung cancer. Oncol Lett. 2013;5(4):1211–1218. | |
Geng J, Sun J, Lin Q, et al. Methylation status of NEUROG2 and NID2 improves the diagnosis of stage I NSCLC. Oncol Lett. 2012;3(4):901–906. | |
Heller G, Weinzierl M, Noll C, et al. Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clin Cancer Res. 2012;18(6):1619–1629. | |
Hua F, Fang N, Li X, Zhu S, Zhang W, Gu J. A meta-analysis of the relationship between RARbeta gene promoter methylation and non-small cell lung cancer. PLoS One. 2014;9(5):e96163. | |
Gu J, Wen Y, Zhu S, et al. Association between P(16INK4a) promoter methylation and non-small cell lung cancer: a meta-analysis. PLoS One. 2013;8(4):e60107. | |
Guo S, Tan L, Pu W, et al. Quantitative assessment of the diagnostic role of APC promoter methylation in non-small cell lung cancer. Clin Epigenetics. 2014;6(1):5. | |
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. | |
Bradly DP, Gattuso P, Pool M, et al. CDKN2A (p16) promoter hypermethylation influences the outcome in young lung cancer patients. Diagn Mol Pathol. 2012;21(4):207–213. | |
Harada H, Miyamoto K, Yamashita Y, et al. Methylation of breast cancer susceptibility gene 1 (BRCA1) predicts recurrence in patients with curatively resected stage I non-small cell lung cancer. Cancer. 2013;119(4):792–798. | |
Heller G, Babinsky VN, Ziegler B, et al. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients. Carcinogenesis. 2013;34(3):513–521. | |
Koga T, Takeshita M, Ijichi K, Yano T, Maehara Y, Sueishi K. CHFR aberrant methylation involves a subset of human lung adenocarcinoma associated with poor clinical outcomes. Hum Pathol. 2013;44(7):1382–1390. | |
Wu D, Xiong L, Wu S, Jiang M, Lian G, Wang M. TFPI-2 methylation predicts poor prognosis in non-small cell lung cancer. Lung Cancer. 2012;76(1):106–111. | |
Nadal E, Chen G, Gallegos M, et al. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2013;19(24):6842–6852. | |
Cao J, Song Y, Bi N, et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73(11):3326–3335. | |
Dietrich D, Hasinger O, Liebenberg V, Field JK, Kristiansen G, Soltermann A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn Mol Pathol. 2012;21(2):93–104. | |
Na Y, Lee SM, Kim DS, Park JY. Promoter methylation of Wnt antagonist DKK1 gene and prognostic value in Korean patients with non-small cell lung cancers. Cancer Biomark. 2012;12(2):73–79. | |
Ramnath N, Nadal E, Jeon CK, et al. Epigenetic regulation of vitamin D metabolism in human lung adenocarcinoma. J Thorac Oncol. 2014;9(4):473–482. | |
Lokk K, Vooder T, Kolde R, et al. Methylation markers of early-stage non-small cell lung cancer. PLoS One. 2012;7(6):e39813. | |
de Fraipont F, Levallet G, Creveuil C, et al; Intergroupe Francophone de Cancérologie Thoracique. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin Cancer Res. 2012;18(10):2976–2986. | |
Ko E, Lee BB, Kim Y, et al. Association of RASSF1A and p63 with poor recurrence-free survival in node-negative stage I-II non-small cell lung cancer. Clin Cancer Res. 2013;19(5):1204–1212. | |
Lee SM, Lee WK, Kim DS, Park JY. Quantitative promoter hypermethylation analysis of RASSF1A in lung cancer: comparison with methylation-specific PCR technique and clinical significance. Mol Med Rep. 2012;5(1):239–244. | |
Morán A, Fernández-Marcelo T, Carro J, et al. Methylation profiling in non-small cell lung cancer: clinical implications. Int J Oncol. 2012;40(3):739–746. | |
Cortés-Sempere M, de Miguel MP, Pernía O, et al. IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer. Oncogene. 2013;32(10):1274–1283. | |
Zhu J, Wang Y, Duan J, et al. DNA methylation status of Wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2012;31:80. | |
Li N, Zhang F, Li S, Zhou S. Epigenetic silencing of MicroRNA-503 regulates FANCA expression in non-small cell lung cancer cell. Biochem Biophys Res Commun. 2014;444(4):611–616. | |
Hochhauser D, Glynne-Jones R, Potter V, et al. A phase II study of temozolomide in patients with advanced aerodigestive tract and colorectal cancers and methylation of the O6-methylguanine-DNA methyltransferase promoter. Mol Cancer Ther. 2013;12(5):809–818. | |
Dobler CC, Crawford AB. Bronchoscopic diagnosis of endoscopically visible lung malignancies: should cytological examinations be carried out routinely? Intern Med J. 2009;39(12):806–811. | |
Van’t Westeinde SC, van Klaveren RJ. Screening and early detection of lung cancer. Cancer J. 2011;17(1):3–10. | |
Ahrendt SA, Chow JT, Xu LH, et al. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst. 1999;91(4):332–339. | |
van der Drift MA, Prinsen CF, Knuiman GJ, Janssen JP, Dekhuijzen PN, Thunnissen FB. Diagnosing peripheral lung cancer: the additional value of the Ras-association domain family 1A gene methylation and Kirsten rat sarcoma 2 viral oncogene homolog mutation analyses in washings in nondiagnostic bronchoscopy. Chest. 2012;141(1):169–175. | |
Dietrich D, Kneip C, Raji O, et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int J Oncol. 2012;40(3):825–832. | |
Darwiche K, Zarogoulidis P, Baehner K, et al. Assessment of SHOX2 methylation in EBUS-TBNA specimen improves accuracy in lung cancer staging. Ann Oncol. 2013;24(11):2866–2870. | |
Hubers AJ, van der Drift MA, Prinsen CF, et al. Methylation analysis in spontaneous sputum for lung cancer diagnosis. Lung Cancer. 2014;84(2):127–133. | |
Leng S, Do K, Yingling CM, et al. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res. 2012;18(12):3387–3395. | |
Powrozek T, Krawczyk P, Kucharczyk T, Milanowski J. Septin 9 promoter region methylation in free circulating DNA-potential role in noninvasive diagnosis of lung cancer: preliminary report. Med Oncol. 2014;31(4):917. | |
Balgkouranidou I, Chimonidou M, Milaki G, et al. Breast cancer metastasis suppressor-1 promoter methylation in cell-free DNA provides prognostic information in non-small cell lung cancer. Br J Cancer. 2014;110(8):2054–2062. | |
Li W, Deng J, Jiang P, Zeng X, Hu S, Tang J. Methylation of the RASSF1A and RARbeta genes as a candidate biomarker for lung cancer. Exp Ther Med. 2012;3(6):1067–1071. | |
Ponomaryova AA, Rykova EY, Cherdyntseva NV, et al. Potentialities of aberrantly methylated circulating DNA for diagnostics and post-treatment follow-up of lung cancer patients. Lung Cancer. 2013;81(3):397–403. | |
Kneip C, Schmidt B, Seegebarth A, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011;6(10):1632–1638. | |
Ilse P, Biesterfeld S, Pomjanski N, Fink C, Schramm M. SHOX2 DNA methylation is a tumour marker in pleural effusions. Cancer Genomics Proteomics. 2013;10(5):217–223. | |
Bruno P, Gentile G, Mancini R, et al. WT1 CpG islands methylation in human lung cancer: a pilot study. Biochem Biophys Res Commun. 2012;426(3):306–309. | |
Carvalho RH, Haberle V, Hou J, et al. Genome-wide DNA methylation profiling of non-small cell lung carcinomas. Epigenetics Chromatin. 2012;5(1):9. | |
Huang T, Jiang M, Kong X, Cai YD. Dysfunctions associated with methylation, microRNA expression and gene expression in lung cancer. PLoS One. 2012;7(8):e43441. | |
Jia Y, Yang Y, Brock MV, Zhan Q, Herman JG, Guo M. Epigenetic regulation of DACT2, a key component of the Wnt signalling pathway in human lung cancer. J Pathol. 2013;230(2):194–204. | |
Lee SM, Park JY, Kim DS. Methylation of TMEFF2 gene in tissue and serum DNA from patients with non-small cell lung cancer. Mol Cells. 2012;34(2):171–176. | |
Lee SM, Park JY, Kim DS. Wif1 hypermethylation as unfavorable prognosis of non-small cell lung cancers with EGFR mutation. Mol Cells. 2013;36(1):69–73. | |
Li B, Lu Q, Song ZG, et al. Functional analysis of DNA methylation in lung cancer. Eur Rev Med Pharmacol Sci. 2013;17(9):1191–1197. | |
Liu WB, Han F, Jiang X, et al. ANKRD18A as a novel epigenetic regulation gene in lung cancer. Biochem Biophys Res Commun. 2012;429(3–4):180–185. | |
Liu WB, Han F, Jiang X, et al. Epigenetic regulation of ANKRD18B in lung cancer. Mol Carcinog. Epub November 19, 2013. | |
Liu WB, Han F, Du XH, et al. Epigenetic silencing of Aristaless-like homeobox-4, a potential tumor suppressor gene associated with lung cancer. Int J Cancer. 2014;134(6):1311–1322. | |
Liu WB, Jiang X, Han F, et al. LHX6 acts as a novel potential tumour suppressor with epigenetic inactivation in lung cancer. Cell Death Dis. 2013;4:e882. | |
Lu KH, Li W, Liu XH, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461. | |
Meng W, Ye Z, Cui R, et al. MicroRNA-31 predicts the presence of lymph node metastases and survival in patients with lung adenocarcinoma. Clin Cancer Res. 2013;19(19):5423–5433. | |
Sandoval J, Mendez-Gonzalez J, Nadal E, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4140–4147. | |
Sato T, Arai E, Kohno T, et al. DNA methylation profiles at precancerous stages associated with recurrence of lung adenocarcinoma. PLoS One. 2013;8(3):e59444. | |
Sato T, Arai E, Kohno T, et al. Epigenetic clustering of lung adenocarcinomas based on DNA methylation profiles in adjacent lung tissue: its correlation with smoking history and chronic obstructive pulmonary disease. Int J Cancer. 2014;135(2):319–334. | |
Scesnaite A, Jarmalaite S, Mutanen P, et al. Similar DNA methylation pattern in lung tumours from smokers and never-smokers with second-hand tobacco smoke exposure. Mutagenesis. 2012;27(4):423–429. | |
Shi J, Marconett CN, Duan J, et al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nat Commun. 2014;5:3365. | |
Shinjo K, Okamoto Y, An B, et al. Integrated analysis of genetic and epigenetic alterations reveals CpG island methylator phenotype associated with distinct clinical characters of lung adenocarcinoma. Carcinogenesis. 2012;33(7):1277–1285. | |
Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22(7):1197–1211. | |
Suzuki M, Shiraishi K, Eguchi A, et al. Aberrant methylation of LINE-1, SLIT2, MAL and IGFBP7 in non-small cell lung cancer. Oncol Rep. 2013;29(4):1308–1314. | |
Tan SX, Hu RC, Tan YL, Liu JJ, Liu WE. Promoter methylation-mediated downregulation of PRDM5 contributes to the development of lung squamous cell carcinoma. Tumour Biol. 2014;35(5):4509–4516. | |
Tang X, Yin X, Xiang T, et al. Protocadherin 10 is frequently downregulated by promoter methylation and functions as a tumor suppressor gene in non-small cell lung cancer. Cancer Biomark. 2012;12(1):11–19. | |
Tekpli X, Zienolddiny S, Skaug V, Stangeland L, Haugen A, Mollerup S. DNA methylation of the CYP1A1 enhancer is associated with smoking-induced genetic alterations in human lung. Int J Cancer. 2012;131(7):1509–1516. | |
Tennis MA, Vanscoyk MM, Wilson LA, Kelley N, Winn RA. Methylation of Wnt7a is modulated by DNMT1 and cigarette smoke condensate in non-small cell lung cancer. PLoS One. 2012;7(3):e32921. | |
Tessema M, Yingling CM, Grimes MJ, et al. Differential epigenetic regulation of TOX subfamily high mobility group box genes in lung and breast cancers. PLoS One. 2012;7(4):e34850. | |
Walter K, Holcomb T, Januario T, et al. DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clin Cancer Res. 2012;18(8):2360–2373. | |
Wilkerson MD, Yin X, Walter V, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS One. 2012;7(5):e36530. | |
Yin D, Jia Y, Yu Y, et al. SOX17 methylation inhibits its antagonism of Wnt signaling pathway in lung cancer. Discov Med. 2012;14(74):33–40. | |
Zhao X, Wang N, Zhang M, Xue S, Shi K, Chen Z. Detection of methylation of the RAR-beta gene in patients with non-small cell lung cancer. Oncol Lett. 2012;3(3):654–658. | |
Kalari S, Jung M, Kernstine KH, Takahashi T, Pfeifer GP. The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene. 2013;32(30):3559–3568. | |
Tanaka N, Toyooka S, Soh J, et al. Frequent methylation and oncogenic role of microRNA-34b/c in small-cell lung cancer. Lung Cancer. 2012;76(1):32–38. | |
Shin KC, Lee KH, Lee CH, Shin IH, Suh HS, Jeon CH. MAGE A1-A6 RT-PCR and MAGE A3 and p16 methylation analysis in induced sputum from patients with lung cancer and non-malignant lung diseases. Oncol Rep. 2012;27(4):911–916. | |
Tan S, Sun C, Wei X, et al. Quantitative assessment of lung cancer associated with genes methylation in the peripheral blood. Exp Lung Res. 2013;39(4–5):182–190. | |
Kontic M, Stojsic J, Jovanovic D, et al. Aberrant promoter methylation of CDH13 and MGMT genes is associated with clinicopathologic characteristics of primary non-small-cell lung carcinoma. Clin Lung Cancer. 2012;13(4):297–303. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.