")
Back to Archived Journals » Journal of Biorepository Science for Applied Medicine » Volume 3 » Issue 1
DNA fingerprinting for sample authentication in biobanking: recent perspectives
Authors Palmirotta R, De Marchis ML , Ialongo C , Majorani C, Alessandroni J, Spila A, Valente MG, Della-Morte D, Ferroni P , Guadagni F
Received 31 March 2015
Accepted for publication 8 June 2015
Published 25 August 2015 Volume 2015:3(1) Pages 35—45
DOI https://doi.org/10.2147/BSAM.S68063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Martin H Bluth
Raffaele Palmirotta,1,2,* Maria Laura De Marchis,1,3,* Cristiano Ialongo,4 Costanza Majorani,5 Jhessica Alessandroni,1 Antonella Spila,1 Maria Giovanna Valente,6 David Della-Morte,1,7 Patrizia Ferroni,1,2 Fiorella Guadagni1,2
1Interinstitutional Multidisciplinary Biobank, IRCCS San Raffaele Pisana, 2Telematic University San Raffaele, 3Department of Cardiovascular, Respiratory, Nephrologic, Geriatric and Anesthesiological Sciences, Sapienza University, 4Department of Internal Medicine, Tor Vergata University, 5Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, 6Fondazione San Raffaele, Ceglie Messapica, 7Department of Systems Medicine, Tor Vergata University, Rome, Italy
*These authors contributed equally to this work
Abstract: The availability of biological samples has assumed a crucial role in the field of biomedical research in order to assess predisposition to complex diseases, identification and validation of new diagnostic biomarkers, drug targets, and improvement of monitoring strategies. To ensure correct collection, storage, and use of biological material, a biobank requires standard operating procedures, not only to guarantee the integrity of the sample, but also for the traceability and uniqueness of the same sample and to avoid potential contamination. Over the past three decades, researchers have increasingly used DNA polymorphisms for biological identification purposes, radically changing forensic investigations. This revolution has led to the birth of new scientific terms, such as “DNA fingerprinting” or “genetic profiling”, and a new field of legal medicine known as “forensic genetics”. Given the significant need for a biobank to devise a system of identification, authentication, and traceability of biological samples, many researchers are applying forensic genetics-based methodologies to the science of biobanking. This review recapitulates the main DNA polymorphisms presently known and the techniques most widely used for biological identification purposes, from standard methods based on short tandem repeats, to use of single nucleotide polymorphisms, through to the more recent insertion/deletion polymorphisms. The experience of several research groups who have applied classical or innovative methods for identification of biological samples in the context of a biorepository is also discussed. Finally, some technical issues are suggested that could facilitate decisions about the most appropriate markers and DNA fingerprint methodology for evaluation of identity and traceability in each disease-based biobank.
Keywords: biobank, biorepository, DNA fingerprint, traceability, standard operating procedures, DNA polymorphisms
Introduction
Establishment of biobanks as a source of biological material for the execution of molecular biology studies is enabling the identification of genetic factors involved in the etiopathogenesis of disease, analysis of their interaction with environmental factors, and development of tailored therapies defined according to the genetic background of the patient.1,2 Accordingly, biobanking is becoming increasingly more complex, given the critical mass of quality samples needed, which translates into the need for large, centralized repositories to be developed wherein samples are collected from a network of donating sites. Therefore, it is crucial to monitor all the steps that biological samples undergo, in order to maintain their integrity and to avoid mislabeling, accidental mixing, and contamination of tubes.3
Since all the preanalytical phases of sample handling may have important implications with regard to subsequent molecular applications, it has become imperative to define a series of accurate standard operating procedures to obtain nucleic acids of excellent quality and to confirm the identity of stored samples.4,5 In particular, when planning large-scale genetic association studies, the traceability of samples and identification of possible contaminants become the most critical points of the process.6–8
Although rapidly expanding, information-based and automation-based technologies can only partially overcome the problem of sample misidentification or cross-contamination, since the main sources of error may derive from inaccurate manual operations, improper storage, and mislabeling of tubes.8–12
A definitive solution to this issue may result from applying forensic genetic methodologies based on analysis of multiallelic molecular markers to certify the authenticity of stored biospecimens.13,14 However, very often, biobanks lack the necessary infrastructure to carry out these specialist procedures, which are very expensive and require specific software for analysis.14 It is therefore necessary to devise low-cost and easily standardized DNA fingerprinting methods, with the robustness required to provide unequivocal authentication of the sample. In the present review, we report on the current knowledge in this field and the main technologies available to develop a quality control system based on analysis of molecular markers in biobanks. To this end, we will examine a number of approaches used by various authors, including the procedure created at our Interinstitutional Multidisciplinary BioBank (Rome, Italy) to easily and quickly verify the identity of a collected biospecimen.3 Readers in need of further details about methods of cryopreservation, storage, retrieval, and distribution of biological materials in biobanks are referred to “Best Practices For Repositories” on the website of the International Society for Biological and Environmental Repositories.15
Molecular markers employed for analysis of biological individuality
Only 1.5% of the human genome (3.2 Gb) encodes for proteins, while another 23.5% is classified as gene-related DNA and comprises many non-coding elements involved in gene regulation, such as promoters, enhancers, repressors, and polyadenylation signals. The extragenic component consists of single copy DNA (approximately 21%), the function of which has not yet been defined, while about half of the human genome is composed of repetitive extragenic elements (54%). Repetitive DNA is, in turn, divided into repetitive sequences distributed throughout the genome (45%) or as tandem repeat DNA (9%).16 The former mainly include four types of elements: short interspersed elements (13%); long interspersed elements (21%), with the Alu and L1 sequences being the most frequent elements; long terminal repeats (8%); and transposons (3%).17–19 The tandem repeat component is constituted by:
- Satellite DNA (5%) – units of 5–171 bp repeated in blocks of hundreds of kilobases, found in regions of centromeric and extracentromeric heterochromatin
- Minisatellite DNA (3%) – consisting of repeated units of 6–64 bp, localized in telomeric and peritelomeric regions (the latter defined as hypervariable DNA)
- Microsatellite DNA (1%) – short sequences of 1–6 nucleotide repeats, usually up to a length of 150 bp.
When performing biological explorations on the genetic variability of a population or a forensic investigation, it is important to select a group of genetic variants with no effect at the phenotypic level. These “neutral” variants, in fact, do not provide selective advantages or disadvantages, so their frequency is not affected by processes of natural selection. In order to have all the markers completely independent, they must be selected in different chromosomes or separated by several centimorgans.
Other deciding factors must be considered before selecting a panel of molecular markers. In particular, variants to be used for DNA profiling studies must be highly polymorphic and, possibly, polyallelic markers.20 Moreover, it is very often necessary to consider combinations of different loci, each with a number of allelic variants. The increase in the number of loci, selecting those with several allelic variants within the studied population, in fact, gives the test a higher discrimination power and therefore a great reliability. Specific databases are available that provide allele frequency data for the human population.21–29
Among the first-generation molecular markers used for human identity testing are the variable numbers of tandem repeats (VNTR), comprising the above-mentioned minisatellite group. This technique, developed by Jeffreys et al, was based on the use of restriction enzymes recognizing and cutting the flanking regions of VNTR sequences and on a Southern blotting analysis using a series of multilocus restriction fragment length polymorphism probes. A different pattern of fragments was obtained, depending on the VNTR length.30 Over time, this method has ceased to be used, given the high execution time and work required, the large quantity of DNA necessary, inability to automate the procedure, and the problems emerging from analysis of mixtures of DNA.13
However, looking at the different targets available for performing a diagnostic test based on DNA fingerprinting, microsatellites undoubtedly have long been the markers of choice. Many of the techniques currently used for individual molecular typing, in fact, are based on panels of microsatellites.
As previously mentioned, DNA regions with repeat units 2–7 bp in length are called microsatellites, simple sequence repeats, or most usually short tandem repeats (STRs).31 Forensically used STRs are usually tetranucleotide repeats, which have few stutter artifacts.32 At the genomic level, a microsatellite sequence recurs approximately every 2 kb, with the number of non-overlapping tandem repeats being 437 per Mb.33 STRs represent a class of molecular markers capable of highlighting a degree of genetic diversity hardly detectable by other types of markers and can be typed by polymerase chain reaction (PCR) amplification. The high level of polymorphism of STR markers is the result of high mutation rates (between 10−2 and 10−6 events per locus per generation), occurring mainly because of two mechanisms, ie, unequal crossing over during meiosis or DNA polymerase “slippage”.34 Very often, it is possible to observe microsatellite loci characterized by more than ten alleles and having more than 60% heterozygosity.
Next, there are two markers characterized by an absence of crossing over that are mainly used for reconstruction of matrilineal and patrilineal descent: mitochondrial DNA (mtDNA) and the Y chromosome, respectively. Given the high level of variability in the mitochondrial D-loop region and the large copy number of circular genomes (the number of copies depending on the cell involved ranges from 100 to more than 1,000), mtDNA is a very informative tool in DNA profiling studies, allowing analysis of very small or degraded samples.35
The unusual characteristics of the Y chromosome, including haploidy, male specificity, lack of recombination, and the presence of many kind of polymorphisms, make its study useful, especially in kinship and paternity testing, analysis of a mixture of male and female DNA, and other types of genetic traceability investigations, alternatively or in combination with autosomal DNA profiling.36
In the field of genetic traceability, a key role is played by single nucleotide polymorphisms (SNPs), a molecular marker class characterized by differences at the single nucleotide level. An SNP is a single base pair variation at a specific locus, usually consisting of two alleles (where the rare allele frequency is ≥1%). More than 1.4 million SNPs have been identified in the human genome, indicating a high degree of polymorphism.33 The SNP-based genotyping process can be easily automated, offering a highly reproducible method and the opportunity to apply the procedure also to degraded DNA samples. However, being biallelic markers, they are less informative than STRs, so a larger panel of markers should be considered to maintain the same power of discrimination (PD).37 An advantage in adopting an SNP-based genotyping platform could derive from the fact that these polymorphisms are often found to be the etiological agent in many common human diseases and are becoming of particular interest in pharmacogenomics. Accordingly, in population-related biorepositories, such a quality control system could also provide a suitable resource for investigating the genetic components of a given disease.12
Another class of markers that has recently emerged in the field of genetic studies is that of insertion/deletion polymorphisms. These biallelic variants are created by the insertion or deletion of one or more nucleotides at more than 2,000 genomic loci widely distributed throughout the genome.38 Furthermore, insertion/deletion polymorphisms have a low mutation rate, a reliable discrimination power, involve non-labor-intensive genotyping methods, and possible automation of analysis, even starting from degraded DNA.3,37,39
Current methods of biological identification through DNA polymorphisms
The number of repeats in STR markers can be highly variable between individuals, and this particular characteristic endows STRs with a high PD, making them better candidates for use in forensic applications, in which degraded material or a small amount of DNA is common. Because of their smaller size, STR alleles can also be more easily separated from other chromosomal locations to ensure that closely linked loci are not chosen.13,40
The development of STR-based multiplex PCR, separation of more than ten STR loci in a single analytical procedure, and automated DNA fragment analysis provide the opportunity to obtain a DNA profile from almost any source of biological material.
STR markers were first described as effective tools for human identity testing in the early 1990s.41,42 Research on STR candidates for identification of new loci and population variation were initiated by the Forensic Science Service and the Royal Canadian Mounted Police as a joint effort with European laboratories. The first Forensic Science Service multiplex applied to forensic casework included four loci, ie, TH01, VWA, FES/FPS, and F13A1.43 This was followed by a second-generation multiplex including the loci TH01, VWA, FGA, D8S1179, D18S51, and D21S11.44 The UK National DNA Database was launched in April 1995 utilizing second-generation multiplex loci and the amelogenin sex-typing test.45,46
Since 1996, with the support of the Combined DNA Index System, STR typing has allowed creation of databases centered on evaluation of 13 loci.22,47 In fact, a genotype obtained from analysis of these loci allows identification of the individual to which the DNA belongs with a sufficient statistical power. These 13 loci are CSF1P0, FGA, TH01, TPOX, VWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, and D21S11.23,24 Amelogenin gene primers are also included in the commercial kits to allow sex identification. In females, the locus of amelogenin is on the X chromosome (XX) and it is represented by a single peak, whereas in males amelogenin is represented by two peaks (one for the X chromosome and the other for the Y chromosome).
Several kits for STR typing are now commercially available, allowing characterization of an increasing number of loci. Among them, the most widely used are PowerPlex ESX and ESI 16 or 17 (Promega Corporation), AmpFlSTR NGM and NGM Select (Applied Biosystems), and ESSplex and ESSplex SE (Qiagen). However, the technical choice should always be made on an individual basis, depending on the number of loci to be analyzed and their PD.48,49
The procedure used for STR profile analysis can be summarized in three major phases. The first is DNA extraction. The extracted DNA is then typed with a multiplex PCR using the above-mentioned kits. Finally, after amplification, samples are separated by capillary electrophoresis in an automated sequencer to obtain an electropherogram. Over the years, fluorescence detection methods and capillary electrophoresis using ABI 3730, 3500, 3130, and 310 DNA sequencers (Thermo Fisher Scientific) have played a significant role in forensic DNA typing. The electropherogram is commonly analyzed using GeneMapper software (Thermo Fisher Scientific).50,51
Nowadays, STR profiling is commonly applied to determine kinship, including paternity testing, but identification of male DNA components in mixed male/female samples is a challenging problem. This could be overcome with the use of Y-chromosomal STRs (Y-STRs), given that autosomal STR profiling is often not successful in such cases because of preferential PCR amplification of the excess female component.52 Haplotypes from sets of non-recombining male-specific Y-STRs have been used for male identification since the late 1990s, especially since the advent of commercial kits containing up to 17 well defined Y-STRs.52–54 The most widely used database, containing several Y haplotypes and their frequency estimates, is the Y-STR Haplotype Reference Database.25,55,56
mtDNA analysis, used in population studies, evolutionary biology, and anthropology, is carried out in a region called the displacement loop, which represents a non-coding control zone. The two hypervariable segments (HV1 and HV2) are amplified, sequenced, and then compared with the reference (“Anderson”) sequence, also known as the Cambridge Reference Sequence.57 Analysis of mtDNA uses specific databases to determine the haplotype frequency in a population. The most common used are the EMPOP and MITOMAP.26,27,58–60 However, it is necessary to specify that the PD of the Y-STR and mtDNA is lower in comparison with STRs.
Genotyping provides a measurement of the genetic variation between members of a species, among which SNPs are the most common type of genetic variation (National Center for Biotechnology Information database of single nucleotide polymorphisms).28 Since SNPs are evolutionarily conserved, these polymorphisms have been proposed as candidate markers for use in association studies in place of microsatellites, and different panels of SNPs have been designed and validated for population and human identity studies.12,61 SNP-microarray platforms can also provide a genetic fingerprint for testing complex forensic DNA mixtures.62
Commercial kits based on insertion/deletion panel analysis such as the Investigator DIPplex kit (Qiagen) and the DIPplex PCR amplification kit (Mentype), have recently been developed and validated in human identification studies.63,64 Thus, combining the advantageous features of both SNPs and STRs, insertion/deletion-based DNA profiling has proved to be a useful tool for quality control in population studies, biobanking projects, paternity cases, and relationship and disaster victim testing.3,37,39,63
Most of the above-described polymorphisms are currently analyzed by capillary electrophoresis using automatic sequencing equipment. More recently, the availability of next-generation sequencing technologies has started to revolutionize all fields of molecular genetics.65 This method offers many advantages when compared with the classic Sanger sequencing technique, including the ability to rapidly detect all variants and mutations in an entire genome, and the “deep sequencing” approach, the reading of the target sequence with an extended coverage, ie, repeated hundreds of times. Application of next-generation sequencing to forensic genetics might offer considerable advantages, including the large number of polymorphisms (STR, mtDNA, SNPs, insertions/deletions) that may be combined into a single next-generation sequencing assay or increased simplification of interpretation of mixtures using the deep sequencing approach.66
After several debates, the first next-generation sequencing kits for human identification, ie, the HID-Ion AmpliSeq identity panel, HID-Ion AmpliSeq ancestry panel, Ion Torrent, HID STR 10-plex (Thermo Fisher Scientific), and ForenSeq DNA signature prep kit (Illumina) are now commercially available.66,67
Statistical framework
DNA analyses for individual identification are essentially based on the comparison of genetic profiles. Comparison may give rise to:
- Genetic compatibility (match) – sample and reference share the same genotype and there are no differences between the two
- Genetic incompatibility – comparison of genotypes between sample and reference shows that differences can be explained only by the origin of biological material from different individuals
- Inconclusiveness – not enough information to draw conclusions.
Of the three possible conclusions, only the first one requires statistical treatment. Mathematical statistical models based on knowledge of the genetic markers used for the analysis, population genetics, and the laws of probability are currently used for interpretation of genetic compatibility.13
The first and most basic task is to obtain the frequency of the locus of a certain genotype in the population to which the donor belongs. Since genotypes arise from combination of alleles, it follows that genotypic frequencies can be inferred from the allelic frequencies observed at the corresponding locus. In particular, if any genetic drift is shown to affect the independent inheritance of alleles within the population of interest, then the inference can be computed by means of the Hardy–Weinberg equilibrium:
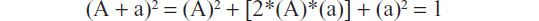
where A and a, given a male Aa and a female Aa, are the observed frequencies of the two alleles of a biallelic locus. In this particular case, “a” is the allele with the lower frequency, so it is usually referred to as the minor allele frequency. In contrast, if a drift is known to exist, like that due to non-random mating, some corrections have to be applied to the formula given above. In particular, the inferred frequency of homozygotes can be adjusted using the parameter theta (θ), which accounts for inbreeding:
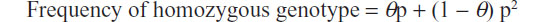
where p is the allele frequency. It must be noted that, albeit small (0.01< θ <0.03), such a correction has a noticeable impact over the inferred genotypic frequencies, so that it must be carefully chosen.68 For an AB heterozygote, the adjustment is Pr = 2papb (1 − q). Of note, if the repository’s population of donors includes samples from related individuals, then the computations to infer both homozygous and heterozygous genotypic frequencies should be corrected to account for that.69,70
Once the frequency of a particular genotype has been calculated, the next task is to compute the probability of a given profile. In this case, if the loci of the profile are independent from each other in inheriting a certain genotype, then the probability to obtain a given profile corresponds to the combination of the probabilities of the genotypes of each locus. Since such a probability is given by a simple multiplicative procedure, it is often referred to as the “product rule”, especially in the forensic field, as follows:69 combined probability of loci = product rule = Πn freq (n observed genotypes).
The value obtained by the product rule measures the probability that two unrelated individuals may present the same profile by chance. For this reason, it is also referred to as the random match probability (RMP):
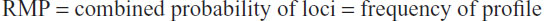
Going back to our statement earlier in this section about specificity for the donor, such a quantity can be seen as the error committed in matching a specimen to a certain donor on the basis of the actual profile observed. Hence, if a profile has a given RMP, then its specificity for the donor is represented by the PD:

The higher the RMP, the lower will be the error committed in matching a specimen to a certain donor.
In situations where it is reasonable to consider the specimen as a mixture of contributors rather than being from a single donor (as in the case of suspected contamination or specimens of a neoplastic nature), it is useful to express the RMP as the probability that the specimen comes from the donor whose profile appears in the mixture being tested. Such information is delivered by the likelihood ratio (LR):
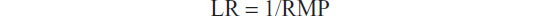
In other words, the more the observed profile is specific for the donor, the more likely it is that the specimen comes from that single donor. Wherever more than one profile is recognized in a mixture (more than one donor), appropriate modifications can be made to the LR calculation.70 In a more general way, the individuality of a mixture’s contributor can be handled by means of the so-called “allele-based” approach instead of the “genotype-based” approach described so far. In this regard, the information provided by the RMP and PD can be delivered by the random man not excluded probability and the PD, respectively.71,72 Noteworthy is that the allelic approach is thought to provide such information without any particular assumption about the number of possible contributors, since the calculations do not involve genotypes arising from possible combinations of the observed alleles.
Software used in forensics includes DNA View and EasyDNA.73–75 Other statistical applications can be searched for on the Short Tandem Repeat DNA Internet DataBase site.24
Fingerprinting of samples in biobanks: state of the art
Despite the relevance of the topic and the impact of biobanks on biomedical research, not all studies describing standard operative procedures for use in biobanks emphasize traceability and methods to be used for recognition of biological samples.8,76
Some groups have approached the problem of traceability using validated classical forensic genetics systems. For example, Azari et al used a system based on the 13 STR loci in the Combined DNA Index System along with sex determination using the amelogenin gene for authentication of human cell lines in the National Cell Bank of Iran.77 To avoid the problem of high disposal costs and the need for expensive equipment, the method includes singlex amplification of each STR, polyacrylamide gel separation, and silver stain detection. A mixture of two PCR products with different non-overlapping sizes is applied to a single lane of polyacrylamide gel (one sample, seven lanes). Although the system shows a certain degree of complexity due to its manual execution, it is robust and efficient, and has an average RMP of approximately one in a trillion (1012) among unrelated individuals.77
On the other hand, Cardoso et al used the AmpFlSTR Identifiler PCR amplification kit and the AmpFlSTR second-generation multiplex plus PCR amplification kit for genetic authentication of blood spots on three different types of support medium (absorbent paper, lint, or non-colored cotton fabric) used in the Banco de ADN, University of the Basque Country, Spain.14 Although the use of this procedure was limited in terms of verification of the integrity of nucleic acids in the various types of medium over several years of storage, the authors noted that using the commercial kits employed in forensic genetics is expensive and labor-intensive.
Recently, Kelly et al from the Coriell Institute for Medical Research investigated the feasibility of performing STR analysis for traceability and biological identification in biobanks of DNA preparations isolated directly from blood collected in the PAXgene blood RNA system (PreAnalytiX, Qiagen).78 This system uses stabilizing agents that inhibit degradation of RNA in blood samples kept for several days at room temperature.79 For this reason, the system is widely used in sample collection for biobank storage and multicenter studies, but has the disadvantage to be depleted of DNA. The results obtained by Kelly et al show that the little amount of DNA extracted from blood samples treated as detailed above is of sufficient quality and quantity to be suitable for analysis with both the PowerPlex 18D System commercial kit (Promega) and the home-made Coriell 6-Plex fluorescent PCR assay. The latter was developed by the Coriell Cell Repositories for detection of six highly polymorphic tetranucleotide microsatellites, ie, FES/FPS, vWA31, D22S417, D10S526, D5S592, and VWA31, and includes primers for the amelogenin gene for sex determination.80 This system is able to discriminate between samples with a matching probability of approximately one in three million.
In a recent study performed at the Integrated Biobank of Luxembourg, Kofanova et al developed a system based on multiplex PCR amplification and subsequent separation on agarose gel for evaluation of the VNTRs of six minisatellites (Apo-B1, COL2A1, D17S5, D1S80, D2S44, and PAH) and one additional locus for sex determination using detection of the SRY gene on the Y chromosome, reaching a DP of 106.81 This method is basically an improvement of a protocol developed in 2005 at the Leibniz Institute DSMZ (German Collection of Microorganisms and Cell Cultures), in which the same VNTR markers were used by developing singlex PCR.29,82 The authors, who have considerable long-term experience with quality control in biobanks, emphasize that this system is rapid, robust, and capable of verifying the identity of a biospecimen.9,76,81,83
A different approach was pursued by Cross et al, who conceived and validated a panel of 36 heterochromosomic SNPs and a sex marker to discriminate biological samples in the Marshfield Clinic Personalized Medicine Research Project.12,84 The design of the panel was based on the assumption that the best method for detecting biological individuality in a biobank is the use of molecular markers developed for forensic applications. These, however, although useful to identify and track a biological sample, do not provide any information on the disease associated with the same sample. Based on these considerations, Cross et al developed a panel of 36 polymorphisms related to the predisposition to various common diseases, including metabolic, neoplastic, cardiovascular, neurodegenerative, and genetic diseases.12 Polymorphisms were detected using a matrix-assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometer and Sequenom Typer 3.4 software for allele determination. This system has some strengths but also several potential limitations. The method is easily reproducible in a laboratory with common expertise of molecular medicine and the number of polymorphisms used for this assay provides a good discrimination power. However, due to the nature of the polymorphisms used, which are necessarily related to specific diseases, this method can be successfully applied only to populations of Caucasian ethnicity. In fact, some polymorphisms have a minor allele frequency below 10% in those of African and Asian ethnicity and consequently decrease the PD and applicability of this assay to these populations. Another limitation is that the clinical relevance of some polymorphisms included in the assay may be overestimated and other polymorphisms not considered could have a strong correlation with certain diseases.12
A MALDI-TOF mass spectrometry platform was also used on archival formalin-fixed, paraffin-embedded tissues for testing the new Sample ID PlusH kit from Sequenom Bioscience, which is based on 47 non-functional SNPs.85 The authors, after comparing the use of the kit and classical STRs, argue that this SNP genotyping platform is a promising and suitable approach for sample identification by next-generation sequencing when processing low amounts of DNA from formalin-fixed, paraffin-embedded tissues.85 Similarly, in a genome wide association study performed on 14,000 cases of seven common diseases and 3,000 controls in the British population, the researchers’ group, the Wellcome Trust, used a panel of 38 SNPs to obtain a molecular fingerprint of biological samples.86
With the aim of proposing a valid alternative to the current STR-based standard for cell line authentication, Liang-Chu et al recently developed the SNPtrace panel, a simple and cost-effective commercial assay that simultaneously assesses 96 SNPs.87 This system shows a discrimination power and an ability to detect intraspecies cross-contamination comparable with those of STR profiling. The main advantages are related to lower cost and the improved performance displayed in analyzing samples characterized by microsatellite instability.87 Other authors have addressed the problem of early detection of contamination and mistaken interpretation of experimental results using an immunological approach. Beaumont and Bedsou performed immunological fingerprinting based on measurement of the serological titer of three human polyclonal antibodies specific for Epstein–Barr virus, Bordetella pertussis, and Chlamydia pneumoniae.83 These microorganisms were selected for their capacity to induce long-lasting humoral immunity in humans. For this purpose, Biobanque de Picardie researchers have developed a triplex peptide fingerprinting enzyme-linked immunosorbent assay using selected synthetic peptides as antigens for detection of specific serological antibodies. Specific software was also developed to evaluate the intra-assay coefficient of variation for each immunoglobulin G level in different samples. This method appears to be a specific tool for quality control, and is reliable and easy to use. However, despite a discriminatory power of 95%, we consider that it cannot be used for identification of a specific subject, given that the antibody titers vary in time in the same individual.
Using a combination of 53 insertion/deletion polymorphisms and two markers for sex determination, Mathot et al developed a fluorescent assay based on a multiplex ligation-dependent genome amplification technique.37 In this technique, genomic DNA fragmented using restriction digestion is subsequently circularized and amplified using universal primers. The method is low-cost because only one probe is required per target, and has fast running times (<5 hours). Use of insertion/deletion polymorphisms has the great advantage of avoiding the discrepancy occurring between normal and cancer tissues when targeting STR in cancer patients displaying loss of heterozygosity or microsatellite instability.
As already described by Fearon and Vogelstein, in the course of the complex process of progression of neoplastic disease, tumors characterized by chromosomal instability, which present an accumulation of mutations in the APC, P53 and KRAS genes, frequently have a loss of genetic material, such as loss of heterozygosity, occurring in the repetitive regions.88 Another pathway is characterized by mutational events involving the DNA mismatch repair gene and is characterized by microsatellite instability.89 In both cases, DNA instability can be highlighted by analysis of the same mononucleotide or dinucleotide microsatellites; however, tetranucleotide repeats, employed in commercial kits used for forensic and paternity analysis, are also involved in several types of neoplasia, including solid tumors and leukemias.90–92
Nevertheless, to fully perform such an investigation, a sample handling robotic platform, capillary electrophoresis automated sequencing, and high qualified personnel were required, making its realization difficult for most of the traditional biobanks.
In 2013, with the aim of implementing the standard operative procedures of the Interinstitutional Multidisciplinary BioBank, we developed a rapid, reliable, low-cost, and simple DNA fingerprinting tool for routine use in quality control for biorepository samples.3 The method includes a double multiplex Alu insertion/deletion genotyping panel (eight Alu insertion/deletion polymorphisms and two sex markers on DXZ4 and SRY loci) suitable for biological identification and sample contamination assessment. The strength of this study is represented by the simplicity of execution capabilities through simple PCR amplification and agarose gel electrophoresis, without the need for expensive equipment. Use of insertion and deletion has proved to be highly useful for biological samples of cancer, regardless of the presence or absence of microsatellite instability. Furthermore, the average RMP and the PD, although not sufficiently discriminatory for strictly legal forensic investigations, provide a satisfactory standard of informativeness for biorepository purposes. Finally, a rapid economic analysis, despite the non-automated nature of this method, indicated an execution time of one day and an approximate cost of 3 USD. This last point should be compared with the conclusions of Glock et al at the Blood Donation Centre of the Austrian Red Cross for Vienna.11 In their study performed in 2002, they used the commercial AmpFISTR Profiler Plus kit to ensure the correspondence of the donor between plasma and whole blood samples. Regardless of the medicolegal implications for the blood donation center and the chronological distance between the two studies, economic evaluation indicates that analysis of a number of samples can be completed within 24 hours, by using a single capillary sequencer, at a cost of approximately 14 USD per sample.11
Conclusion
Despite the large number of studies conducted in this field, the wide availability of commercial kits, and specific international guidelines, the applicability of these methods in biobanks is often not practicable for several reasons.8,14 First, the expanding number of biobanks can not afford the acquisition of the equipment and software necessary for these kind of tests. A number of biobanks have a limited budget and often it is not possible to purchase the infrastructure, such as an automatic sequencer or a mass spectrometer, whose cost is comparable to that of several -80°C freezers.93 Second, the specific reagents required are extremely expensive, especially for large-scale projects.14 Considering the prices of the different commercial kits, it is easy to estimate the economic burden for a biological bank, which shifts the choice between commercial kits and low-cost “home-made” methods in favor of the latter.3,11,81 Finally, a highly specialized approach is required since these techniques involve different professional figures, including molecular biologists and geneticists.3
The situation becomes even worse when the choice of a specific molecular methodology needs to be applied to a particular biorepository. Indeed, as described above, the method of molecular genotyping must be carefully considered depending on the types of biospecimens stored in the biobank. For example, issues related to neoplastic biospecimens with variable degrees of microsatellite instability must be considered when using STRs.90,91 Moreover, in biobanks dedicated to specific diseases, the choice of an SNP-based fingerprinting method could be statistically biased if polymorphisms related to the pathology of the examined samples are selected.12 In any case, it will always be difficult to use markers with allele frequencies that do not depend on ethnic differences.12,81 The use of biological samples for forensic identification panels can present some ethical issues that should be evaluated in each specific case and in each specific country. With regard to this complex issue, the reader is referred to the considerations described by the International Society for Biological and Environmental Repositories and the Combined DNA Index System.15,22 In this review, we have attempted to report the main currently used methods of traceability and biological individuality. In addition, we have described the most important studies utilizing fingerprint techniques for biobanking purposes. Our hope is that further studies may move the current problems forward to develop less expensive and more reliable technological approaches.
Acknowledgments
This study was partially supported by the European Social Fund, under the Italian Ministry of Education, University and Research, PON03PE_00146_1/10 BIBIOFAR to FG (CUP B88F12000730005) and was partially supported by a grant (MERIT RBNE08NKH7) to the San Raffaele Foundation Ceglie Messapica (http://www.fondazionesanraffaele.com/). We wish to thank Dr Nadia Lopez for her excellent assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Steinberg K, Beck J, Nickerson D, et al. DNA banking for epidemiologic studies: a review of current practices. Epidemiology. 2002; 13(3):246–254. | |
Gion M, Fabricio AS. New frontiers in tumor marker studies: from biobanking to collaboration in translational research. Int J Biol Markers. 2011;26(2):73–74. | |
Palmirotta R, De Marchis ML, Ludovici G, et al. A reliable and reproducible technique for DNA fingerprinting in biorepositories: a pilot study from BioBIM. Int J Biol Markers. 2013;28(4):e398–e404. | |
Palmirotta R, Ludovici G, De Marchis ML, et al. Pre-analytical procedures for DNA studies: the experience of the Interinstitutional Multidisciplinary BioBank (BioBIM). Biopreserv Biobank. 2011;9(1):35–45. | |
Palmirotta R, De Marchis ML, Ludovici G, et al. Impact of pre-analytical handling and timing for PBMCs isolation and RNA studies. The experience of the Interinstitutional Multidisciplinary BioBank (BioBIM). Int J Biol Markers. 2012;27(2):e90–e98. | |
Della-Morte D, Guadagni F, Palmirotta R, et al. Genetics of ischemic stroke, stroke-related risk factors, stroke precursors and treatments. Pharmacogenomics. 2012;13(15):1741–1757. | |
Savonarola A, Palmirotta R, Guadagni F, Silvestris F. Pharmacogenetics and pharmacogenomics: role of mutational analysis in anti-cancer targeted therapy. Pharmacogenomics J. 2012;12(4):277–286. | |
Reid Y, Mintzer J. The current state of cell contamination and authentication – and what it means for biobanks. Biopreserv Biobank. 2012; 10(3):236–238. | |
Nanni U, Betsou F, Riondino S, et al. SPRECware: software tools for Standard PREanalytical Code (SPREC) labeling – effective exchange and search of stored biospecimens. Int J Biol Markers. 2012;27(3):e272–e279. | |
Nanni U, Spila A, Riondino S, et al. RFID as a new ICT tool to monitor specimen life cycle and quality control in a biobank. Int J Biol Markers. 2011;26(2):129–135. | |
Glock B, Reisacher RB, Schöck MA, et al. DNA profiling: a valuable tool for quality control of sample logistics including occurrences of suspected sample confusion in a blood donation centre. Vox Sang. 2002;82(3):137–140. | |
Cross DS, Ivacic LC, McCarty CA. Development of a fingerprinting panel using medically relevant polymorphisms. BMC Med Genomics. 2009;2:17. | |
Butler JM. Advanced Topics in Forensic DNA Typing: Methodology. San Diego, CA, USA: Elsevier Academic Press; 2012. | |
Cardoso S, Valverde L, Odriozola A, Elcoroaristizabal X, de Pancorbo MM. Quality standards in biobanking: authentication by genetic profiling of blood spots from donor’s original sample. Eur J Hum Genet. 2010;18(7):848–851. | |
International Society for Biological and Environmental Repositories (ISBER). Vancouver, BC, Canada. Available from http://www.isber.org/. Accessed May 15, 2015. | |
Tagliabracci A, Alessandrini F, Mazzarini L, Onofri V, Onori N, Turchi C. [Introduction to forensic genetics-investigations of personal identification and paternity] Introduzione alla genetica forense-Indagini di identificazione personale e di paternità. Milano, Italy: Springer-Verlag Italia; 2010. Italian. | |
Li WH, Gu Z, Wang H, Nekrutenko A. Evolutionary analyses of the human genome. Nature. 2001;409(6822):847–849. | |
Kass DH, Batzer MA. Genome organization: human. Available from: http://onlinelibrary.wiley.com/doi/10.1038/npg.els.0001889/otherversions. Accessed June 27, 2015. | |
Strachan T, Read A. Human Molecular Genetics. 4th ed. London, UK: Garland Science/Taylor and Francis Group; 2011. | |
Doveri S, Lee D, Maheswaran M, Powell W. Molecular markers - history, features and applications. In: Kole C, Abbott AG, editors. Principles and practices of plant genomics. Volume 1: Genome mapping. Enfield, HN: Science Publishers; 2008:23–67. | |
Kidd KK. The ALlele FREquency Database [updated 2012]. New Haven, CT, USA: Yale University; 1999. Available from: http://alfred.med.yale.edu/alfred/index.asp. Accessed March 26, 2015. | |
FBI Federal Bureau of Investigation. Combined DNA Index System (CODIS) [updated 2014]. United States Government. Available from: http://www.fbi.gov/about-us/lab/biometric-analysis/codis. Accessed March 26, 2015. | |
International Society for Forensic Genetics. Mainz, Germany; 2015. Available from: http://www.isfg.org. Accessed March 26, 2015. | |
Butler JM, Reeder DJ. Short Tandem Repeat DNA Internet Database [updated 2015]. Biochemical Science Division, National Institute of Science and Technology; 1997. Available from: http://www.cstl.nist.gov/strbase/index.htm. Accessed March 26, 2015. | |
Willuweit S, Roewer L. Y-chromosome STR Haplotype Reference Database 2000–2015 [updated 2015]. Available from: http://www.yhrd.org. Accessed March 26, 2015. | |
European DNA Profiling. Forensic mtDNA Population Database 1999 [updated 2015]. Available from: http://www.empop.org. Accessed March 26, 2015. | |
Lott MT, Leipzig JN, Derbeneva O, et al. mtDNA variation and analysis using MITOMAP and MITOMASTER. Curr Protoc Bioinformatics. 2013;1(123):1.23.1–1.23.26. | |
National Center for Biotechnology Information. Database of single nucleotide polymorphisms [updated 2015]. Bethesda, MD, USA: US National Library of Medicine. Available from: http://www.ncbi.nlm.nih.gov/snp. Accessed March 26, 2015. | |
[Deutsche Sammlung von Mikoorganismen Und Zellkulturen. Braunschweig; Jena, Germany: Leibniz Institute. Available from: http://www.dsmz.de/. Accessed March 26, 2015. | |
Jeffreys AJ, Wilson V, Thein SL. Hyper variable mini satellite regions in human DNA. Nature. 1985;314(6006):67–73. | |
Jin L, Zhong Y, Chakraborty R. The exact numbers of possible microsatellite motifs. Am J Hum Genet. 1994;55(3):582–583. | |
Kirby LT. DNA Fingerprinting: An Introduction. New York, NY, USA: WH Freeman; 1990. | |
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. | |
Brohede J, Ellegren H. Microsatellite evolution: polarity of substitution within repeats and neutrality of flanking sequences. Proc Biol Sci. 1999;266(1421):825–833. | |
Barreto G, Vago AR, Ginther C, Simpson AJG, Pena SDJ. Mitochondrial D-loop signatures produced by low-stringency single specific primer PCR constitute a simple comparative human identity test. Am J Hum Genet. 1996;58(3):609–616. | |
Jobling MA, Pandya A, Tyler-Smith C. The Y chromosome in forensic analysis and paternity testing. Int J Legal Med. 1997;110(3):118–124. | |
Mathot L, Falk-Sörqvist E, Moens L, Allen M, Sjöblom T, Nilsson M. Automated genotyping of biobank samples by multiplex amplification of insertion/deletion polymorphisms. PLoS One. 2012; 7(12):e52750. | |
Weber JL, David D, Heil J, Fan Y, Zhao C, Marth G. Human diallelic insertion/deletion polymorphisms. Am J Hum Genet. 2002;71(4):854–862. | |
Pereira R, Phillips C, Alves C, et al. A new multiplex for human identification using insertion/deletion polymorphisms. Electrophoresis. 2009;30:3682–3690. | |
Jobling MA, Gill P. Encoded evidence: DNA in forensic analysis. Nat Rev Genet. 2004;5(10):739–751. | |
Edwards A, Civitello A, Hammond HA, Caskey CT. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet. 1991;49(4):746–756. | |
Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics. 1992;12(2):241–253. | |
Kimpton CP, Fisher D, Watson S, et al. Evaluation of an automated DNA profiling system employing multiplex amplification of four tetrameric STR loci. Int J Legal Med. 1994;106(6):302–311. | |
Kimpton CP, Oldroyd NJ, Watson SK, et al. Validation of highly discriminating multiplex short tandem repeat amplification systems for individual identification. Electrophoresis. 1996;17(8):1283–1293. | |
GOV.UK. DNA database documents. UK Government Service and Information. Published March 20, 2013. Available from https://www.gov.uk/government/collections/dna-database-documents. Accessed March 26, 2015. | |
Werrett DJ. The national DNA database. Forensic Sci Int. 1997; 88:33–42. | |
Hares DR. Expanding the CODIS core loci in the United States. Forensic Sci Int Genet. 2012;6(1):e52–e54. | |
Butler JM. Genetics and genomics of core short tandem repeat loci used in human identity testing. J Forensic Sci. 2006;51(2):253–265. | |
Butler JM, Hill CR. Biology and genetics of new autosomal STR loci useful for forensic DNA analysis. Forensic Sci Rev. 2012;24(1):15–26. | |
GeneMapper® Software, appliedbiosystems.com [homepage on the Internet]. Thermo Fisher Scentific Inc. [updated 2015]. Available from: http://www.appliedbiosystems.com/absite/us/en/home/support/software/dna-sequencing/genemapper.html. Accessed March 26, 2015. | |
Zheng XZ, Hui P, Chang B, et al. STR DNA genotyping of hydatidiform moles in South China. Int J Clin Exp Pathol. 2014;7(8):4704–4719. | |
Brisighelli F, Blanco-Verea A, Boschi I, et al. Patterns of Y-STR variation in Italy. Forensic Sci Int Genet. 2012;6(6):834–839. | |
Raziel A, Dell’Ariccia-Carmon A, Zamir A. Reduction of Powerplex(®) Y23 reaction volume for genotyping buccal cell samples on FTA(TM) cards. J Forensic Sci. 2015;60(1):152–156. | |
Coble MD, Hill CR, Butler JM. Haplotype data for 23 Y-chromosome markers in four US population groups. Forensic Sci Int Genet. 2013;7(3):e66–e68. | |
Parvathy SN, Geetha A, Jagannath C. Haplotype analysis of the polymorphic 17 YSTR markers in Kerala nontribal populations. Mol Biol Rep. 2012;39(6):7049–7059. | |
Olofsson JK, Mogensen HS, Buchard A, Børsting C, Morling N. Forensic and population genetic analyses of Danes, Greenlanders and Somalis typed with the Yfiler® Plus PCR amplification kit. Forensic Sci Int Genet. 2015;16C:232–236. | |
Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. | |
Mikkelsen M, Fendt L, Röck AW, et al. Forensic and phylogeographic characterisation of mtDNA lineages from Somalia. Int J Legal Med. 2012;126(4):573–579. | |
Roewer L, Parson W. Internet Accessible Population Databases: YHRD and EMPOP. In: Siegel JA, Saukko PJ, editors. Encyclopedia of Forensic Sciences. 2nd ed. Amsterdam, The Netherlands: Elsevier Ltd; 2013. | |
Parson W, Dürb A. EMPOP – a forensic mtDNA database. Forensic Sci Int Genet. 2007;1(2):88–92. | |
Phillips C, Fang R, Ballard D, et al. Evaluation of the Genplex SNP typing system and a 49plex forensic marker panel. Forensic Sci Int Genet. 2007;1(2):180–185. | |
Voskoboinik L, Ayers SB, LeFebvre AK, Darvasi A. SNP-microarrays can accurately identify the presence of an individual in complex forensic DNA mixtures. Forensic Sci Int Genet. 2015;16C:208–215. | |
LaRue BL, Ge J, King JL, Budowle B. A validation study of the Qiagen Investigator DIPplex® kit; an INDEL-based assay for human identification. Int J Legal Med. 2012;126(4):533–540. | |
Friis SL, Børsting C, Rockenbauer E, et al. Typing of 30 insertion/deletions in Danes using the first commercial indel kit-Mentype® DIPplex. Forensic Sci Int Genet. 2012;6(2):e72–e74. | |
Del Chierico F, Ancora M, Marcacci M, Cammà C, Putignani L, Conti S. Choice of next-generation sequencing pipelines. Methods Mol Biol. 2015;1231:31–47. | |
Børsting C, Morling N. Next generation sequencing and its applications in forensic genetics. Forensic Sci Int Genet. February 14, 2015. [Epub ahead of print.] | |
Fordyce SL, Mogensen HS, Børsting C, et al. Second-generation sequencing of forensic STRs using the Ion Torrent™ HID STR 10-plex and the Ion PGM™. Forensic Sci Int Genet. 2015;14:132–140. | |
Buckleton J, Triggs CM, Walsh SJ. Forensic DNA Evidence Interpretation. Boca Raton, FL, USA: CRC Press; 2004. | |
Balding DJ, Nichols RA. DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Sci Int. 1994;64(2–3):125–124. | |
Buckleton J, Triggs CM. Relatedness and DNA: are we taking it seriously enough? Forensic Sci Int. 2005;152(2–3):115–119. | |
Brenner CH. The mythical “exclusion” method for analysing DNA mixtures – does it make any sense at all? Proceedings of the American Academy of Forensic Sciences. 2011;17:79. | |
Buckleton J, Curran J. A discussion of the merits of random man not excluded and likelihood ratios. Forensic Sci Int Genet. 2008; 2(4):343–348. | |
dna-view.com [homepage on the Internet]. Forensic Mathematics [updated April 9,2015]. Available from: http://dna-view.com/. Accessed May 15, 2015. | |
Familias home page. [Updated 2015]. Available from: http://www.familias.name/index.html. Accessed May 15, 2015. | |
Fung WK, Hu Y-Q. EasyDNA. Statistical DNA Forensics: Theory, Methods and Computation [updated 2013]. Wiley, 2008. Available from: http://www.saasweb.hku.hk/EasyDNA/. | |
Betsou F. Clinical biospecimens: reference materials, certified for nominal properties? Biopreserv Biobank. 2014;12(2):113–120. | |
Azari S, Ahmadi N, Tehrani MJ, Shokri F. Profiling and authentication of human cell lines using short tandem repeat (STR) loci: report from the National Cell Bank of Iran. Biologicals. 2007;35(3):195–202. | |
Kelly VR, Jones SP, Sammartino HL, Arocena DI, Madore SJ. Donor verification using short tandem repeat (STR) analysis directly from blood collected in PAXgene RNA tubes. Biopreserv Biobank. 2014;12(3):217–219. | |
Duale N, Brunborg G, Rønningen KS, et al. Human blood RNA stabilization in samples collected and transported for a large biobank. BMC Res Notes. 2012;5:510. | |
Coriell Institute for Medical Research. Identity mapping kit [updated 2015]. Camden, NJ, USA. Available from: https://catalog.coriell.org/0/Sections/Search/MSK.aspx?Ref=MSK&PgId[=]202. Accessed March 26, 2015. | |
Kofanova OA, Mathieson W, Thomas GA, Betsou F. DNA fingerprinting: a quality control case study for human biospecimen authentication. Biopreserv Biobank. 2014;12(2):151–153. | |
Dirks WG, Drexler HG. Authentication of scientific human cell lines: easy-to-use DNA fingerprinting. Methods Mol Biol. 2005;290:35–50. | |
Beaumont K, Betsou F. Immunological fingerprinting method for differentiation of serum samples in research-oriented biobanks. Clin Vaccine Immunol. 2010;17(5):735–740. | |
McCarty CA, Wilke RA, Giampietro PF, Wesbrook SD, Caldwell MD. Marshfield Clinic Personalized Medicine Research Project (PMRP): design, methods and recruitment for a population-based biobank. Per Med. 2005,2:49–79. | |
Miller JK, Buchner N, Timms L, et al. Use of Sequenom sample ID Plus® SNP genotyping in identification of FFPE tumor samples. PLoS One. 2014;9(2):e88163. | |
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. | |
Liang-Chu MM, Yu M, Haverty PM, et al. Human biosample authentication using the high-throughput, cost-effective SNPtrace(TM) system. PLoS One. 2015;10(2):e0116218. | |
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. | |
Stigliano V, Assisi D, Cosimelli M, et al. Survival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patients. J Exp Clin Cancer Res. 2008;27:39. | |
Poetsch M, Petersmann A, Woenckhaus C, et al. Evaluation of allelic alterations in short tandem repeats in different kinds of solid tumors – possible pitfalls in forensic casework. Forensic Sci Int. 2004; 145(1):1–6. | |
Page K, Graham EA. Cancer and forensic microsatellites. Forensic Sci Med Pathol. 2008;4(1):60–66. | |
Filoglu G, Bulbul O, Rayimoglu G, et al. Evaluation of reliability on STR typing at leukemic patients used for forensic purposes. Mol Biol Rep. 2014;41(6):3961–3972. | |
Elliott P, Peakman TC; UK Biobank. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int J Epidemiol. 2008;37(2):234–244. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.