")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Disorders of sex development: advances in genetic diagnosis and challenges in management
Authors Kyriakou A, Lucas-Herald A, McGowan R, Tobias E, Ahmed F
Received 9 January 2015
Accepted for publication 2 March 2015
Published 8 April 2015 Volume 2015:5 Pages 165—177
DOI https://doi.org/10.2147/AGG.S53226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr John Martignetti
Andreas Kyriakou,1 Angela K Lucas-Herald,1 Ruth McGowan,2 Edward S Tobias,2 S Faisal Ahmed1
1Developmental Endocrinology Research Group, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK; 2Medical Genetics, Southern General Hospital, Glasgow, UK
Abstract: Disorders of sex development (DSD) are a group of rare conditions that usually present with atypical genitalia in the newborn period or as delayed puberty in an adolescent. Although a concern about the development of external genitalia may exist in one in 300 newborn infants, discrete genetic conditions that underlie DSD are generally rarely identified. It is likely that this diagnostic gap exists for a number of reasons and these include an inadequate knowledge of the pathogenesis and underlying mechanisms that lead to DSD, variation in assessment and in-depth phenotyping of these rare conditions, inadequate availability of quality accredited laboratories and, lastly, limited awareness of the value of a molecular genetic diagnosis for improving short-term and long-term care of the affected person.
Keywords: DSD, genetics, novel, rare disease, genomics, NGS
Introduction to the management issues in disorders of sex development (DSD)
DSD are a group of rare conditions that are generally characterized by an abnormality of the chromosomal, gonadal or phenotypic features that typically define sex development. Such conditions usually present with atypical genitalia in the newborn period or as delayed puberty in an adolescent. Although truly ambiguous genitalia on expert examination are relatively rare, reported to occur in about 1:4,500 births,1 a concern about the development of the external genitalia may, however, exist in one in 300 newborn infants.2 Discrete genetic conditions that underlie DSD are generally rarely identified. For instance, congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, which is the commonest cause of 46,XX DSD, has an approximate incidence of one in 10,000 to one in 14,000 in all infants. Whilst over 95% of cases of 46,XX DSD are due to this condition, the likelihood of identifying a discrete genetic condition in cases of 46,XY DSD is, at best, 50%.3 It is likely that this diagnostic gap exists for a number of reasons and these include an inadequate knowledge of the pathogenesis and underlying mechanisms that lead to DSD, variation in assessment and in-depth phenotyping of these rare conditions, inadequate availability of quality accredited laboratories and, lastly, limited awareness of the value of a molecular genetic diagnosis for improving short-term and long-term care of the affected person. Besides narrowing the diagnostic gap, the other challenges that face genetics include the development of a cost effective and efficient method of processing large scale genetic data in such a way that not only does it benefit individual patients in explaining inter-individual disease variation but it can also be integrated with other rapidly developing technologies (including transcriptomics, proteomics, and metabolomics as well as novel methods of targeted phenotyping such as cellular and organ imaging), so that the pathology as well as the management of each person with a DSD can be understood at an individual level.
The review is subdivided into four main sections: the first section covers the pathway of the genetic control and the pathophysiology of sex development as well as the current classification of DSD and the initial diagnostic investigations. Emphasis is placed on the advances in genetic and genomic technologies for the clinical diagnosis and on the ethical challenges in management. The final section covers the future directions in the field of DSD, including the modern aspects of electronic media and communication, the networks of clinical and research expertise and the necessity for them to extend and include a wider community of scientists.
Pathogenesis of DSD
DSD are a group of heterogeneous conditions with diverse pathophysiology.4 There are many factors involved in the differentiation of the sex organs into male or female, resulting in a diverse clinical phenotype (Figure 1).
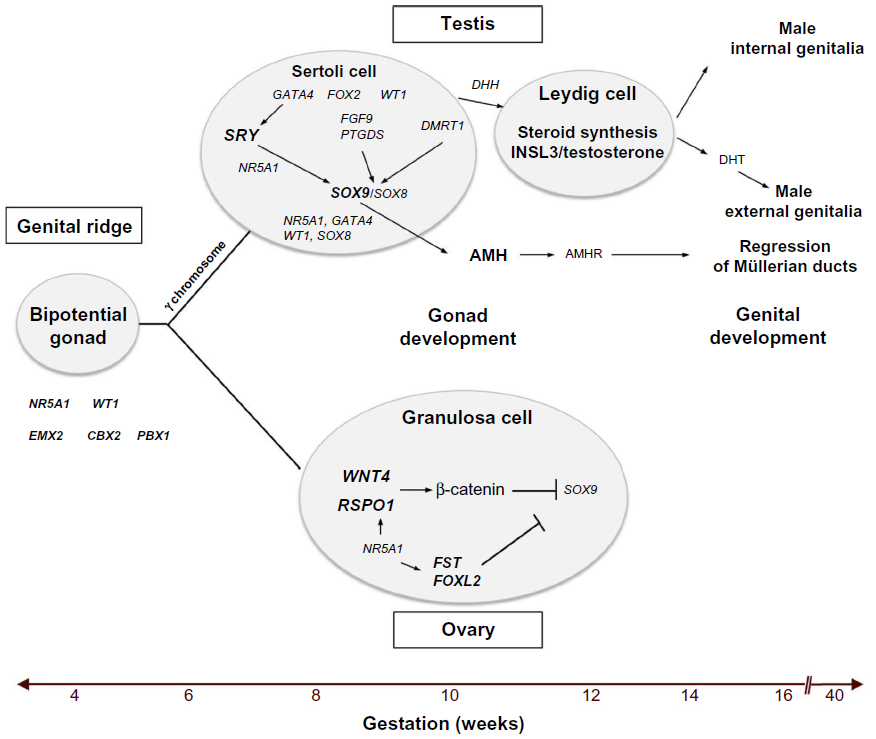 | Figure 1 A summary of the critical molecular and genetic events in mammalian sex determination and differentiation. |
The urogenital ridge, the common precursor of the urinary and genital systems, develops at approximately 4 weeks post-fertilization in the human embryo as a thickening of the mesodermic mesonephros covered by coelomic epithelium.3 Several genes including NR5A1, WT1, EMX2, CBX2 and PBX1 are required for formation of the bipotential gonadal ridge. At 7 weeks, in the XY gonad, SRY is expressed in pre-Sertoli cells resulting in the upregulation of SOX9 expression that is further augmented by the synergistic action of SRY and NR5A1, leading to the initiation of definitive Sertoli cell differentiation.5,6 The transcriptional activation by SRY and NR5A1 is mediated by the binding of these two proteins to the testis-specific enhancer of SOX9 core (TESCO) region, which lies approximately 13 kb upstream of SOX9.7 Once SOX9 levels reach a critical threshold, several positive regulatory loops are initiated, including autoregulation of SOX9 expression and formation of feed-forward loops via FGF9 or PGD2 signaling.5,6 During testicular development, SOX9 functions, by regulating the production of AMH from Sertoli cells, and possibly by repressing genes involved in ovarian development such as WNT4 and FOXL2.6,8,9 The DMRT1 transcription factor may also be involved in this process.3
Once formed, Sertoli cells induce the development of fetal Leydig cells, via a hedgehog-signaling pathway, which at 8–9 weeks of development produces androgens and INSL3.10 Testosterone and AMH cause regression of Müllerian structures and differentiation of the Wolffian duct into the epididymis, vas deferens, and seminal vesicles, whereas INSL3 is required for testicular descent. In 46,XY males, testosterone is converted to 5-dihydrotestosterone (DHT) by the enzyme 5-AR, resulting in the development of male external genitalia.11,12 The activity of DHT is mediated by the nuclear transcription factor, AR, which has high affinity for DHT.
In the XX gonad, the absence of SRY, resulting in the inability of SOX9 expression to reach a critical threshold, together with the expression of factors such as RSPO1/WNT4 signaling, FST and FOXL2 lead to formation of the ovary, at least in part through suppression of the activity of “testis” genes.8,9 In the 46,XX female, the Wolffian duct regresses and the Müllerian duct is maintained and forms the oviduct, uterus, cervix, and upper part of the vagina. The absence of androgens leads to the development of female genitalia.
The use of genomic analyses in recent years (including, for example, high-resolution single nucleotide polymorphism [SNP] array with copy-number estimation using Robust Multichip Analysis), has led to the identification of genomic alterations affecting several of the genes that participate in the control of gonadal development and to the delineation of their transcriptional regulatory regions. For instance, genomic rearrangements (such as duplication and triplication) affecting SOX9 regulatory elements, located significantly further upstream (>500 kb) than the TESCO enhancer region, were found in patients with 46,XX sex reversal.13–15
In addition, genomic rearrangements affecting the regulatory region of SOX3, and leading to its increased or ectopic expression, were detected in three people with 46,XX sex reversal.16 The X chromosomal SOX3 gene, believed to be the ancestor of the SRY gene, is not normally expressed in the developing gonad. Therefore, when ectopically expressed, it might substitute for SRY in driving testicular development.7 In addition, a probable regulatory element lying downstream of the GATA4 gene was identified when a 35 kb deletion was detected in a person with 46,XY complete gonadal dysgenesis.17 Similarly, in those with 46,XY DSD, microarray analysis detected an intragenic deletion of WWOX18 and a deletion at least 42 kb upstream of the DMRT1 gene.19 In fact, array analyses have now identified many DNA duplications and deletions in people with DSD, several of which are located close to known sex development genes and are suspected to have caused disruption of gene regulatory regions.7
It is clear, therefore, that there is potential for disruption of the sex organ differentiation process at multiple different stages. The clinical phenotype will depend on the nature of disruption.
Classification of DSD
The current system for the classification of DSD was introduced in the Chicago Consensus in 2005.20 There are three broad groups: sex chromosome DSD, 46,XX DSD, and 46,XY DSD.
Sex chromosome DSD
Sex chromosome DSD includes conditions such as 47,XXY (Klinefelter syndrome and variants), 45,X (Turner syndrome and variants), 45,X/46,XY (mixed gonadal dysgenesis) and 46,XX/46,XY (chimerism). These are often identified antenatally, frequently as an incidental finding, with confirmation of the diagnosis after birth.21 Antenatal diagnosis allows for focused evaluation of the other complications often associated with these disorders, for example, cardiac anomalies in Turner syndrome. It also provides the opportunity to offer counselling to families prior to the birth.
46,XX DSD
46,XX DSD encompasses disorders of gonadal development, such as gonadal dysgenesis and disorders secondary to androgen excess. Androgen excess during pregnancy may be endogenous (secondary to an adrenal adenoma, dermoid cyst, Sertoli-Leydig tumor, sex cord stromal tumor or metastatic carcinoma) or exogenous (secondary to danazol, progestins or potassium sparing diuretics).22 Exogenous steroids taken during pregnancy can also cause posterior fusion of the labia, clitoral enlargement, and increased degrees of androgenization.23
The commonest known genetic condition that leads to 46,XX DSD is CAH due to 21α-hydroxylase deficiency and this occurs in approximately one in 10,000 to one in 14,000 infants.24 Rarer conditions include 46,XX testicular DSD which refers to a male with testes and male genitalia, and 46,XX ovotesticular DSD which refers to individuals that have both ovarian and testicular tissue in the gonads, usually as ovotestes, but less commonly as separate gonads.
46,XY DSD
46,XY DSD has three broad categories: disorders of gonadal development, disorders in androgen synthesis or action and other causes, including hypogonadotropic hypogonadism, cryptorchidism, and isolated hypospadias. Girls with 46,XY DSD will most likely have androgen insensitivity syndrome (AIS), gonadal dysgenesis or a biochemical disorder of androgen synthesis.
Diagnosis
Initial investigations should include a thorough history and clinical examination of the infant. Clinical examination should not simply focus on the external genitalia, but also seek to determine if there are any dysmorphic features or evidence of further developmental anomalies. Careful inspection and palpation of the external genitalia should then be undertaken, with the aid of scoring systems, such as the external masculinization score for a more objective approach.25
After initial examination, first line investigations should include a karyotype, ultrasound examination for Müllerian structures, and serum levels of AMH and 17-hydroxyprogesterone. Ideally, results of these tests should be available within 48 hours to allow for sex assignment as early as possible.26 Additional investigations should include serum testosterone, cortisol, androstenedione, gonadotropins, and urinalysis. Serial measurements of urea and electrolytes are useful, in order to identify cases of CAH with salt wasting, although this may present later. The requirements for further investigations will be guided by the results of these initial tests.4
Advances in genetic and genomic technologies for clinical diagnosis of DSD
Genetic testing is recognized as a key element in the investigation of individuals with a suspected DSD.26 Molecular diagnosis is useful in the management of DSD, potentially providing information about long-term fertility, adrenal health, and germ-line tumor risk as well as an understanding of the etiology and recurrence risks for affected individuals and their parents.
The first line investigation of a possible DSD includes quantitative fluorescence-polymerase chain reaction and karyotyping, with respective turnaround times of 1–2 days and 5 days.23 These investigations are crucial where there is uncertainty about sex assignment but also are an important guide for management where phenotypic sex is apparent. Sex chromosome DSD may be identified when a karyotype is performed and in this situation it is diagnostic in itself, obviating the need for further molecular analysis. The most common sex chromosome DSD is Turner syndrome (45,X) followed by Klinefelter syndrome (47,XXY) and, less commonly, other forms of sex chromosome mosaicism may be identified. In the latter situation, a karyotype of genital or other tissue may be useful, or further analysis performed, to detect the presence of Y chromosome material. Prenatal studies consistently show that the majority of fetuses with confirmed 45,X/46,XY mosaicism are phenotypically normal at birth.27 A proportion of these may be at risk of late-onset “Turner syndrome-like” sequelae. However, there is no correlation between the severity of phenotype and the ratio of 45,X and 46,XY cell lines in the blood.28 Small supernumerary marker chromosomes (sSMC) can be associated with a normal karyotype but also with a Turner syndrome karyotype (mos 45,X/46,X,+mar). The majority of these sSMC are derived from sex chromosomes, generally either sSMC(X) ring or sSMC(Y), inverted duplication/isodicentric chromosomes.29 However, their exact chromosome break points are rarely defined. Future more precise characterization of sSMC(X) or sSMC(Y) using molecular cytogenetic techniques may provide greater understanding of their cause and effect.
Detailed chromosome analysis using array comparative genomic hybridization (aCGH) allows the identification of microdeletions or microduplications, below the threshold of visibility by standard karyotype analysis (5 Mb) and is now widely available in optimally resourced centers. As standard practice, the routine genetic investigation of learning disability now includes aCGH, with a 15% rate of identification of a clinically significant microdeletion or microduplication. It is now recognized that in syndromic and non-syndromic DSD, recurrent, clinically significant regions of copy number variation (CNV) may be found in approximately a fifth of individuals.30 In 46,XX DSD due to disorders of Müllerian development, aCGH variations may be present in over a quarter of the cases.31 Furthermore, specific genomic regions of CNV may correlate with atypical genitalia, cryptorchidism, and hypospadias. These regions of CNV may be de novo, occurring as a new event in the affected individual, or may be inherited and thus have implications for parents with regard to future recurrence risk and for the wider family. It is recognized that the variable phenotype associated with certain DSD genes, such as NR5A1, explains why a region of CNV spanning this gene may be inherited from an apparently unaffected mother who in fact had premature ovarian failure.32
The identification of novel genes involved in DSD as well as insight into developmental mechanisms may be facilitated using aCGH. For instance, investigation of individuals with myotubular myopathy and 46,XY DSD with this technique led to the identification of a commonly deleted region, which spanned a single gene, MAMLD1 (CXorf6).33 Subsequently, nonsense mutations have been identified in Japanese boys with penoscrotal hypospadias and further studies have confirmed that mutation of MAMLD1 may contribute to a small proportion of 46,XY DSD.34
Traditional methods of molecular investigation of DSD include Sanger sequencing, often combined with multiplex ligation-dependent probe amplification to identify abnormalities in the dosage of a gene, such as a gene deletion or duplication. Investigation of 46,XY males would often include stepwise analysis of the AR gene, with mutations detected in approximately 30% of individuals with partial androgen insensitivity syndrome (PAIS), followed by SRY and NR5A1 testing in individuals with gonadal dysgenesis.35,36 Mutations in MAMLD1, HSD17B3, and SRD5A2 and duplication of DAX1 are likely to account for a smaller proportion (5%–10%) of molecular diagnoses but their relative contributions to 46,XY DSD is less clear. The investigation of 46,XY DSD females (mostly AIS) is clearer, with mutations in the AR gene found in over 80% of individuals.35 Similar techniques remain the mainstay of investigation of 46,XX DSD females, of whom the majority have CAH due to compound heterozygous, or homozygous, mutations in CYP21A2 gene.37 Such a stepwise approach to genetic analysis in a combination of people with 46,XY and 46,XX DSD is reported to reveal a genetic alteration, including chromosomal rearrangement and gene mutations, in 64% of cases.38
The heterogeneous nature, particularly of XY DSD, has led to the grouping of genes into panels that can still be analyzed by these traditional methods. Advances in genetic technology, however, combined with an improved understanding of the complex network of genes involved in gonadal development, have led to the use of next generation sequencing (NGS) to permit more rapid analysis of a larger number of genes. Compared to standard fluorescent dideoxynucleotide Sanger sequencing, NGS, also known as massively parallel sequencing, permits at least 1,000 times more DNA sequence to be analyzed per run, in the diagnostic laboratory. The technology is advancing extremely rapidly, with regard to both the machines and the sequencing chemistry that can be used.39 The advantages and limitations of NGS and of other types of genetic analysis are summarized in Table 1.
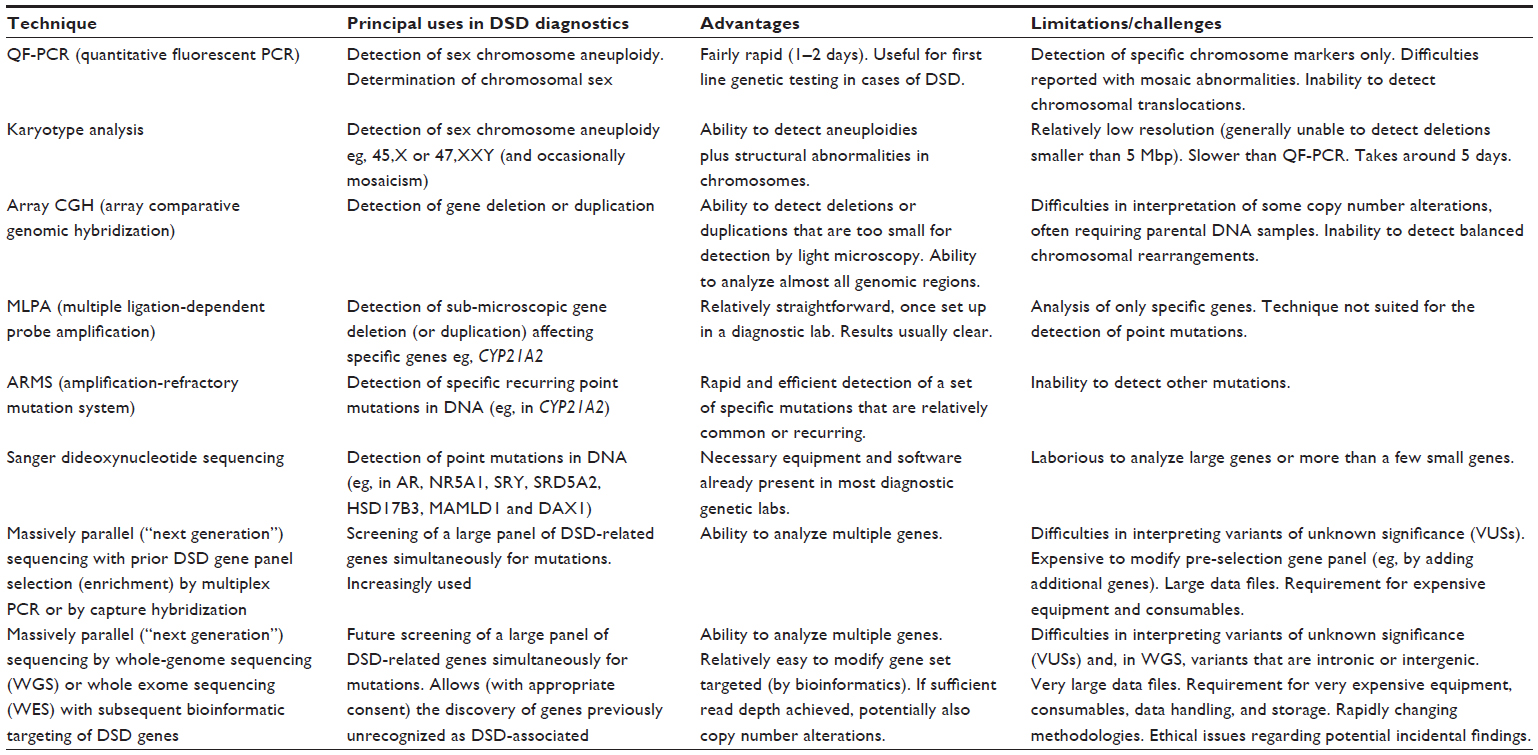 | Table 1 A comparison of the principal uses, advantages, and limitations of the more commonly used diagnostic genetic analysis techniques |
At the present time, there are several different forms of NGS technologies. Two of the most popular technologies in the UK are the so-called Ion Semiconductor method used by the Life Technologies machines (including the Ion Torrent PGM and the Ion Proton) and the method utilized by Illumina (in the MiSeq, NextSeq 500, HiSeq 2500, and HiSeq X machines). The Life Technologies method detects the pH changes that result from the release of individual protons upon the addition of individual nucleotides. In contrast, the Illumina method employs fluorescence analysis as its means of detecting nucleotide addition. It is, in fact, Illumina that is providing the DNA sequencing for the 100,000 Genomes Project of Genomics England.
Detection of sequence changes within a set or panel of many DSD-related genes could be achieved by pre-selecting DNA sequences (by using a customized hybridization method or by a custom-designed multiplex polymerase chain reaction). This pre-selection approach is at present the most commonly used strategy in UK diagnostic laboratories for NGS of gene panels. An alternative approach is to conduct whole genome or whole exome sequencing and then to use bioinformatic selection of the regions of DNA sequence that are related to the set of genes of interest (ie, the target genes). This could be done in two or more stages: for example analyzing an initial set of the most commonly involved genes and then, if no pathogenic is detected, analyzing a wider set of genes, thus minimizing the number of detected variants of uncertain significance (Tables 2 and 3).
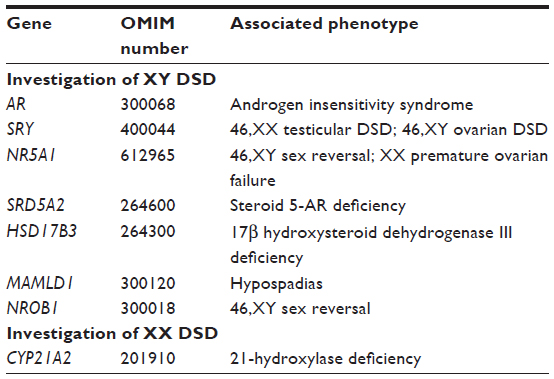 | Table 2 Primary list of DSD-related genes for simultaneous mutation analysis |
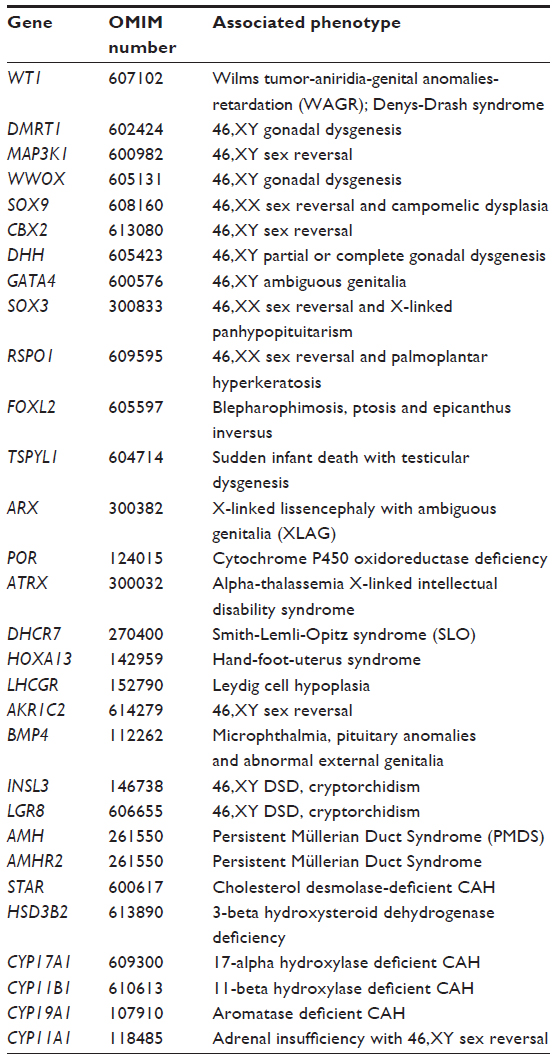 | Table 3 Extensive panel of DSD-related genes for simultaneous mutation analysis, if no pathogenic variants are detected from the primary list of genes |
At present, unfortunately, while whole genome sequencing provides the most even coverage of the gene sequences (ie, with least likelihood of excluding gene sequences of interest) and is being used in a number of research laboratories worldwide, this approach is currently generally too costly for routine diagnostic purposes. Moreover, the clinical significance of variants located within approximately 98% of the genome is currently generally uninterpretable, although the ongoing international ENCODE project (ENCODE; encyclopedia of DNA elements) may clarify the significance of non-coding regions of the genome in the future.40
In research laboratories, NGS by whole exome or whole genome sequencing can provide not only the means by which to detect mutations within a large set of known DSD-related genes simultaneously, but also to identify new causative genes. In clinical diagnostic laboratories, however, the pre-selected gene set (or gene panel) approach is currently most widely utilized in the UK for the simultaneous mutation analysis of multiple genes. A recent report of an exome sequencing approach with filters for a large set of 64 genes known to be associated with a DSD, revealed an abnormality in about 30% of cases of 46,XY DSD41 whereas the stepwise sequencing approach using a limited panel of nine genes identified an abnormality in 65% of cases of 46,XY DSD.38 It is possible that the reported diagnostic yield may depend on the clinical approach that is utilized, but there is a need to clearly understand the health as well as economic benefit of these two different approaches.
Despite the power of NGS, there are nevertheless several obstacles that usually require to be overcome in a diagnostic laboratory, prior to the introduction of NGS methodology.39 These include the purchase of expensive equipment and information technology infrastructure that can process and store enormous data files (eg, FastQ files) and the acquisition of bioinformatics expertise. A data analysis pipeline must be generated that is able to reliably map the millions of sequence reads to the genome, identify sequence variants, and then filter and extract a small number of likely pathogenic mutations from a very large dataset of detected sequence variants. A particular challenge is the determination of pathogenicity of missense mutations (amino acid substitutions), despite a range of available computer algorithms for this purpose. These include not only the popular SIFT and Polyphen2 algorithms but also Align-GVGD, MutPred, nsSNPAnalyzer, Panther, PhD-SNP, SNAP, and SNPs&GO.39 A further challenge is deciding upon the extent of variant information that should be reported to the patients themselves, although this is currently being addressed by wide discussion and by published European and US guidelines.42 Finally, it is important that, for clinical reporting of mutations, there is a multi-disciplinary group (including laboratory scientists and experienced clinicians with specialized knowledge) who together can assess the significance of detected variants, in conjunction with available detailed clinical phenotypic and biochemical data.41
Increasing economic constraints on health spending generates a necessity to justify the clinical utility of investigation of affected individuals. Newer technologies are becoming more affordable but, even where available, at present their expense may restrict their wider application. In developed countries, obtaining a molecular diagnosis for a DSD is still only possible in less than half of affected individuals. Genomic or exomic sequencing therefore, offers the opportunity to identify additional genes and mechanisms associated with the development of these conditions, to improve diagnostic rates and, ultimately, to have a positive impact on management.
Current and emerging therapies – ethical challenges
Optimal care for infants and adolescents with DSD requires an experienced multidisciplinary team. The team should include specialists in endocrinology, surgery and/or urology, clinical psychology/psychiatry, radiology, nursing, and neonatology. For infants with suspected DSD, this team has a role in providing patients and parents with information regarding diagnosis, sex assignment, and treatment options. In addition, the clinical team should have links to a wider multidisciplinary team that consists of specialists from adult endocrinology, plastic surgery, gynecology, clinical genetics, clinical biochemistry, adult clinical psychology and social work and, if possible, also links to a clinical ethics forum.26
The presence at birth of a congenital anomaly affecting a child’s genital appearance or future fertility potential is challenging for both parents and health care providers. Shortly after birth major decisions are made, including sex assignment and genital surgery, without knowledge of orientation, interests, or needs of the future adolescent and adult. The decisions taken are most of the time irreversible while, at the same time, interfering in highly sensitive domains, affecting reproduction, sexuality, physical and mental integrity, and the formation of a person’s self-identity.43,44
The Chicago Consensus of 2005 stated that sex assignment cannot solely be based on genital appearance but should include consideration of the diagnosis, surgical options, need for lifelong replacement therapy, the potential for fertility, views of the family, and circumstances relating to cultural practices.20 Although karyotype and external appearance of genitalia continue to be associated with the choice of assigned sex,45 the results from a recent analysis of the International DSD (I-DSD) Registry revealed that the practice of assigning male sex in newborns with 46,XY DSD seems to be increasing.46 This change in practice may be influenced by improvements in long-term surgical outcome in men with 46,XY DSD with conditions such as micropenis and potential for penile reconstruction or neophallus construction.47,48 The knowledge of current trends in the practice of sex assignment increases the necessity for improving the knowledge about the genetic etiology in 46,XY DSD. In addition, it has implications for the long-term management of domains such as hypogonadism, subfertility, gonadal tumorigenesis, sexual function, gender identity, and quality of life. It is also possible that there may be more cases of complex hypospadias and micropenis that may require expert input for reconstructive surgery later in life.46,48,49
It is likely that practice has also been influenced by those cases of XY DSD who were raised as girls and who developed gender dysphoria. Gender identity shows fluidity among persons born with PAIS, 5-AR deficiency and 17β-HSD3 deficiency.43,50 Testosterone influences the development of a male gender role and probably increases the chance of a male gender identity development. However, even in circumstances in which the person has been prenatally exposed to high levels of testosterone and reared as girl from birth, a female adult gender identity is the rule rather than the exception.51 Psychosexual outcomes in DSD have been most extensively studied in 46,XX individuals with CAH.52 The vast majority of those raised as girls develop and maintain a female gender identity.53 However, a less strong female identification and gender dysphoria are more common in this group than in women without CAH51,54–56 and this may affect their quality of life.57
Early management in DSD has been based on the need to establish sex of rearing. Thereafter, medical and surgical treatment has been used to reinforce this decision. Feminizing surgery comprises clitoroplasty, labioplasty, and vaginoplasty. It is performed to allow menstrual flow, permit vaginal intercourse, and achieve a pleasing cosmetic result.58 The current recommendation suggests for restriction of genital surgery to those girls with severe masculinization of the external genitalia.20,59
There is no evidence to determine the best timing for clitoral reduction or vaginoplasty and there are no long-term studies comparing the functional, cosmetic, and psychological outcomes of early versus delayed (after puberty) reconstructive surgery. Some parents may consider early genital surgery as a mechanism that could protect their child from the risk of future stigma, however a thorough discussion of the controversies around undertaking or withholding early genital surgery is imperative in informing decisions.26,44 Supporting evidence for early surgery suggests that high rates of stress in mothers, social stigmatization, and isolation of the child and adverse psychosexual development could be avoided by early surgical treatment.58,60,61 Women, who had undergone surgery, when asked about the optimal timing of surgery, preferred scheduling the feminizing procedure in early childhood.62–64
On the other hand, altered physical sex development does not usually represent a surgical emergency for newborns. Psychological support to parents being given the diagnosis of DSD to their newly born child may be more important than discussing surgical procedures with them. These conditions present significant parenting stress and reduced adaptive coping capacity, irrespective of the degree of genital ambiguity.65 It is unclear whether surgical correction of atypical external genitalia is necessary or sufficient for well-being; quality of life, particularly with respect to sexual function, also needs to be taken into account.44 Damage of the neuronal network is reported to be highly likely in clitoral reduction, despite improvement in surgical technique, thus affecting sexual function in adult life.66–69 The cosmetic result in adulthood is also influenced by changes during puberty, including pubic hair and labial fat development. In the case of CAH, steroid replacement may induce reduction of the clitoris after a few months of treatment. Vaginal dilation is first line therapy for the girls with vaginal hypoplasia to permit menstruation70 but can be deferred where there is no uterus, until such time that the girl is sexually active. Moreover, most surgical interventions are irreversible and may restrict later options for sex reversal. In conditions associated with significant risk of gender dysphoria early surgery may also compromise future options.
DSD is associated with increased risk for germ-cell cancer (GCC). GCC is more prevalent (30%–50%) in conditions associated with gonadal dysgenesis as compared to 46,XY disorders of hormone synthesis or action (<1%–15%), where testis development is normal.71–74 An abdominal or inguinal position of the gonads represents an additional risk factor.75 The informed decision of the parent/patient for performing gonadectomy is based on the GCC risk and the sex of rearing.76
In boys with partial gonadal dysgenesis, Leydig cell function may be sufficient for spontaneous pubertal development77–79 and, therefore, the current practice is to leave the gonads in place. In view of the increased risk of GCC in undescended testes, laparoscopy and early orchidopexy is performed at 6–12 months of age allowing for regular palpation, ultrasound, and biopsy. In girls with partial gonadal dysgenesis, the possibility of androgen production during puberty, the expected infertility and the high risk of GCC, usually lead to the decision of gonadectomy during early or late childhood.76
Although past practice was the prophylactic removal of the gonads in females with complete androgen insensitivity syndrome (CAIS) as soon as the diagnosis is confirmed, it is more common now, as in PAIS, to leave the gonads in place until puberty is complete, because it allows spontaneous pubertal development without estrogen replacement and allows the adolescent to be involved in the discussions around surgery. Given the practice of prophylactic gonadectomy in most cases in the past, data on GCC risk in adults are still lacking, but have been estimated to range from 0.8%–22%.71
A small group of adult women with CAIS choose to retain their gonads after puberty, despite the malignancy risk, avoiding surgery and life-long replacement therapy and also raising the possibility, in light of advances in fertility treatment, of preservation of fertility potential.80 The testes in AIS are structurally normal, and spermatogonia have been reported in resected testes in several series, though they pose an uncertain fertility potential.81,82 The difficulty with conserving intra-abdominal gonads is that there are no available reliable screening tests that detect early malignant change. Understanding the molecular etiology underlying normal gonadal development allowed the development of markers to estimate the risk for GCC.76 It is expected that, with increased accessibility of diagnostic facilities, and with more children with these conditions being raised as males, additional data will become available in the future.
The increasing knowledge about sex determination and differentiation and modern society’s decreasing rigidity about the nature of sexual identity and gender roles have led to a thorough reappraisal of former treatment practice in DSD. Involving patient support groups in the assessment of medical interventions and seeking for more evidence on the long-term outcome of quality of life are both essential.44,83 The children should participate in decisions that affect them now or later and they should be comprehensively informed about their condition.57 Children who are informed and educated about their condition will have better opportunities to develop adaptive coping skills, including development of a positive self-image and expectations for a fulfilling adult life.
Future directions
Registries and data sharing
Given that discrete conditions that are associated with DSD are all very rare, meaningful research into these rare conditions that has led to major evidence-based changes in clinical practice are limited. Following the Chicago Consensus in 2005, which highlighted the need for greater data sharing across geographical boundaries, a group of experts created a web-based registry which was initially funded by the European Society for Paediatric Endocrinology and a European Union (EU) 7th Framework grant (EuroDSD) and is currently supported by the Medical Research Council of the UK as the I-DSD Registry (http://www.i-dsd.org). This registry has users from 47 countries from all six habitable continents. Of these countries, there are 37 centers that have entered almost 1,500 cases. The registry also supports the development of new modules, as it is currently undertaking for EU-TAIN (Treatment for adrenal insufficiency in neonates and infants; http://www.tain-project.org), another EU funded project. Through its standardized collection of data, the registry has the ability to perform conclusive secondary research46,84 and has the potential to act as a gateway to a virtual research environment that integrates other datasets and paves the way to primary research. To maximize stakeholder involvement, the registry has recently introduced the option for participants to access some of the data that have been entered on them by their clinicians. It is likely that greater involvement of these patient participants in the I-DSD Registry will lead to more patient-focused research as well as an improved wider understanding of complex research questions.
Networks of clinical and research expertise
Although many centers manage conditions such as DSD through a multidisciplinary team, it is increasingly realized that such expertise is not available at many centers and there is a need to throw the net wider. Registries such as I-DSD are not simply a virtual biobank of data on people with DSD, but are also a registry of experts who manage a range of rare conditions. Thus, a rare disease registry can also facilitate the development of a virtual network of clinical and research expertise. There is a need for formal networks at several levels. Recently, the European Cooperation of Science and Technology has funded the development of a network of clinical researchers, scientists, and patient groups (DSDnet; http://www.dsdnet.eu). It is anticipated that this formal network will lay down the framework as well as the priorities for research that needs to be performed over the next decade. It is also anticipated that this body will also define the standards of care that should be maintained in centers of expertise in DSD. Thus, it is possible that DSDnet evolves into a European reference network for DSD that relies on the I-DSD Registry for data curation as well as dissemination of information to a wider international as well as a virtual network of clinicians, scientists as well as patients and participants.
Handling complex genetic information
With increasing availability of complex genetic analyses including aCGH, NGS as well as whole exome and genome sequencing, many patients will undergo these analyses. With the increasing complexity of genetic and bioinformatic analysis and the greater emphasis and requirement for clear communication with patients and carers, it is likely that the diagnostic and prognostic genetic pathway that starts and ends with the patients will require a wider group of experts. A DSD diagnostic group such as that described recently41 or created by the Scottish DSD Network (http://www.sdsd.scot.nhs.uk) and which consists of pediatric endocrinologists, clinical and molecular geneticists and steroid biochemists, who have links to bioinformatic support, will become increasingly routine. In cases where there is uncertainty at a regional level, there is a need for the international DSD community to develop a panel of experts that can advise and assist. This requires a new way of working and a level of collaboration that has not previously existed in the field of DSD. However, given the experience of recent initiatives such as I-DSD, DSDnet, and the DSD Working Group of the European Society for Paediatric Endocrinology (http://www.eurospe.org) it is likely that this will succeed. These networks need to extend and include the wider community of scientists so that new cost efficient but reliable tools can be developed for functional confirmation of novel genetic variants that have not been previously linked to a DSD phenotype.
Transcriptomics and its potential to clarify the phenotype
One of the major findings of projects such as ENCODE40 was that the human transcriptome is far larger and more complex than the genome, especially with the presence of alternative splicing of genes and the presence of thousands of non-coding ribonucleic acids (RNAs) that have the ability to modify RNA silencing and post-transcriptional regulation of gene expression. Sexual dimorphism of gene transcription is a very common phenomenon in the mammal and in the mouse and may affect two thirds of the transcriptome in some tissues.85 Although it is possible that some of this sexual dimorphism is due to sex chromosome differences, it is possible that some of these differences are due to androgen effects. The presence of an androgen-dependent transcriptome has been identified in genital skin fibroblasts86 as well as peripheral blood mononuclear cells.87 An assessment of an androgen-dependent transcriptome may be of benefit in a number of clinical scenarios but its translation into clinical practice will require major bioinformatic input.
Imaging
In the past, most radiological research focused on improving the technical quality of imaging. However, substantial advances in magnetic resonance spectroscopy, diffusion weighted imaging, dynamic contrast enhanced methods and radiochemistry means that cellular and molecular processes such as angiogenesis, apoptosis, signal transduction and metabolic pathways can now be imaged and this is already becoming increasingly routine in the management of androgen dependent prostate cancer.88 The prospect of improving our understanding of the underlying pathology, linking it to the genetic variation and understanding the longitudinal evolution of the clinical phenotype as well as its response to therapy will advance considerably as these technologies become more accessible.
Health economic assessment of interventions in DSD
Rare diseases may pose a burden on the affected person and the family89 and whilst parents of children with DSD clearly report experiencing difficulties in coping,65 this has rarely been quantified. In addition, some affected people and their clinical and non-clinical carers may not necessarily consider the condition a disorder but simply a difference in development which does not have a bearing on day to day function. Given that in most cases, interventions involve surgical, medical, and psychological interventions with a limited evidence base, there is a need to understand the health economic benefit of these interventions. This assessment should also be extended to the use of new diagnostic and therapeutic technologies so that the clinical utility of these complex procedures can be quantified.
Conclusion
In conclusion, DSD represents a group of diverse, rare conditions with a very wide phenotypic spectrum where a clear molecular genetic diagnosis remains elusive in the majority. These conditions pose multiple therapeutic challenges. It is likely that innovation in “omic” technologies will lead to improved understanding of the underlying abnormality as well as explain the variability in the phenotype. However, a real improvement in the health of those who are affected requires a change in practice of the scientific community so that data sharing in clinical practice and scientific endeavor becomes normal practice in this large group of rare conditions.
Acknowledgments
SFA is supported by a Medical Research Council partnership award G1100236. The work was also assisted by a UK Wellcome Trust ISSF grant to EST and SFA and also by a University of Glasgow Medical Fund award and Teaching Excellence Award to EST.
Disclosure
The authors do not have any conflict of interest.
References
Thyen U, Lanz K, Holterhus PM, Hiort O. Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm Res. 2006;66(4):195–203. | |
Ahmed SF, Dobbie R, Finlayson AR, et al. Prevalence of hypospadias and other genital anomalies among singleton births, 1988–1997, in Scotland. Arch Dis Child Fetal Neonatal Ed. 2004;89(2):F149–F151. | |
Ahmed SF, Bashamboo A, Lucas-Herald A, McElreavey K. Understanding the genetic aetiology in patients with XY DSD. Br Med Bull. 2013;106:67–89. | |
Lucas-Herald A, Ahmed SF. Sex assignment in disorders of sex development. eLS. Chichester: John Wiley & Sons, Ltd; 2012. | |
Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol Rev. 2007;87(1):1–28. | |
Sekido R, Lovell-Badge R. Sex determination and SRY: down to a wink and a nudge? Trends Genet. 2009;25(1):19–29. | |
Ohnesorg T, Vilain E, Sinclair AH. The genetics of disorders of sex development in humans. Sex Dev. 2014;8(5):262–272. | |
Uhlenhaut NH, Jakob S, Anlag K, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139(6):1130–1142. | |
Schlessinger D, Garcia-Ortiz JE, Forabosco A, Uda M, Crisponi L, Pelosi E. Determination and stability of gonadal sex. J Androl. 2010; 31(1):16–25. | |
Barsoum IB, Bingham NC, Parker KL, Jorgensen JS, Yao HH. Activation of the Hedgehog pathway in the mouse fetal ovary leads to ectopic appearance of fetal Leydig cells and female pseudohermaphroditism. Dev Biol. 2009;329(1):96–103. | |
Woodward M, Patwardhan N. Disorders of sex development. Surgery (Oxford) 2010;28:396–401. | |
Biason-Lauber A. Control of sex development. Best Pract Res Clin Endocrinol Metab. 2010;24(2):163–186. | |
Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. 2011;364(1):91–93. | |
Benko S, Gordon CT, Mallet D, et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. 2011;48(12):825–830. | |
Vetro A, Ciccone R, Giorda R, et al. XX males SRY negative: a confirmed cause of infertility. J Med Genet. 2011;48(10):710–712. | |
Sutton E, Hughes J, White S, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. 2011;121(1):328–341. | |
White S, Ohnesorg T, Notini A, et al. Copy number variation in patients with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS One. 2011;6(3):e17793. | |
White S, Hewitt J, Turbitt E, et al. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur J Hum Genet. 2012;20(3):348–351. | |
Barbaro M, Balsamo A, Anderlid BM, et al. Characterization of deletions at 9p affecting the candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur J Hum Genet. 2009;17(11):1439–1447. | |
Hughes IA, Houk C, Ahmed SF, Lee PA; Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148–162. | |
Pasterski V, Prentice P, Hughes IA. Impact of the consensus statement and the new DSD classification system. Best Pract Res Clin Endocrinol Metab. 2010;24(2):187–195. | |
Auchus RJ, Chang AY. 46, XX DSD: the masculinised female. Best Pract Res Clin Endocrinol Metab. 2010;24(2):219–242. | |
Ahmed SF, Rodie M. Investigation and initial management of ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24(2):197–218. | |
Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005; 365(9477):2125–2136. | |
Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85(1):120–124. | |
Ahmed SF, Achermann JC, Arlt W, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf). 2011;75(1):12–26. | |
Lazebnik N, Filkins KA, Jackson CL, Linn KB, Doshi NN, Hogge WA. 45,X/46,XY mosaicism: the role of ultrasound in prenatal diagnosis and counselling. Ultrasound Obstet Gynecol. 1996;8(5):325–328. | |
Telvi L, Lebbar A, Del Pino O, Barbet JP, Chaussain JL. 45,X/46,XY mosaicism: report of 27 cases. Pediatrics. 1999;104(2 Pt 1):304–308. | |
Liehr T, Mrasek K, Hinreiner S, et al. Small supernumerary marker chromosomes (sSMC) in patients with a 45,X/46,X,+mar karyotype – 17 new cases and a review of the literature. Sex Dev. 2007;1(6):353–362. | |
Tannour-Louet M, Han S, Corbett ST, et al. Identification of de novo copy number variants associated with human disorders of sexual development. PLoS One. 2010;5(10):e15392. | |
McGowan R, Tydeman G, Shapiro D, et al. DNA Copy Number Variations are Important in the Complex Genetic Architecture of Müllerian Disorders. Fertil Steril. Epub February 20, 2015. | |
Harrison SM, Granberg CF, Keays M, Hill M, Grimsby GM, Baker LA. DNA Copy Number Variations in Patients with 46,XY Disorders of Sex Development. J Urol. 2014;192(6):1801–1806. | |
Ogata T, Laporte J, Fukami M. MAMLD1 (CXorf6): a new gene involved in hypospadias. Horm Res. 2009;71(5):245–252. | |
Kalfa N, Fukami M, Philibert P, et al. Screening of MAMLD1 mutations in 70 children with 46,XY DSD: identification and functional analysis of two new mutations. PLoS One. 2012;7(3):e32505. | |
Ahmed SF, Cheng A, Dovey L, et al. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000; 85(2):658–665. | |
Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008;2(4–5):200–209. | |
White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245–291. | |
Laino L, Majore S, Preziosi N, et al. Disorders of sex development: a genetic study of patients in a multidisciplinary clinic. Endocr Connect. 2014;3(4):180–192. | |
Tobias ES, McElreavey K. Next generation sequencing for disorders of sex development. Endocr Dev. 2014;27:53–62. | |
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. | |
Baxter RM, Arboleda VA, Lee H, et al. Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J Clin Endocrinol Metab. 2015;100(2):E333–E344. | |
Wright CF, Middleton A, Burton H, et al. Policy challenges of clinical genome sequencing. BMJ. 2013;347:f6845. | |
Sandberg DE, Gardner M, Cohen-Kettenis PT. Psychological aspects of the treatment of patients with disorders of sex development. Semin Reprod Med. 2012;30(5):443–452. | |
Wiesemann C, Ude-Koeller S, Sinnecker GH, Thyen U. Ethical principles and recommendations for the medical management of differences of sex development (DSD)/intersex in children and adolescents. Eur J Pediatr. 2010;169(6):671–679. | |
Suresh D, Crawford J, Axelrad ME, et al. Assessing sex assignment concordance with genotype and phenotype. Int J Pediatr Endocrinol. 2013;2013(1):7. | |
Kolesinska Z, Ahmed SF, Niedziela M, et al. Changes over time in sex assignment for disorders of sex development. Pediatrics. 2014;134(3):e710–e715. | |
Callens N, Hoebeke P. Phalloplasty: A panacea for 46,XY disorder of sex development conditions with penile deficiency? Endocr Dev. 2014;27:222–233. | |
Sircili MH, e Silva FA, Costa EM, et al. Long-term surgical outcome of masculinizing genitoplasty in large cohort of patients with disorders of sex development. J Urol. 2010;184(3):1122–1127. | |
van der Zwan YG, Callens N, van Kuppenveld J, et al. Long-term outcomes in males with disorders of sex development. J Urol. 2013;190(3):1038–1042. | |
de Vries AL, Doreleijers TA, Cohen-Kettenis PT. Disorders of sex development and gender identity outcome in adolescence and adulthood: understanding gender identity development and its clinical implications. Pediatr Endocrinol Rev. 2007;4(4):343–351. | |
Dessens AB, Slijper FM, Drop SL. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav. 2005;34(4):389–397. | |
Stout SA, Litvak M, Robbins NM, Sandberg DE. Congenital adrenal hyperplasia: classification of studies employing psychological endpoints. Int J Pediatr Endocrinol. 2010;2010:191520. | |
Cohen-Kettenis PT. Psychosocial and psychosexual aspects of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2010;24(2):325–334. | |
Pasterski V, Zucker KJ, Hindmarsh PC, et al. Increased Cross-Gender Identification Independent of Gender Role Behavior in Girls with Congenital Adrenal Hyperplasia: Results from a Standardized Assessment of 4- to 11-Year-Old Children. Arch Sex Behav. Epub September 20, 2014. | |
Hines M, Brook C, Conway GS. Androgen and psychosexual development: core gender identity, sexual orientation and recalled childhood gender role behavior in women and men with congenital adrenal hyperplasia (CAH). J Sex Res. 2004;41(4):75–81. | |
Meyer-Bahlburg HF, Dolezal C, Baker SW, Ehrhardt AA, New MI. Gender development in women with congenital adrenal hyperplasia as a function of disorder severity. Arch Sex Behav. 2006;35(6):667–684. | |
Jürgensen M, Lux A, Wien SB, Kleinemeier E, Hiort O, Thyen U. Health-related quality of life in children with disorders of sex development (DSD). Eur J Pediatr. 2014;173(7):893–903. | |
Wolffenbuttel KP, Crouch NS. Timing of feminising surgery in disorders of sex development. Endocr Dev. 2014;27:210–221. | |
Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(9):4133–4160. | |
Warne GL, Raza J. Disorders of sex development (DSDs), their presentation and management in different cultures. Rev Endocr Metab Disord. 2008;9(3):227–236. | |
Ediati A, Juniarto AZ, Birnie E, Drop SL, Faradz SM, Dessens AB. Body Image and Sexuality in Indonesian Adults with a Disorder of Sex Development (DSD). J Sex Res. 2015;52(1):15–29. | |
Nordenström A, Frisén L, Falhammar H, et al. Sexual function and surgical outcome in women with congenital adrenal hyperplasia due to CYP21A2 deficiency: clinical perspective and the patients’ perception. J Clin Endocrinol Metab. 2010;95(8):3633–3640. | |
Wisniewski AB, Migeon CJ, Malouf MA, Gearhart JP. Psychosexual outcome in women affected by congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Urol. 2004;171(6 Pt 1):2497–2501. | |
Fagerholm R, Santtila P, Miettinen PJ, Mattila A, Rintala R, Taskinen S. Sexual function and attitudes toward surgery after feminizing genitoplasty. J Urol. 2011;185(5):1900–1904. | |
Duguid A, Morrison S, Robertson A, et al. The psychological impact of genital anomalies on the parents of affected children. Acta Paediatr. 2007;96(3):348–352. | |
Crouch NS, Liao LM, Woodhouse CR, Conway GS, Creighton SM. Sexual function and genital sensitivity following feminizing genitoplasty for congenital adrenal hyperplasia. J Urol. 2008;179(2):634–638. | |
Jürgensen M, Kleinemeier E, Lux A, et al. Psychosexual development in adolescents and adults with disorders of sex development – results from the German Clinical Evaluation Study. J Sex Med. 2013;10(11):2703–2714. | |
Minto CL, Liao LM, Woodhouse CR, Ransley PG, Creighton SM. The effect of clitoral surgery on sexual outcome in individuals who have intersex conditions with ambiguous genitalia: a cross-sectional study. Lancet. 2003;361(9365):1252–1257. | |
Lesma A, Bocciardi A, Corti S, Chiumello G, Rigatti P, Montorsi F. Sexual function in adult life following Passerini-Glazel feminizing genitoplasty in patients with congenital adrenal hyperplasia. J Urol. 2014;191(1):206–211. | |
Ismail-Pratt IS, Bikoo M, Liao LM, Conway GS, Creighton SM. Normalization of the vagina by dilator treatment alone in Complete Androgen Insensitivity Syndrome and Mayer-Rokitansky-Kuster-Hauser Syndrome. Hum Reprod. 2007;22(7):2020–2024. | |
Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27(5):468–484. | |
Berra M, Liao LM, Creighton SM, Conway GS. Long-term health issues of women with XY karyotype. Maturitas. 2010;65(2):172–178. | |
Sakai N, Yamada T, Asao T, Baba M, Yoshida M, Murayama T. Bilateral testicular tumors in androgen insensitivity syndrome. Int J Urol. 2000; 7(10):390–392. | |
Schober J, Nordenström A, Hoebeke P, et al. Disorders of sex development: summaries of long-term outcome studies. J Pediatr Urol. 2012;8(6):616–623. | |
Lip SZ, Murchison LE, Cullis PS, Govan L, Carachi R. A meta-analysis of the risk of boys with isolated cryptorchidism developing testicular cancer in later life. Arch Dis Child. 2013;98(1):20–26. | |
Cools M, Looijenga LH, Wolffenbuttel KP, T’Sjoen G. Managing the risk of germ cell tumourigenesis in disorders of sex development patients. Endocr Dev. 2014;27:185–196. | |
Cools M, Pleskacova J, Stoop H, et al. Gonadal pathology and tumor risk in relation to clinical characteristics in patients with 45,X/46,XY mosaicism. J Clin Endocrinol Metab. 2011;96(7):E1171–E1180. | |
Lindhardt Johansen M, Hagen CP, Rajpert-De Meyts E, et al. 45,X/46,XY mosaicism: phenotypic characteristics, growth, and reproductive function – a retrospective longitudinal study. J Clin Endocrinol Metab. 2012;97(8):1540–1549. | |
Martinerie L, Morel Y, Gay CL, et al. Impaired puberty, fertility, and final stature in 45,X/46,XY mixed gonadal dysgenetic patients raised as boys. Eur J Endocrinol. 2012;166(4):687–694. | |
Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence. Clin Endocrinol (Oxf). 2012;76(6):894–898. | |
Hannema SE, Scott IS, Rajpert-De Meyts E, Skakkebaek NE, Coleman N, Hughes IA. Testicular development in the complete androgen insensitivity syndrome. J Pathol. 2006;208(4):518–527. | |
Cools M, van Aerde K, Kersemaekers AM, et al. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J Clin Endocrinol Metab. 2005;90(9):5295–5303. | |
Amaral RC, Inacio M, Brito VN, et al. Quality of life of patients with 46,XX and 46,XY disorders of sex development. Clin Endocrinol (Oxf). 2015;82(2):274–279. | |
Cox K, Bryce J, Jiang J, et al. Novel associations in disorders of sex development: findings from the I-DSD Registry. J Clin Endocrinol Metab. 2014;99(2):E348–E355. | |
Yang X, Schadt EE, Wang S, et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16(8):995–1004. | |
Holterhus PM, Deppe U, Werner R, et al. Intrinsic androgen-dependent gene expression patterns revealed by comparison of genital fibroblasts from normal males and individuals with complete and partial androgen insensitivity syndrome. BMC Genomics. 2007;8:376. | |
Holterhus PM, Bebermeier JH, Werner R, et al. Disorders of sex development expose transcriptional autonomy of genetic sex and androgen-programmed hormonal sex in human blood leukocytes. BMC Genomics. 2009;10:292. | |
Gravina GL, Festuccia C, Bonfili P, et al. Strategies for imaging androgen receptor signaling pathway in prostate cancer: implications for hormonal manipulation and radiation treatment. Biomed Res Int. 2013;2013:460546. | |
Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371:2039–2041. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.