")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Discovery of nonnucleoside inhibitors of polymerase from infectious pancreatic necrosis virus (IPNV)
Authors Bello-Pérez M, Falco A , Galiano V , Coll J, Perez L, Encinar JA
Received 14 April 2018
Accepted for publication 14 May 2018
Published 30 July 2018 Volume 2018:12 Pages 2337—2359
DOI https://doi.org/10.2147/DDDT.S171087
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Melissa Bello-Pérez,1 Alberto Falcó,1 Vicente Galiano,2 Julio Coll,3 Luis Perez,1 José Antonio Encinar1
1Molecular and Cell Biology Institute (IBMC), Miguel Hernández University (UMH), Elche, Spain; 2Department of Physics and Computer Architecture, Miguel Hernández University (UMH), Elche, Spain; 3Department of Biotechnology, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Madrid, Spain
Introduction: Infectious pancreatic necrosis virus (IPNV) causes serious losses in several fish species of commercial interest. IPNV is a non-enveloped double-stranded RNA virus with a genome consisting of two segments A and B. Segment B codes for the VP1 protein, a non-canonical RNA-dependent RNA polymerase that can be found both in its free form and linked to the end of genomic RNA, an essential enzyme for IPNV replication.
Materials and methods: We take advantage of the knowledge over the allosteric binding site described on the surface of the thumb domain of Hepatitis C virus (HCV) polymerase to design new non-nucleoside inhibitors against the IPNV VP1 polymerase.
Results: Molecular docking techniques have been used to screen a chemical library of 23,760 compounds over a defined cavity in the surface of the thumb domain. Additional ADMET (absorption, distribution, metabolism, excretion, and toxicity) filter criteria has been applied.
Conclusion: We select two sets of 9 and 50 inhibitor candidates against the polymerases of HCV and IPNV, respectively. Two non-toxic compounds have been tested in vitro with antiviral capacity against IPNV Sp and LWVRT60 strains in the low µM range with different activity depending on the IPNV strain used.
Keywords: IPNV, HCV, antiviral drugs, non-nucleoside inhibitors, RdRp, molecular docking
Introduction
Since the discovery of the first vaccine against smallpox in 1796 by Edward Jenner, society has relied almost entirely on the development of new vaccines to battle viral diseases. However, the world’s capacity for vaccine development is currently already falling behind the rate of emergence and reemergence of dangerous viral diseases. Indeed, viral diseases are particularly troublesome, because effective treatments against most of them are lacking. For instance, approximately half the short list of US Food and Drug Administration-approved antiviral drugs are aimed at HIV1 and those remaining target only six more viruses.1 Furthermore, resistance to existing antimicrobials is emerging, along with new viral pathogens.2 Consequently, urgent efforts are necessary to accelerate novel antiviral drug discovery. One such technology that has allowed for the in silico screening of large amounts of compounds for targeting specific sites within functionally significant proteins is the use of bioinformatic tools. Through the use of increasingly potent computational hardware systems combined with the ever-increasing availability of detailed information on the molecular structure of relevant proteins and extensive chemical libraries, significant advances have been made in both clinical and veterinary fields.3,4
Infectious pancreatic necrosis virus (IPNV) was the first virus to be isolated from fish, and is now recognized as the known causative agent of IPN disease, which mainly affects cultured salmonids.5 IPNV belongs to the family Birnaviridae and is a member of the genus Aquabirnavirus. IPNV is the prototype member of the genus Aquabirnavirus of the family Birnaviridae. The most characteristic macro- and histopathological symptoms of this disease are exophthalmia, skin hyperpigmentation, abdominal and pyloric petechial hemorrhages, erratic swimming, and necrosis of both the kidney and pancreas.5,6
Infection outbreaks by IPNV can cause high mortality in first-feeding fry and postsmolts,7,8 consequently incurring high economic losses to the aquaculture industry.6,9,10 The mortality rate is very variable (10%–90%) and affects youngest fish to a greater extent, reaching 45%, 35%, and 7% in 1-, 2-, and 4-month-old fish, respectively.11 Interestingly, while currently unlisted in the Model Aquatic Health Code of the World Organization for Animal Health, the presence of IPNV is continuously being detected worldwide in both aquacultured12–17 and wild fish, including several nonsalmonid species.15,17–19 Apart from the fact that this virus is transmitted both vertically and horizontally,20,21 fish that recover or are asymptomatically infected often become carriers of the virus throughout their lives,11,22 contributing to its broad spread. Unfortunately, there is no therapy for this disease, so current protective measures are aimed at avoiding and alleviating its incidence. Such approaches have included less stressful handling of animals, use of IPN-resistant fish lines, improved management procedures, and the use of vaccination programs. In any case, the spread of the virus has been shown to be unpredictable, and there is still room to improve the protection conferred by existing vaccines, such as reducing their cost and making them more suitable to all life stages.9
IPNV is an unenveloped virus with an icosahedral and single-shelled capsid (T=13 symmetry) of about 60 nm in diameter, which consists of two proteins (VP2 and VP3). Its linear dsRNA genome is bisegmented (segment A 3,097 nucleotide [nt], segment B, 2,784 nt), uncapped, and unpolyadenylated.10 Segment A is bicistronic. Among its two open reading frames (ORFs), the largest one, ORF L, codes for the proteins VP2-4 as a 106-kDa polyprotein (NH2-pVP2-VP4-VP3-COOH) which is co-translationally cleaved by the viral protease VP4. The precursor pVP2 belongs to the major capsid protein VP2 (being most abundant overall),23 of which VP3 is a minor capsid protein that complexes with the dsRNA genome.10 In turn, the other segment-A ORF (ORF S) is not present in all isolates. ORF S overlaps the amino-terminal end of ORF L and encodes VP5. VP5 is variable in size (3.3–17 kDa) and a nonstructural protein that is not essential for viral infectivity, but may contribute to the virulence of the strain by presumably triggering an antiapoptotic mechanism.24 Segment B contains a single ORF that encodes the VP1 protein, which is a noncanonical RNA-dependent RNA polymerase (RdRp; 94 kDa). This protein, which can be found in its free form or indistinctively linked to the end of the genomic RNA (VPg),25 lacks the hallmark catalytic GDD signature in the region corresponding to the presumptive motif VI of infectious bursal disease virus.26 However, it presents a spatially rearranged LDD motif (residues 653–655 from Protein Data Bank [PDB] 2YI8).27 VP1 also has enzymatic activity, such as that possessed by guanylyl and methyl transferase.28
Taking advantage of the knowledge obtained from previous studies on the allosteric binding site described on the surface of the thumb domain of hepatitis C virus (HCV) polymerase,29 we herein explored a similar site in IPNV VP1 polymerase, allowing for the discovery of new antiviral drugs. This work describes the molecular docking results for a chemical library selected against a cavity site in the thumb domain of the RdRp of different IPNV strains, the successive filters applied for candidate compounds, and preliminary biological assays aimed at assessing antiviral capacity and specificity against two different IPNV strains for two of the selected candidates.
Materials and methods
Chemical compounds for antiviral assays
The compounds with the PubChem IDs 3274414 and 39834288 were purchased from the chemical supplier Ambinter (supplier references Amb10836885 and Amb674545, respectively) (Ambinter c/o Greenpharma Orléans, France).
Protein structure for IPNV RNA-dependent RNA–polymerase VP1 and chemical libraries
To date, five resolved structures have been deposited in the PDB for the VP1 protein of the Jasper strain of IPNV (UniProt code P22173): 2YI8, 2YI9, 2YIA, 2YIB, and 3ZED.30,31 However, no structures of this protein are yet deposited for the Sp (UniProt code P22174) or LWVRT60 (UniProt code A0A1B2AQF1) strains. Therefore, three-dimensional (3-D) structural models of the VP1 protein from both strains were generated by homology modeling in automated mode, using the 2YIB structure as a template.32 Briefly, a template search with BLAST and HHblits was performed against the Swiss-Model Template Library (SMTL; last update December 6, 2017, last included PDB release December 1, 2017). The target sequence was searched with BLAST33 against the primary amino-acid sequence contained in the SMTL. A total of 28–30 templates were found in each case. An initial HHblits profile was built using the procedure outlined in Remmert et al,34 followed by one iteration of HHblits against NR20. The profile obtained was then searched against all profiles of the SMTL. A total of 140–163 templates were found in each case. For each template identified, its quality was predicted from features of the target-template alignment. Templates of the highest quality were then selected for model building. Models were built based on the target-template alignment using ProMod II. Coordinates conserved between the target and the template were copied from the template to the model. Insertions and deletions were remodeled using a fragment library. Side chains were then rebuilt. Finally, the geometry of the resulting model was regularized using a force field. In cases where loop modeling with ProMod II35 did not yield satisfactory results, an alternative model was built with Modeller.36 For these molecular docking studies, amino-acid sequences 31–36 and 122–157 were electronically removed in both models and template. Structures 2BRK and 2BRL of HCV NS5 RdRp from Di Marco et al29 were used additionally, thereby employing nine structures for overall structural refinement. Visualization of the structures and preparation of the figures were carried out with PyMol 2.0 software.
For these experiments, a chemical library of 23,764 compounds was built using the option available at the PubChem site (https://pubchem.ncbi.nlm.nih.gov/search/search.cgi; in the “Identity/similarity” section) for searching structurally similar compounds to a given template. In our case, the structures of compounds 1 (PubChem ID 4369534) and 2 (PubChem ID 4369535), which were described to interact with the polymerase of HCV and to inhibit its activity,29 were used as query templates. Compounds were subsequently searched with at least 70% structural identity, thus generating a chemical library to be tested in molecular docking experiments. The PubChem web application used herein to allowed for the 3-D chemical structure of all compounds retrieved to be downloaded as spatial data files.
Molecular docking procedures
Before carrying out molecular docking experiments, PDBQT files of both the protein (receptor) and the ligands of our chemical library were calculated.37,38 Next, the five structures and two models of VP1 proteins were subjected to a geometric optimization process using the repair function of the FoldX algorithm.39 Molecular docking experiments were performed using AutoDock Vina software version 1.1.240 and targeted to a grid with dimensions of 24×24×24 points centered around the cavity generated by the amino acids Glu557, Asn580, Ans624, Pro625, and Pro550 of HCV NS5 RdRp and likewise around the amino acids Trp500, Arg503, Pro495, Val494, Leu492, Leu392, Ala396, and His428 of the VP1 protein of IPNV. AutoDock Vina was set up on a lusitania2.cenits.es Linux cluster (Research, Technological Innovation, and Supercomputing Center of Extremadura, Cáceres, Spain). AutoDock Vina generates for each tested ligand a conformer docked to the binding site in the protein and calculates the Gibbs free-energy variation of the binding process. Compounds with lower ΔG (kcal/mol) outperform a first-screening filter as potential candidates for inhibitors.
Calculation of pharmacokinetic parameters and potential toxicity of inhibitor candidates
Physicochemical parameters for the best-docked compounds were calculated as described previously37,38 using DataWarrior version 4.7.2.41 ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties were calculated with the AdmetSAR web application42 and DataWarrior.41 The same applications were used to calculate these parameters for the drugs included in the DrugBank database,43 and they are available at the website http://dockingfiles.umh.es/drugbank/DrugBanklist.asp.
Cell culture and viral strains
The Chinook salmon-embryo cell line CHSE214 was purchased from the (European Collection of Authenticated Cell Cultures, Public Health England, Salisbury, UK) (91041114). It was maintained at 20°C in a 5% CO2 atmosphere in Roswell Park Memorial Institute (RPMI) 1640 (Dutch modification) medium containing 10% FBS (Sigma-Aldrich, St Louis, MO, USA), 2 mM glutamine (Thermo Fisher Scientific, Waltham, MA, USA) and 50 μg/mL gentamicin (Thermo Fisher Scientific). Both Sp and LWVRT60 strains of IPNV were grown in CHSE214 cells at 14°C. When cytopathic effects were widespread, supernatants from infected cell cultures were clarified by centrifugation, filtered (0.22 μm), and stored in aliquots at −80°C. Virus titers were determined by end-point dilution in confluent CHSE214-cell monolayers grown in 96-well plates at 14°C in infection medium (ie, growth medium supplemented with 2% instead of 10% FBS). The Reed–Muench method44 was used to calculate 50% tissue-culture infective dose (TCID50)/mL. Average TCID50/mL (and SD) for each batch was obtained from three different titrations.
Cytotoxicity assays
The potential toxicity of selected compounds on CHSE cells was analyzed by measuring changes in cell viability with MTT (Sigma-Aldrich) assays. Briefly, confluent cell monolayers in 96-well plates were treated with different concentrations of each type of compound in infection media for 24 hours (100 μL/well). Then, 0.5 mg/mL MTT from tenfold-concentrated stocks in PBS (stored at −20°C) in fresh media (100 μL/well) was used to replace treatments. MTT solutions were incubated with cells under the same conditions for an additional 4 hours. Finally, media were carefully removed and the colored formazan product dissolved in 100 μL dimethyl sulfoxide (DMSO; Merck, Kenilworth, NJ, USA) and measured at 570 nm vs reference absorbance at 620 nm with a SpectroStar Omega absorbance microplate reader (BMG LabTech, Ortenberg, Germany). OD is expressed in percentages relative to the control group consisting of untreated cells. Additional controls included corresponding compounds and solvents (DMSO for the PubChem 3274414 compound and DMSO:acetone 1:1 for the PubChem 39834288 compound up to a maximum final concentration of 0.5% v:v) at an equivalent concentration to that used at each compound concentration tested at 1, 5, 10, 20, and 50 μM. Cell viability was calculated by the formula: 100× (treated-cell absorbance/control-cell absorbance). All experiments were performed in triplicate, and results are shown as mean with SD calculated from three different experiments.
Antiviral assays
To test the influence of the selected compounds on IPNV infectivity, IPNV was added at a 0.01 multiplicity of infection in 100 μL RPMI 1640 (Dutch modification) medium supplemented with 2% FBS (infection medium) to confluent CHSE-cell monolayers grown in 96-well plates and incubated for 2 hours at 4°C (adsorption period). Then, infected cell monolayers were washed twice with PBS and compounds (PubChem ID 3274414 was dissolved in DMSO and PubChem ID 39834288 in DMSO: acetone 1:1) were added to corresponding wells in 100 μL infection medium at different concentrations of 1, 5, 10, 20, and 50 μM. The final concentration of organic solvent never exceeded 0.5% (v:v). Infected cells were further incubated with the treatments for 24 hours at 14°C. After incubation, cells were collected for quantification of the virus by reverse-transcription quantitative polymerase chain reaction (RT-qPCR). In parallel and as a control for each concentration of compound used, infected cells were also treated with the corresponding solvents at a concentration equivalent to each concentration of compound used. Positive and negative infection controls were also included. All conditions were performed in tetraplicate (RT-qPCR was performed by pooling the cells of all replicates).
RNA isolation, cDNA synthesis, and RT-qPCR assays
Levels of each IPNV strain replicating in infected CHSE cells were evaluated by determining their content in viral transcripts. Therefore, RT-qPCR was performed on cDNA from CHSE-cell RNA previously used in the antiviral assays. Therein, RNA from the aforementioned collected cells was isolated using an E.Z.N.A.® Total RNA kit (Omega Bio-tek, Norcross, GA, USA) following the manufacturer’s guidelines. RNA concentration was assessed using a NanoDrop 1,000 spectrophotometer (Thermo Fisher Scientific) by measuring absorbance at 260 nm. Samples were stored at −80°C until use.
In order to obtain cDNA reverse transcriptase (Moloney murine leukemia virus; Thermo Fisher Scientific), 1 μg RNA from each sample was used, as previously described.45 RT-qPCR reactions were carried out using the ABI 7300 Real-Time PCR System (Thermo Fisher Scientific) with SYBR® Green Master Mix PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). The total volume of each reaction was 20 μL and included 2 μL cDNA, 900 nM each primer, and 10 μL SYBR green PCR master mix. Nontemplate controls were performed for each gene analysis. Cycling conditions were 95°C for 10 minutes, followed by 40 cycles of 1 minute at 65°C, 15 seconds at 95°C, and finally an extension of 1 minute at 60°C and 15 seconds at 95°C. Results were obtained by normalizing the expression of the target gene respective to that of the endogenous reference using a variation of Livak and Schmittgen’s method46 by the formula 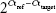 . The endogenous gene used in this study was elongation ef1a. Primers used are shown in Table 1. All reactions were performed in duplicate. Results are presented as percentages of inhibition of IPNV infectivity relative to values obtained by an equivalent amount of corresponding solvent, and correspond to means with SD calculated from four different experiments.
. The endogenous gene used in this study was elongation ef1a. Primers used are shown in Table 1. All reactions were performed in duplicate. Results are presented as percentages of inhibition of IPNV infectivity relative to values obtained by an equivalent amount of corresponding solvent, and correspond to means with SD calculated from four different experiments.
 | Table 1 Primers used in this study |
Results and discussion
Exploring allosteric binding-site cavity from IPNV VP1 protein
Di Marco et al29 reported the crystal structure of HCV RdRp (genotype 1b, strain BK) in a complex with two nonnucleoside inhibitors occupying a cavity (Figure 1A) that appeared in the thumb domain for deletion mutants lacking the 55 amino acids at the C-terminus (ΔC55). Both compounds inhibited not only the purified full-length and truncated C-terminal ΔC55 enzyme in a low-nanomolar range but also the replicon system, although with minor affinity (15–30 times lower). This cavity is not accessible in the crystal structure of the full-length protein, and arises after displacement of the α-helix, which implies that the interaction between thumb and fingers is weak enough to allow for a slightly open structure of the polymerase.29 This observation suggests that a priori this domain in IPNV VP1 polymerase might also be displaced similarly by the interaction of molecules with its corresponding homologous cavity. Therefore, potential antiviral drugs against IPNV could be designed against that cavity without generating deletion mutants.
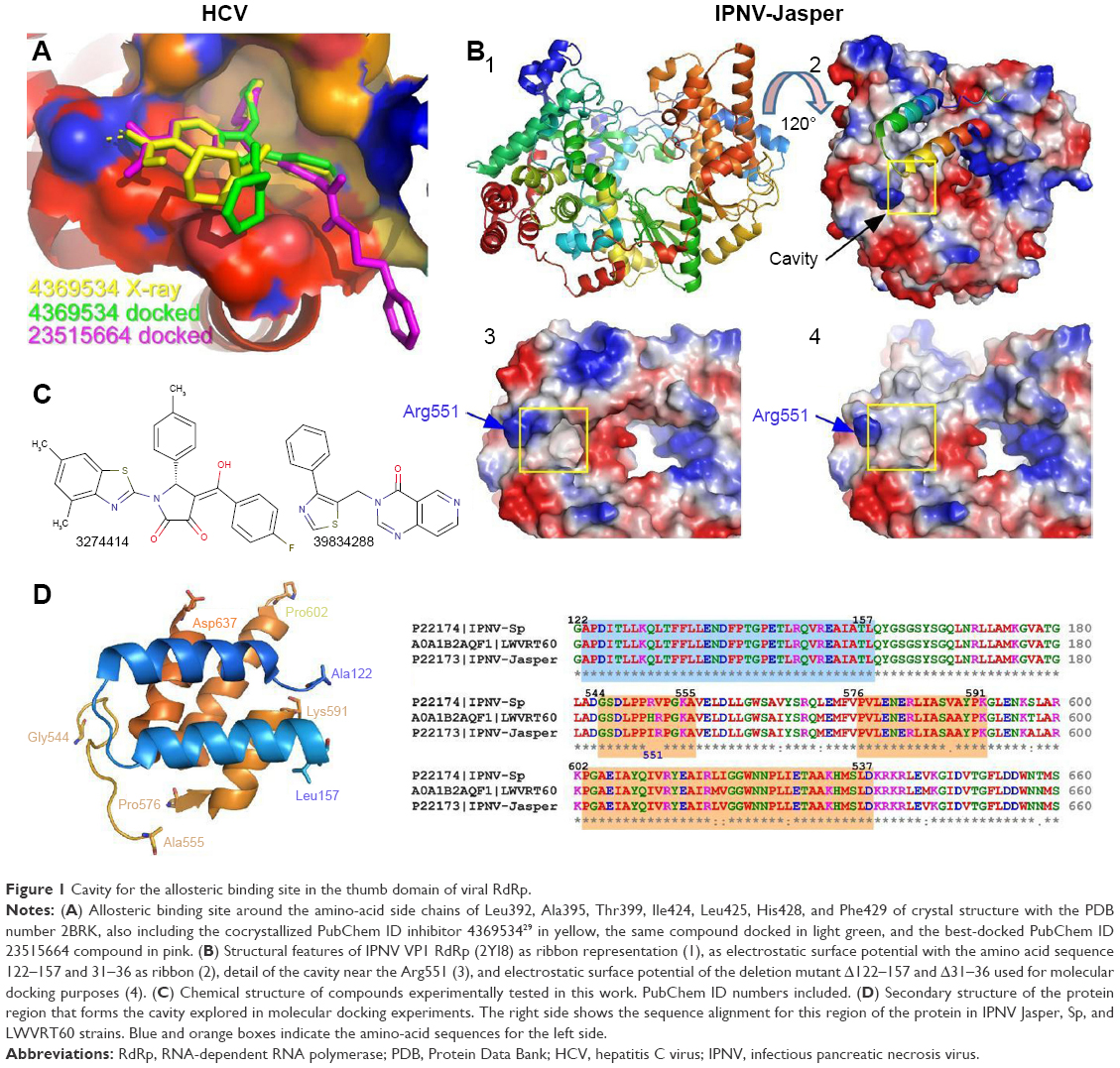 | Figure 1 Cavity for the allosteric binding site in the thumb domain of viral RdRp. |
Five high-resolution structures are known for the VP1 polymerase of the Jasper strain of IPNV. It can be observed that unlike HCV polymerase, there is a partially accessible cavity even if residues 122–157 of the thumb domain in HCV polymerase are present (Figure 1B). This cavity becomes more evident for the same structure when amino acids 122–157 are electronically removed (Figure 1B). Although no high-resolution structures of the VP1 protein are available for the Sp and LWVRT60 IPNV strains, those available in our laboratory show high sequence identity (88.7%–99.5%) with Jasper VP1. This is especially true for the three domains involved in the definition of the cavity (Figure 1D). For this reason, homology modeling of the VP1 protein using the structures of the Jasper strain as a template was carried out.38 In both the available structures and resulting models, the amino-acid sequences 122–157 and 31–36 were electronically deleted and molecular docking experiments carried out on the resulting structures.
Analysis of compounds docked to cavity on surface of thumb domain
Initially, an in silico screening of our chemical library was carried out on HCV polymerase, in order to check the predictive capacity of AutoDock Vina with the compound cocrystallized by Di Marco et al, define thresholds for the variation of Gibbs free energy (ΔG, kcal/mol) in the screening process, and find compounds with smaller ΔG values, and then potentially higher affinity, than those tested by Di Marco et al.29 Collectively, such data would allow for a targeted approach in the design of potential inhibitors of the IPNV VP1 protein, as was the main objective of this study.
Figure 1A depicts compound one in yellow (PubChem 4369534), present in the crystal structure with the PDB number 2BRK.29 The same compound highlighted in light green is shown superimposed after molecular docking calculations (ΔG = −8.75 kcal/mol). Docked and experimental conformation of 4369534 compound had an root-mean-square deviation of 5.141 Å. Finally, one of the optimal compounds with respect to binding was calculated from our chemical library (PubChem 23515664), and is depicted in pink. Docking calculations for compound 2 (PubChem 4369535) also predicted a conformer interacting in such a cavity, in agreement with that obtained through cocrystallization-derived structures29 (not shown) and with a similar ΔG value (−8.60 kcal/mol). After analysis of the docking data for our chemical library against HCV structures (2BRKΔ31–36, Δ122–157 and 2BRLΔ31–36, Δ122–157) we found 200 compounds (not shown) with ΔG values ≤−9.0 kcal/mol (the ΔG value chosen as threshold based on previous calculated results for Di Marco et al’s active compounds; Figure 2), and among these 26 compounds with ΔG values ≤−9.5 kcal/mol. Calculated ΔG values for compounds that primarily bind to hydrophobic sites are greater than expected when the binding site is more hydrophilic. As a consequence, in these cases the Ki calculated from the value of ΔG (Ki= expΔG/RT)38 did not correspond to the experimental values in the subnanomolar range for compounds 1 and 2.
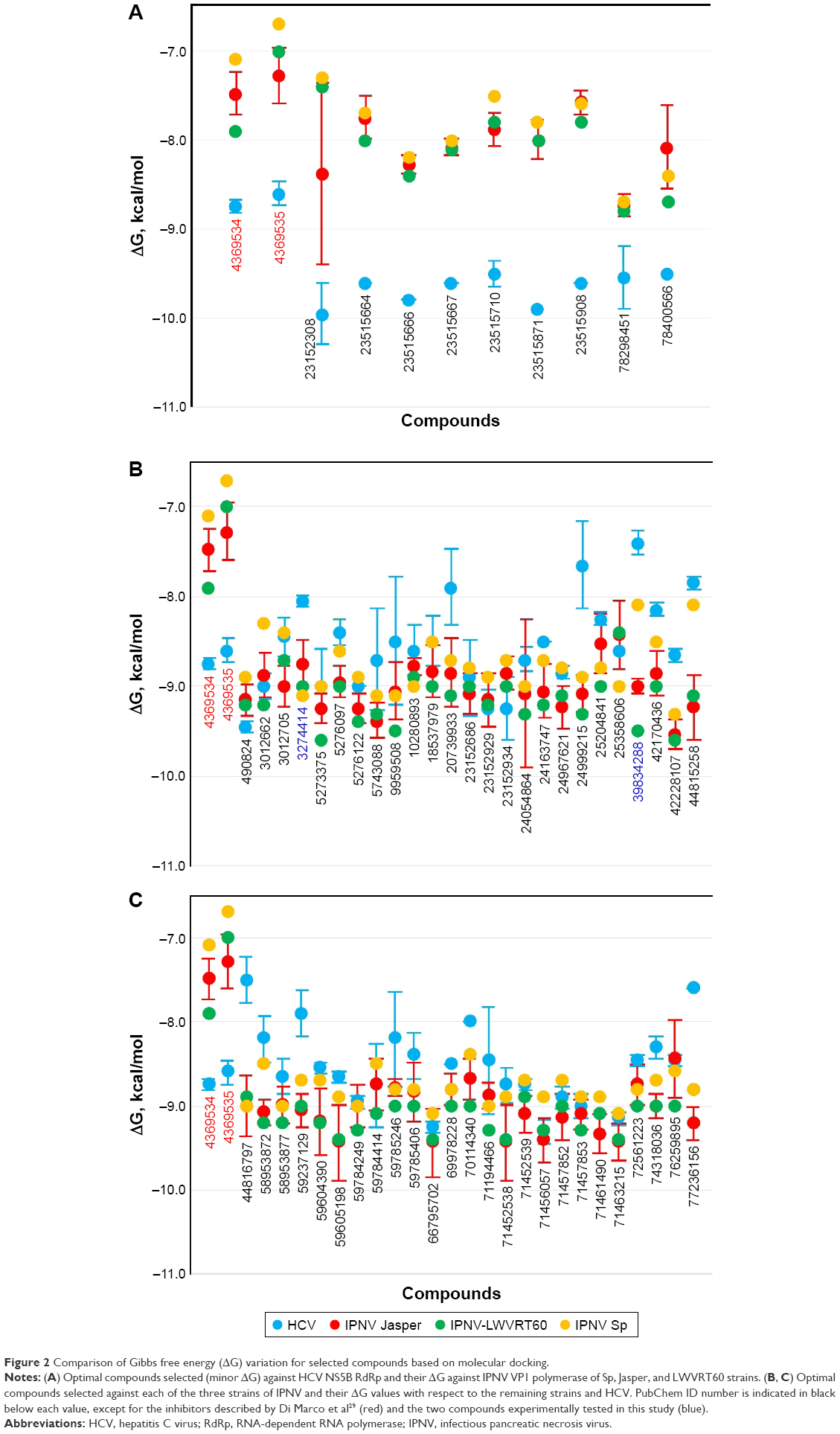 | Figure 2 Comparison of Gibbs free energy (ΔG) variation for selected compounds based on molecular docking. |
Of those 26 compounds with the lowest ΔG values, 17 were discarded, as their ADMET profiles were not optimal. In this sense, the range of optimal values was slightly varied for different parameters with respect to other research by our group.37,38 Briefly, different parameters of the ADMET profile were analyzed (Figure 3) for both approved and experimental drugs included in the DrugBank database43 (these data are available at the website http://dockingfiles.umh.es/drugbank/DrugBanklist.asp). Eight of the nine parameters analyzed in Figure 3 show a Gaussian distribution in a frequency where 80%–90% of the values of these parameters varied by molecular weight, calculated logarithm of partition coefficient, drug score, drug likeness, H-bond acceptor, hydrogen (H)-bond donor, and topological polar surface area. In this manner, it can be observed that a high percentage of these drugs showed values in several parameters that are far from standard as was especially evident for drug score, drug likeness, and topological polar surface area. Moreover, up to 21% of these drugs present more than one violation of Lipinski’s rules.50 According to these data, the extreme values of these Gaussian distributions will be taken into account to be used as a screening filter for potential antiviral drugs against IPNV. Even up to 3 violations of Lipinski’s rules were admitted (See Tables 2, 4 and 6).
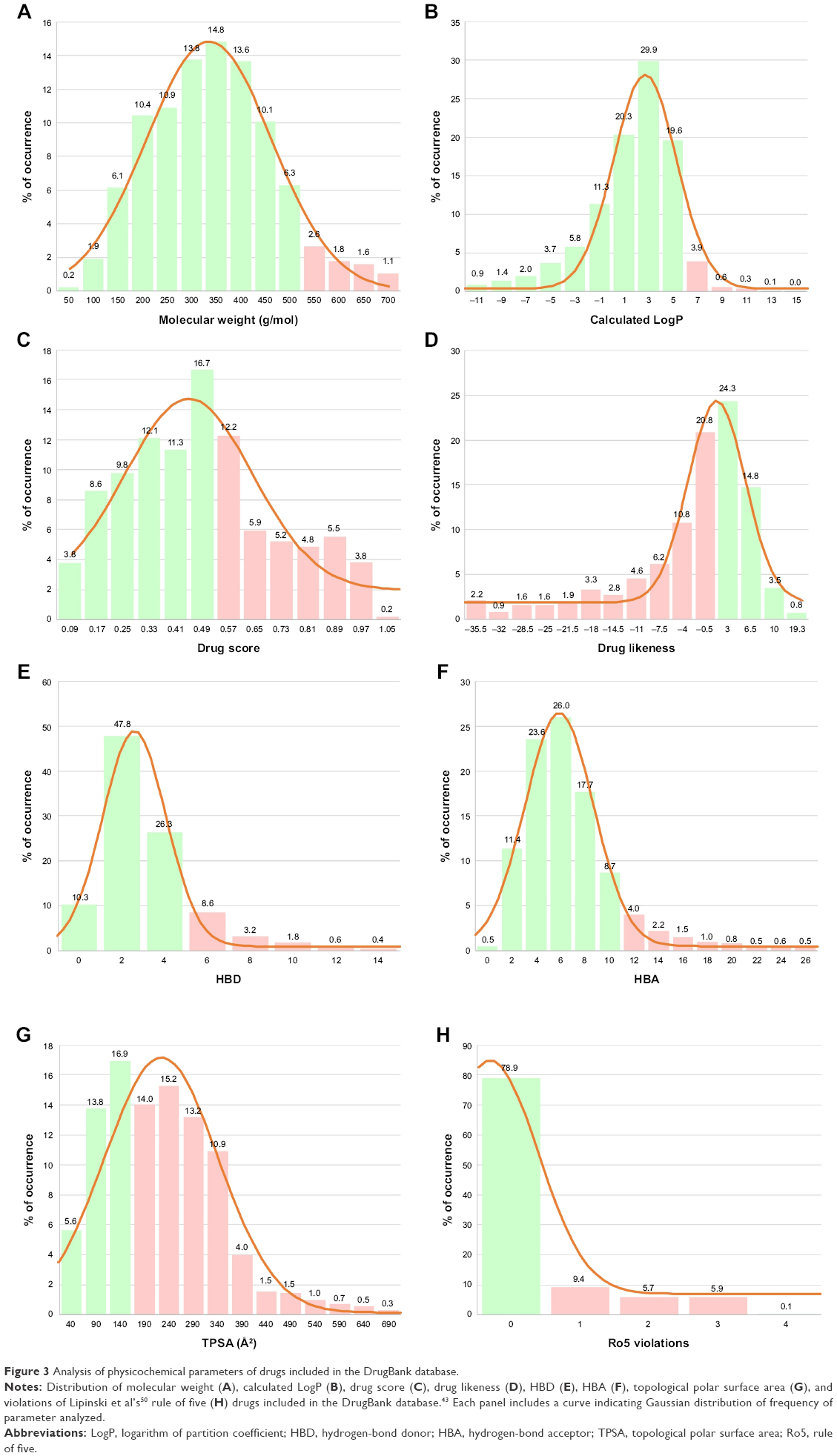 | Figure 3 Analysis of physicochemical parameters of drugs included in the DrugBank database. |
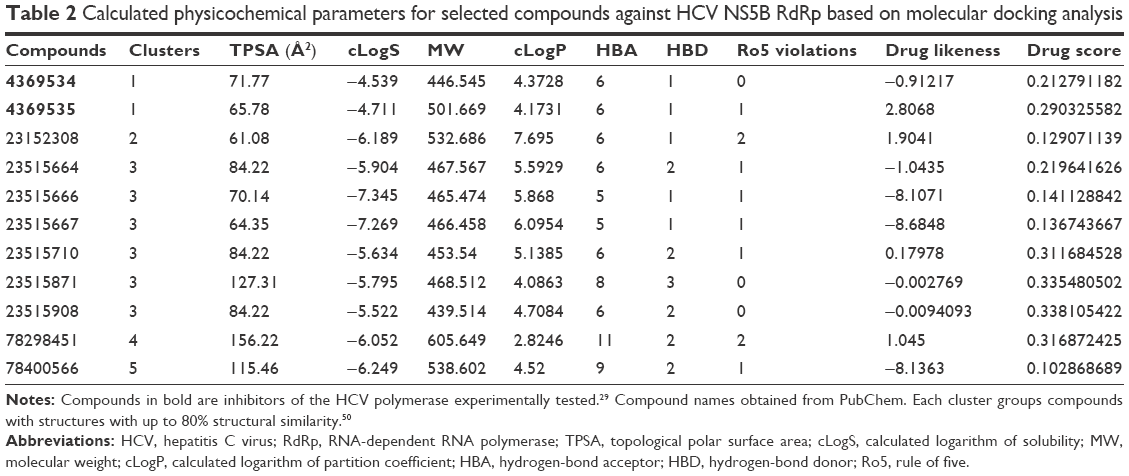 | Table 2 Calculated physicochemical parameters for selected compounds against HCV NS5B RdRp based on molecular docking analysis |
In Figure 2A, calculated ΔG values for selected compounds are compared to compounds 1 (4369534) and 2 (4369535)29 for not only HCV but also all three IPNV strains. As expected, both compounds showed an ΔG value of almost 2 kcal/mol higher for all the IPNV strains, as they were not designed for targeting their polymerases. This occurred similarly with the nine compounds selected against HCV (Figure 2A). These data reflect the differences in the volume of the cavity and in the sequence of amino acids that define it between HCV and IPNV.
In Figure 2B and C, the ΔG values of the selected compounds docked against the IPNV VP1 of all three strains are compared. For each strain, compounds with ΔG values ≤−9 kcal/mol were selected. The ΔG values of these compounds are also shown against HCV. All these compounds were also within the range of values of the ADMET profile (see Tables 3, physicochemical parameters; 5 ADME; 7 toxicity). Although the chemical library used in molecular docking experiments was obtained from the PubChem database, not all deposited compounds are commercially available, and thus only some can be tested experimentally. In fact, this is the third (and very relevant) filter that joins the two previous filters for a final proposal of candidates for antiviral compounds. A total of 50 compounds were proposed as potential antiviral compounds (Figure S1) for the three strains of IPNV. Careful observation of the ΔG values of these 50 compounds for all IPNV strains (Figure 2B and C) reveals that for some the ΔG values can vary up to more than 1 kcal/mol between the Sp and LWVRT60 strains. This was especially true for compounds 3012662, 39834288, 44815258, and 58953872. However, for many others, the differences were negligible. Taking this observation into consideration, in order to assess if different ΔG values between the IPNV strains reflected different rates of inhibition of compounds against the Sp and LWVRT60 strains, two compounds with two different ΔG patterns were selected for testing in vitro. The compounds selected for further in vitro assays were 3274414 and 39834288 (Figure 1C).
 | Table 3 Predicted molecular pharmacokinetic properties of selected compounds against HCV NS5B RdRp |
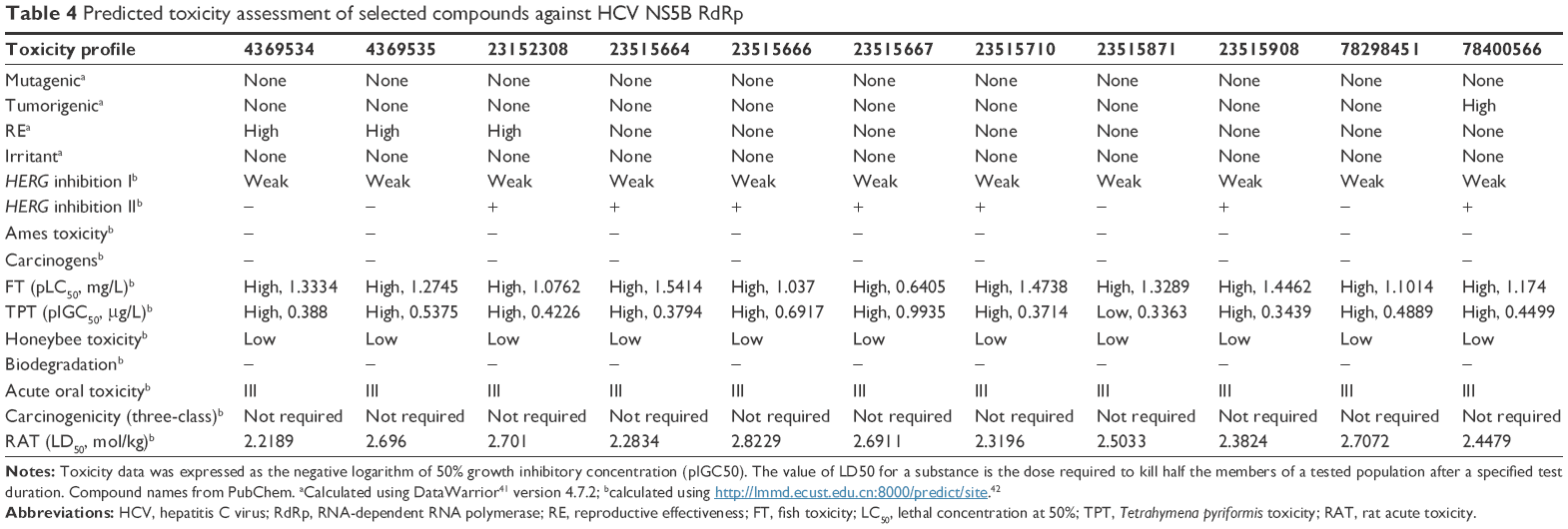 | Table 4 Predicted toxicity assessment of selected compounds against HCV NS5B RdRp |
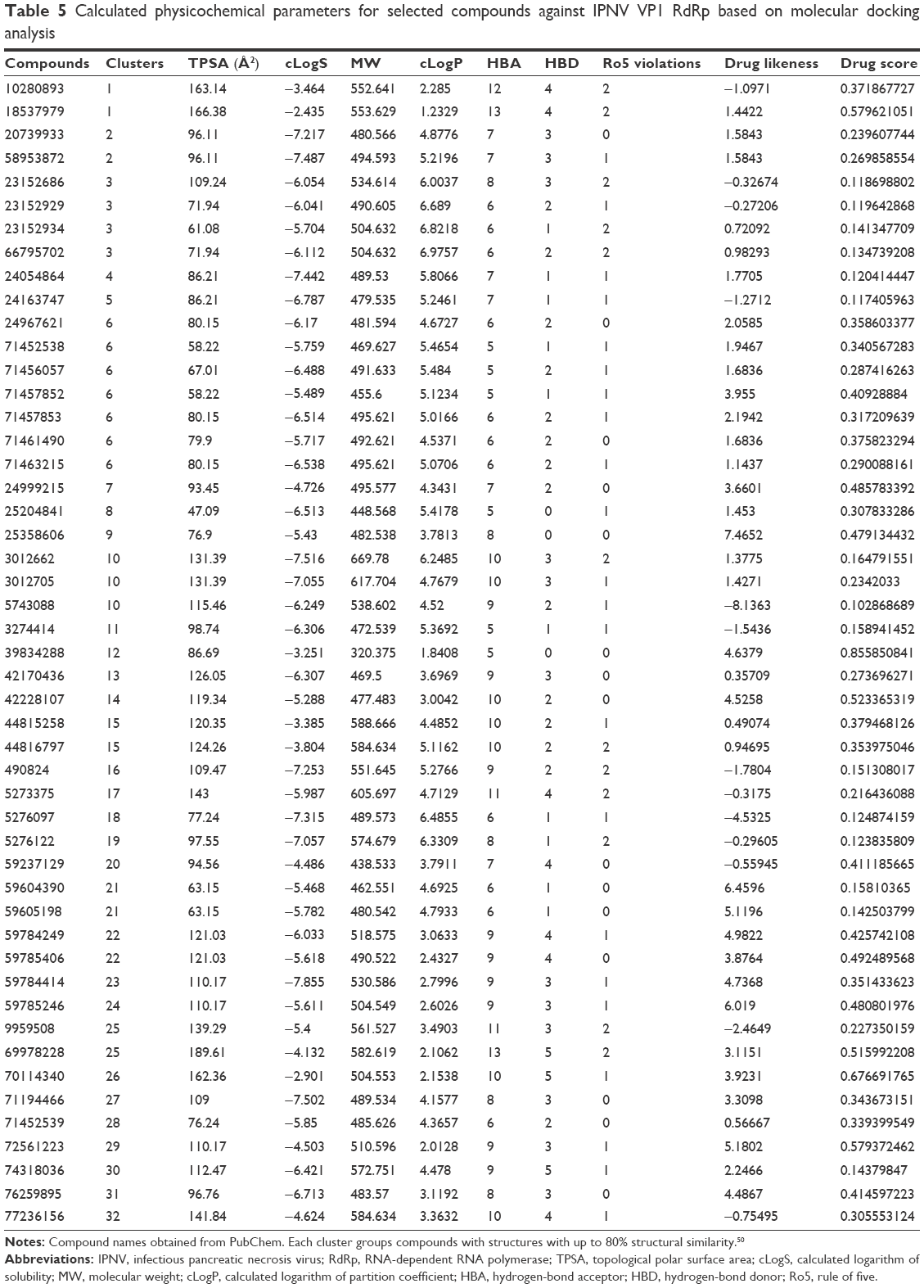 | Table 5 Calculated physicochemical parameters for selected compounds against IPNV VP1 RdRp based on molecular docking analysis |
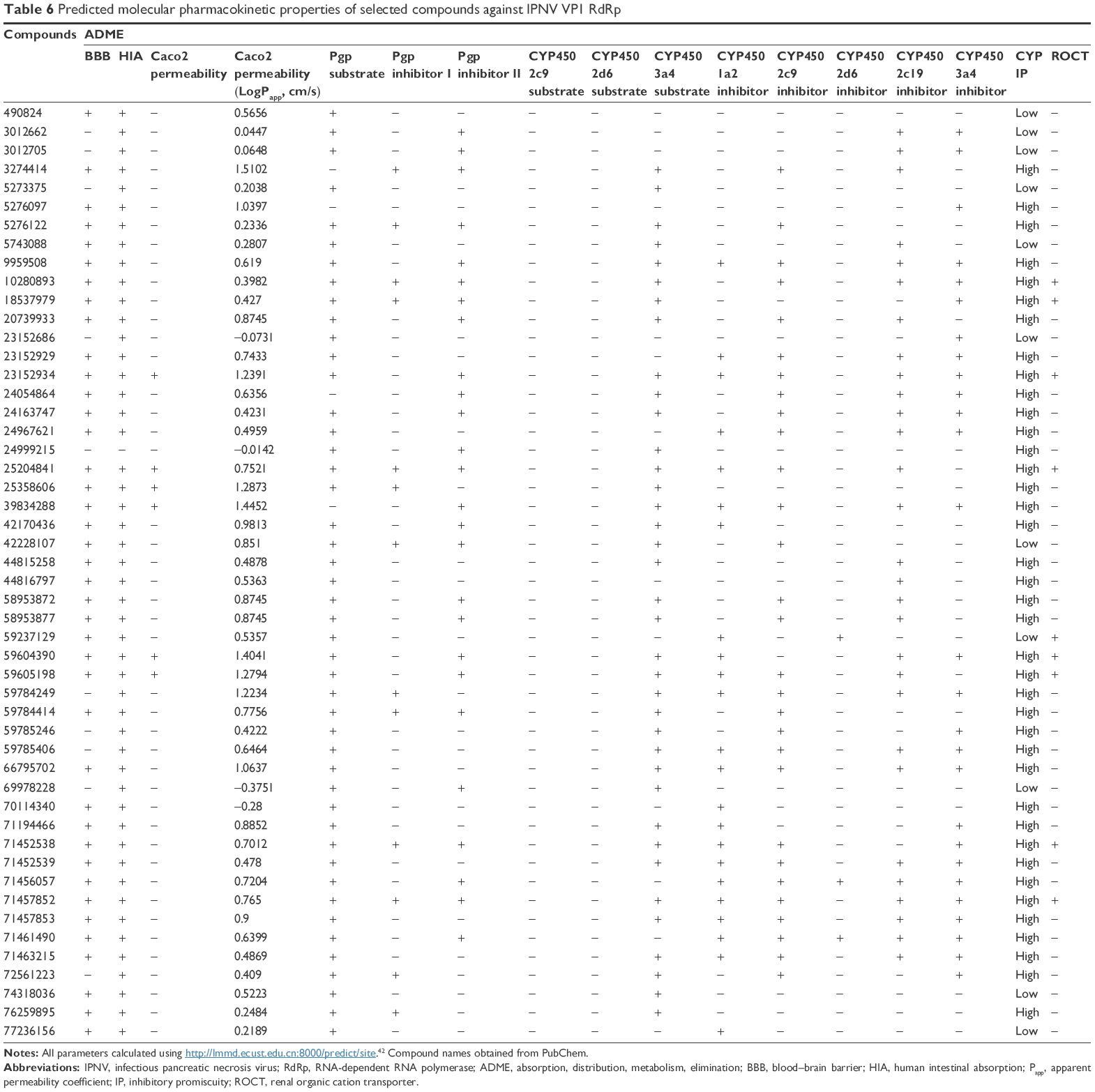 | Table 6 Predicted molecular pharmacokinetic properties of selected compounds against IPNV VP1 RdRp |
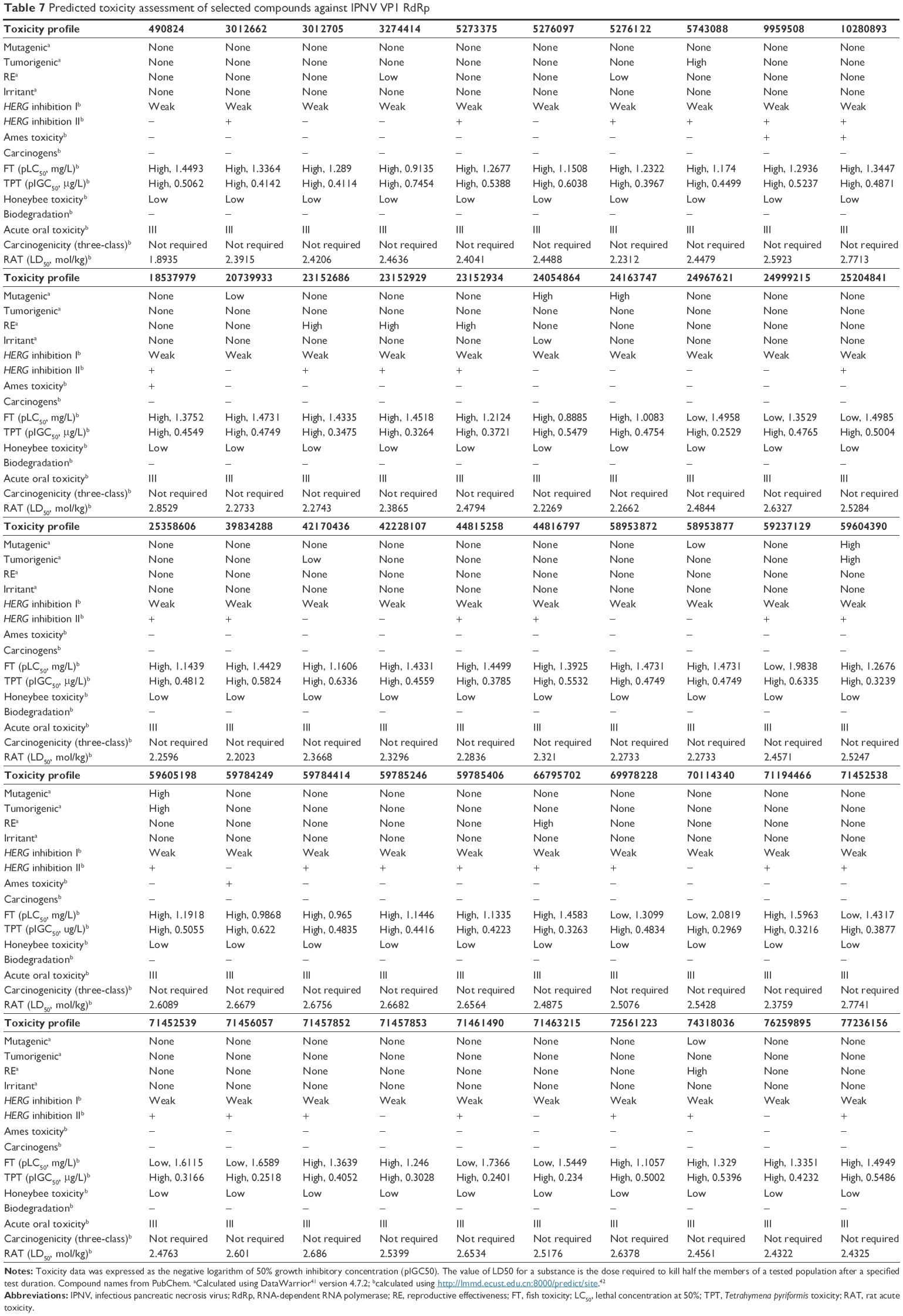 | Table 7 Predicted toxicity assessment of selected compounds against IPNV VP1 RdRp |
Determination of cell viability of CHSE cells after treatment with compounds 3274414 and 39834288
Cellular cytotoxicity induced by the selected experimental compounds was evaluated by an MTT cell-viability assay (Figure 4). For these assays, CHSE cells were treated with a range of concentrations of each compound (0–50 μM). In parallel, treatments with equivalent amounts of the corresponding compound solvents were performed. After 24 hours, cell viability was determined by MTT as described in the “Materials and methods” section. As shown in Figure 5, CHSE cells were more sensitive to 3274414 than 39834288. For treatments with 3274414, no significant toxic effect was observed in the concentration range tested of 0–20 μM (95.4%±4.6% at 20 μM). Cell viability slightly decreased to 74.9%±2.9% for 3274414 at 50 μM. In contrast, at this concentration (the highest tested), cell viability was 88.3%±3.9% for 39834288. In turn, both solvents at any concentration showed cell-viability percentages close to 100%.
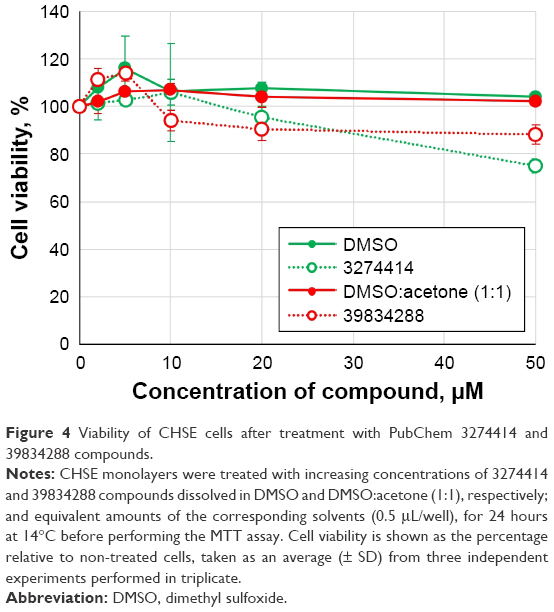 | Figure 4 Viability of CHSE cells after treatment with PubChem 3274414 and 39834288 compounds. |
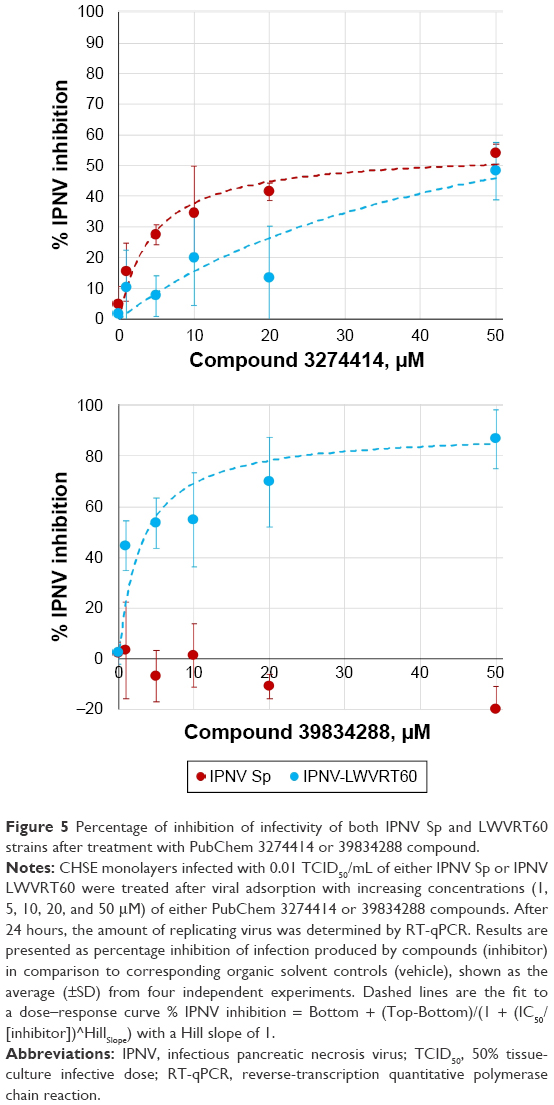 | Figure 5 Percentage of inhibition of infectivity of both IPNV Sp and LWVRT60 strains after treatment with PubChem 3274414 or 39834288 compound. |
Determination of anti-IPNV activity induced by selected compounds
Infected CHSE cells were further incubated with different concentrations of selected compounds (3274414 or 39834288) for 24 hours. Viral loads were subsequently measured as the total abundance of viral transcripts quantified by RT-qPCR. As shown in Figure 5, different inhibition patterns were observed dependent on the compound and IPNV strain used. When the compounds induced antiviral activity, this occurred in a dose-dependent manner.
For the 3274414 compound, both IPNV strains were inhibited following a similar pattern. At the maximum concentration tested (50 μM), the infectivity of both viral strains was reduced by approximately 50% (Figure 5A). The potency of this compound was lower than that induced by mycophenolic acid or ribavirin,48 which at 1 μM both inhibited the infectivity of IPNV Sp up to 90%. Our docking data showed that 3274414–polymerase interactions were only hydrophobic and involved residues Lys554, Ala555, Glu557, and Asn580. The compound showed the same orientation in its binding to the polymerase in all three IPNV strains.
For the 39834288 compound, its antiviral capacity was markedly different (Figure 5B), exhibiting no effect upon the Sp strain. In contrast, its potency was found to be greater than that of compound 3274414 against the LWVRT60 strain, reducing its infectivity by 80% at 20 μM. By adjusting the parameters to a Hill equation, an IC50 of 3.1 μM was calculated. The estimated IC50 value for compound 3274414 was about 15 μM for both Sp and LWVRT60 strains. These IC50 values are comparable with those of other nonnucleoside inhibitors designed to block the RNA-template tunnel of RdRp dengue virus 2.51 The compounds (NITD1, -2, and -29) analyzed therein showed no toxicity up to 50 μM and presented IC50 values of 7.2, 0.7, and 1.5 μM, respectively. In spite of showing inhibitory activity against the recombinant viral polymerase, the antiviral activity of NTD1 and NTD2 compounds in cell cultures could not be demonstrated.51 Selective inhibition of one virus subtype over other subtypes has been previously for small-molecule inhibitors of influenza A virus, although the molecular basis of such specificity remained obscure.52
The interactions of 39834288–polymerase (H-bonds between the compound and the residues Pro552, Glu557, and, Asn623) predicted by molecular docking were similar for the three IPNV strains (Figure 6A). Therefore, the question remained as to how compound 39834288 showed no activity against the Sp strain. The calculation of position for the compounds docked to the cavity in the surface of the thumb was made in the absence of residues 31–36 and 122–157. Therefore, it is understandable that they did not show differences in 39834288–polymerase interactions. However, cell-culture assays for antiviral activity were carried out with the full enzyme. Binding of the inhibitor in the cavity is only possible if the loop 122–157 is displaced, especially for the compound 39834288, which would have large clashes (Figure 6B), as it is located in the full protein (2YI8). As would be expected, based upon 3274414–polymerase docking-calculated interactions, a much smaller rearrangement of loop 122–157 would be required to accommodate compound 39834288. In this sense, the binding energy of loop 122–157 to the domain that forms the cavity (residues Asp523 to Asp691) was calculated using FoldX software.39 These data showed that although there were neither differences between the three strains for the numbers of hydrogen-bonds (Asp124–Ala588, Thr146–Asn624, Tyr159–Glu594, Asp124–Arg612, Asp124–Tyr613, Thr146–Asn624, Gln149–Asn580, and Ile154–Tyr589), saline bridges (Asp124–Arg612) nor the interface area (about 1,060 Å2), there were appreciable differences in the global calculation of binding free-energy variation. The calculated ΔG for the binding of both domains was −36.9 kcal/mol for the LWVRT60 strain and −38.1 kcal/mol for the Sp strain. In other words, the rearrangement of loop 122–155 was more energy-expensive for the polymerase of the Sp strain than for LWVRT60. Such may also explain the observed inability of compound 39834288 to displace this loop in the Sp strain, resulting in negligible activity of its polymerase and in turn its infectivity.
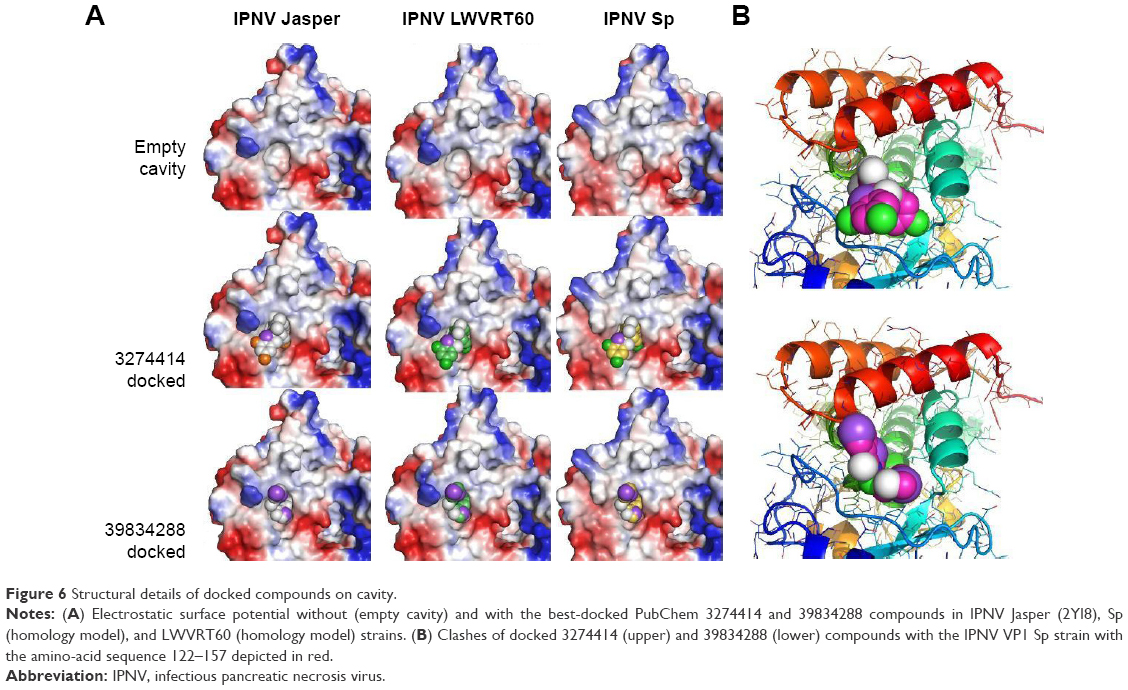 | Figure 6 Structural details of docked compounds on cavity. |
Conclusion
The results presented herein are compatible with the existence of an allosteric regulation site in the IPNV VP1 polymerase. From a library of 23,760 compounds, nine and 50 were predicted as antiviral drug candidates against HCV and IPNV polymerases, respectively (Figure 7). Two nontoxic compounds were tested in vitro, and showed antiviral activity against IPNV in the low-micromolar range.
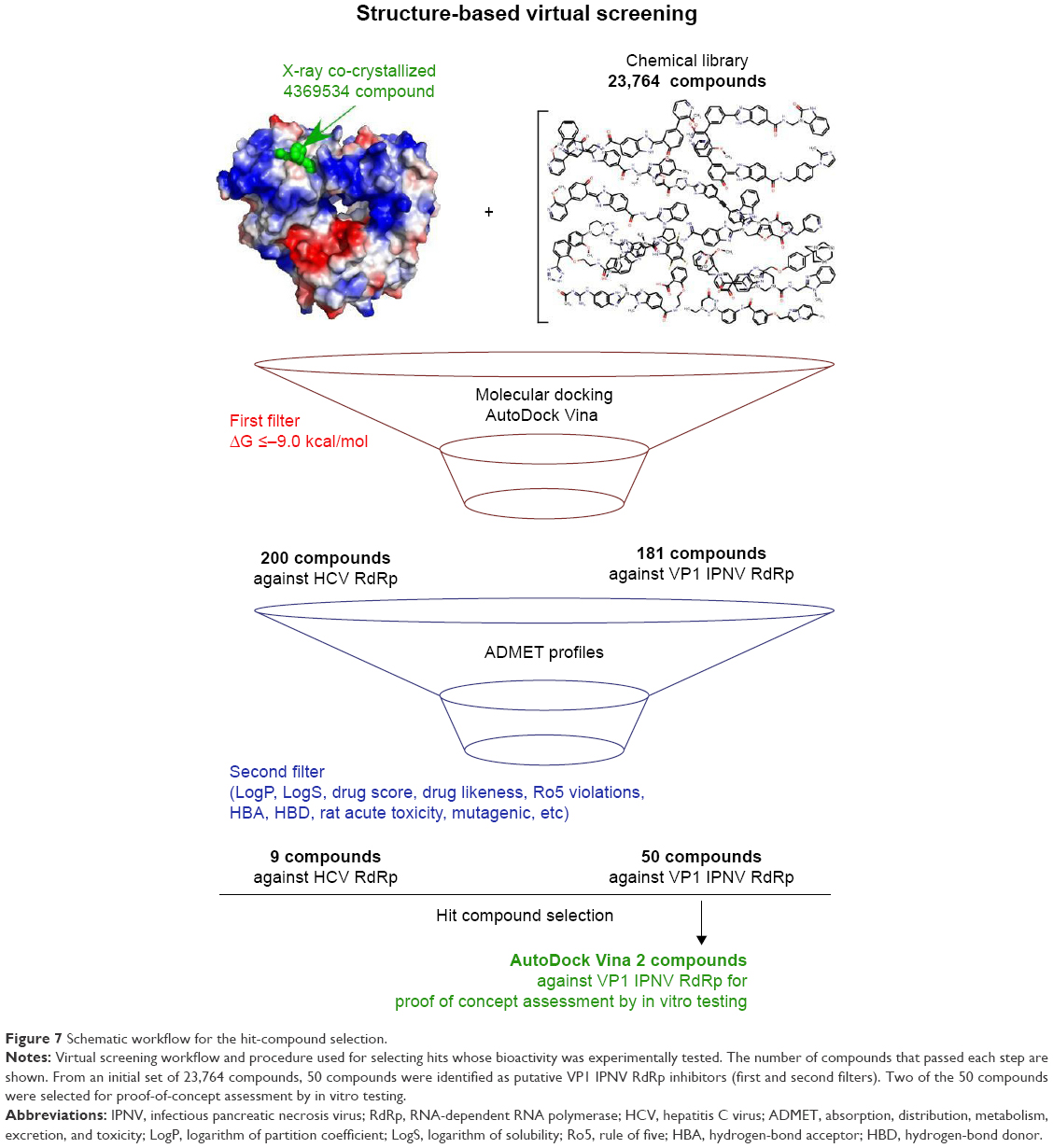 | Figure 7 Schematic workflow for the hit-compound selection. |
Acknowledgments
Special thanks are due to Dr Amparo Estepa Perez, who has passed away, but contributed to financing this work. MBP is financed by the Generalidad Valenciana, fellowship ACIF/2016. We are grateful to Research, Technological Innovation, and the Supercomputing Center of Extremadura (CénitS) for allowing us to use their supercomputing facilities (Lusitania II). This work was supported by the Programa Estatal de Investigación, Desarrollo e Innovación Orientada a los Retos de la Sociedad project AGL2014-51773-C3-1-R of the Ministerio de Economía y Competitividad of Spain. We thank Dr Beatriz Novoa (Instituto de Investigaciones Marinas, Consejo Superior de Investigaciones Científicas, Vigo, Spain) for providing the IPNV LWVRT60 strain. Technical support from Angeles Gómez is also acknowledged. Dr Matthew Mold (Keele University, Newcastle, UK) provided some assistance with the English. We thank the anonymous reviewers for their constructive comments, which helped us to improve the manuscript.
Author contributions
JAE, LP, and AF conceived and designed the experiments and wrote the paper, MBP, JC, and AF conducted the in vitro experiments, JAE and VG conducted the in silico molecular docking experiments and the DrugBank analysis, and LP and JAE were responsible for funding acquisition. All authors contributed to the general discussion of the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
de Clercq E. Antivirals: current state of the art. Future Virol. 2008;3(4):393–405. | ||
Griffiths PD. A perspective on antiviral resistance. J Clin Virol. 2009;46(1):3–8. | ||
Awoonor-Williams E, Walsh AG, Rowley CN. Modeling covalent-modifier drugs. Biochim Biophys Acta. 2017;1865(11 Pt B):1664–1675. | ||
Raghavendra NM, Pingili D, Kadasi S, Mettu A, Prasad S. Dual or multi-targeting inhibitors: the next generation anticancer agents. Eur J Med Chem. 2018;143:1277–1300. | ||
Wolf K, Snieszko SF, Dunbar CE, Pyle E. Virus nature of infectious pancreatic necrosis in trout. Proc Soc Exp Biol Med. 1960;104:105–108. | ||
Munro ES, Midtlyng PJ. Infectious pancreatic necrosis and associated aquatic birnaviruses. In: Woo P, Bruno D, editors. Fish Diseases and Disorders. 2nd ed. Wallingford, UK: CABI; 2011:1–65. | ||
Guy DR, Bishop SC, Brotherstone S, et al. Analysis of the incidence of infectious pancreatic necrosis mortality in pedigreed Atlantic salmon, Salmo salar L., populations. J Fish Dis. 2006;29(11):637–647. | ||
Rønneseth A, Wergeland HI, Devik M, Evensen O, Pettersen EF. Mortality after IPNV challenge of Atlantic salmon (Salmo salar L.) differs based on developmental stage of fish or challenge route. Aquaculture. 2007;271(1–4):100–111. | ||
Dhar A, LaPatra S, Orry A, Allnutt F. Infectious pancreatic necrosis virus. In: Woo P, Cipriano R, editors. Fish Viruses and Bacteria: Pathobiology and Protection. Wallingford, UK: CABI; 2017:1–12. | ||
Delmas B, Mundt E, Vakharia VN, Wu JL. Family – Birnaviridae. In: King AM, Carstens EB, editors. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. London: Academic Press; 2012:499–507. | ||
Lvov DK, Shchelkanov MY, Alkhovsky SV, Deryabin PG. Double-stranded RNA viruses. In: Lvov DK, Shchelkanov MY, Vladimirovich S, Alkhovsky SV, Deryabin PG, editors. Zoonotic Viruses in Northern Eurasia: Taxonomy and Ecology. Boston: Academic Press; 2015:113–133. | ||
Büyükekiz AG, Altun S, Hansen EF, et al. Infectious pancreatic necrosis virus (IPNV) serotype Sp is prevalent in Turkish rainbow trout farms. J Fish Dis. 2018;41(1):95–104. | ||
Holopainen R, Eriksson-Kallio AM, Gadd T. Molecular characterisation of infectious pancreatic necrosis viruses isolated from farmed fish in Finland. Arch Virol. 2017;162(11):3459–3471. | ||
Manríquez RA, Vera T, Villalba MV, et al. Molecular characterization of infectious pancreatic necrosis virus strains isolated from the three types of salmonids farmed in Chile. Virol J. 2017;14(1):17. | ||
Ogut H, Altuntas C, Parlak R. Viral surveillance of cultured rainbow trout in the eastern Black Sea, Turkey. J Aquat Anim Health. 2013;25(1):27–35. | ||
Ruane NM, McCarthy LJ, Swords D, Henshilwood K. Molecular differentiation of infectious pancreatic necrosis virus isolates from farmed and wild salmonids in Ireland. J Fish Dis. 2009;32(12):979–987. | ||
Wallace IS, Mckay P, Murray AG. A historical review of the key bacterial and viral pathogens of Scottish wild fish. J Fish Dis. 2017;40(12):1741–1756. | ||
Crane MS, Hardy-Smith P, Williams LM, et al. First isolation of an aquatic birnavirus from farmed and wild fish species in Australia. Dis Aquat Organ. 2000;43(1):1–14. | ||
Moreno P, Olveira JG, Labella A, et al. Surveillance of viruses in wild fish populations in areas around the Gulf of Cadiz (South Atlantic Iberian Peninsula). Appl Environ Microbiol. 2014;80(20):6560–6571. | ||
Delmas B, Mundt E, Vakharia V, Wu J. Family Birnaviridae. In: King AM, Carstens EB, editors. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. London: Academic Press; 2012:499–507. | ||
Molloy SD, Pietrak MR, Bricknell I, Bouchard DA. Experimental transmission of infectious pancreatic necrosis virus from the blue mussel, Mytilus edulis, to cohabitating Atlantic salmon (Salmo salar) smolts. Appl Environ Microbiol. 2013;79(19):5882–5890. | ||
Munro ES, Gahlawat SK, Acosta F, Ellis AE. In infectious pancreatic necrosis virus carrier Atlantic salmon, Salmo salar L., post-smolts, almost all kidney macrophages ex vivo contain a low level of non-replicating virus. J Fish Dis. 2006;29(1):43–48. | ||
Chevalier C, Lepault J, Erk I, da Costa B, Delmas B. The maturation process of pVP2 requires assembly of infectious bursal disease virus capsids. J Virol. 2002;76(5):2384–2392. | ||
Santi N, Song H, Vakharia VN, Evensen O. Infectious pancreatic necrosis virus VP5 is dispensable for virulence and persistence. J Virol. 2005;79(14):9206–9216. | ||
Magyar G, Chung HK, Dobos P. Conversion of VP1 to VPg in cells infected by infectious pancreatic necrosis virus. Virology. 1998;245(1):142–150. | ||
Duncan R, Mason CL, Nagy E, Leong JA, Dobos P. Sequence analysis of infectious pancreatic necrosis virus genome segment B and its encoded VP1 protein: a putative RNA-dependent RNA polymerase lacking the Gly-Asp-Asp motif. Virology. 1991;181(2):541–552. | ||
Koonin EV, Wolf YI, Nagasaki K, Dolja VV. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat Rev Microbiol. 2008;6(12):925–939. | ||
Dobos P. In vitro guanylylation of infectious pancreatic necrosis virus polypeptide VP1. Virology. 1993;193(1):403–413. | ||
Di Marco S, Volpari C, Tomei L, et al. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J Biol Chem. 2005;280(33):29765–29770. | ||
Graham SC, Sarin LP, Bahar MW, et al. The N-terminus of the RNA polymerase from infectious pancreatic necrosis virus is the determinant of genome attachment. PLoS Pathog. 2011;7(6):e1002085. | ||
Bahar MW, Sarin LP, Graham SC, et al. Structure of a VP1-VP3 complex suggests how birnaviruses package the VP1 polymerase. J Virol. 2013;87(6):3229–3236. | ||
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. | ||
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. | ||
Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–175. | ||
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. | ||
Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. | ||
Encinar JA, Fernández-Ballester G, Galiano-Ibarra V, Micol V. In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols. Drug Des Devel Ther. 2015;9:5877–5895. | ||
Galiano V, Garcia-Valtanen P, Micol V, Encinar JA. Looking for inhibitors of the dengue virus NS5 RNA-dependent RNA-polymerase using a molecular docking approach. Drug Des Devel Ther. 2016;10:3163–3181. | ||
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460–473. | ||
Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. | ||
Reed LJ, Muench H. A Simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27(3):493–497. | ||
Falco A, Chico V, Marroquí L, Perez L, Coll JM, Estepa A. Expression and antiviral activity of a beta-defensin-like peptide identified in the rainbow trout (Oncorhynchus mykiss) EST sequences. Mol Immunol. 2008;45(3):757–765. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the | ||
Sobhkhez M, Joensen LL, Tollersrud LG, Strandskog G, Thim HL, Jørgensen JB. A conserved inhibitory role of suppressor of cytokine signaling 1 (SOCS1) in salmon antiviral immunity. Dev Comp Immunol. 2017;67:66–76. | ||
Marroquí L, Estepa A, Perez L. Inhibitory effect of mycophenolic acid on the replication of infectious pancreatic necrosis virus and viral hemorrhagic septicemia virus. Antiviral Res. 2008;80(3):332–338. | ||
García I, Galiana A, Falcó A, Estepa A, Perez L. Characterization of an infectious pancreatic necrosis (IPN) virus carrier cell culture with resistance to superinfection with heterologous viruses. Vet Microbiol. 2011;149(1–2):48–55. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Niyomrattanakit P, Chen YL, Dong H, et al. Inhibition of dengue virus polymerase by blocking of the RNA tunnel. J Virol. 2010;84(11):5678–5686. | ||
Yuan S, Chu H, Ye J, et al. Identification of a novel small-molecule compound targeting the influenza A virus polymerase PB1-PB2 interface. Antiviral Res. 2017;137:58–66. |
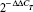 method. Methods. 2001;25(4):402–408.
method. Methods. 2001;25(4):402–408.Supplementary material
 | Figure S1 Molecular structure of compounds selected against the allosteric binding site for HCV NS5B (A) and IPNV VP1 RdRp (B). |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.